
Abstract
We hypothesized that hypoxic preconditioning (PC) modifies the microvasculature in the immature brain and thereby affects the cerebral blood flow (CBF) during a subsequent hypoxic—ischemic (HI) insult. On postnatal day 6 rats were exposed to hypoxia (36°C, 8.0% O2) or normoxia for 3 h. Unilateral HI (unilateral carotid ligation and 8% hypoxia) was induced 24 h later. Cortical CBF was measured with the 14C-iodoantipyrine technique (at the end of HI) or with laser Doppler flowmetry (Perimed PF5001) before and during HI. At 0, 2, 8, and 24 h cerebral cortex was sampled and analyzed with gene arrays (Affymetrix 230 2.0).
Introduction
Consequences of birth asphyxia (hypoxia—ischemia; HI) are associated with neurologic sequelae and life-long disabilities such as cerebral palsy, mental retardation, and epilepsy (Johnston et al, 2002). As a response to HI, energy failure initiates a cascade of deleterious events leading to brain damage. The underlying mechanisms are suggested to involve excitotoxicity, apoptosis, oxidative stress, and inflammation but are, as of yet, not completely understood (Johnston et al, 2002). At present, clinical strategies to treat HI are lacking, and continued research is therefore urgently needed to provide new therapeutic interventions.
Exposure to an interval of sublethal hypoxia renders the immature brain less sensitive in response to a subsequent severe HI insult (Gidday et al, 1994). This phenomenon is termed hypoxic preconditioning (PC), and it induces a robust neuroprotection, which recently has been found to be long-lasting, both regarding histopathological and functional outcome (Gustavsson et al, 2005). Preconditioning initiates immediate stress responses and subsequent alterations in RNA transcription and protein synthesis, which are thought to account for its protective effect, at least in the adult setting (Barone et al, 1998). The basis of these mechanisms is, however, not known.
Even though the vascular response to hypoxia-induced PC in adult animals has not been addressed, tolerance induction by brief ischemia in the adult brain does not alter cerebral blood flow (CBF) during or after ischemia (Matsushima and Hakim, 1995; Chen et al, 1996; Barone et al, 1998). In the immature brain, PC induces vascular genes (Bernaudin et al, 2002). Preconditioning also delays the rate of energy depletion seen during HI (Brucklacher et al, 2002), suggesting an effect either directly on energy metabolism or on CBF. There are no published reports on the effects of PC on CBF in the developing brain.
Furthermore, findings in response to PC suggest that nitric oxide (NO), produced by the endothelial nitric oxide synthase isoform (eNOS), mediates protection in the immature brain, as inhibition of eNOS was shown to block the PC-induced effect (Gidday et al, 1999) whereas specific neural NOS (nNOS) and inducible (iNOS) inhibitors were ineffective. However, there is no information on the effect of NOS inhibitors on CBF in the setting of PC and HI.
Induction of PC hypoxia in the immature brain correlates with expression of transcription factor HIF-1α and promotes expression of target genes such as vascular endothelial growth factor, erythropoietin, glucose transporters, and glycolytic enzymes (Bergeron et al, 2000). Vascular endothelial growth factor is a mediator of angiogenesis (Carmeliet and Jain, 2000) and is involved in the regulation of capillary density in rats exposed to chronic hypoxia (Boero et al, 1999; Pichiule and LaManna, 2002). There is, however, no information on whether such a response is rapid enough to affect capillary density and CBF as soon as 24 h after PC.
The purpose of this study was to investigate the vascular response to hypoxic PC by evaluating (1) the effect of PC on CBF during HI using two independent methods of measurement; (2) vascular gene expression at different time points after PC; (3) the effect of NO synthase inhibition on CBF during HI after PC; and (4) structural changes in vascular density 24 h after PC.
Materials and methods
Subjects
Sprague—Dawley rats, obtained from Charles River Laboratories (Sulzfeld, Germany), were either ordered pregnant or bred at Göteborg University local animal care facility EBM (Göteborg, Sweden). Animals were housed in accordance to standard guidelines. Experimental protocols were approved by the Regional Animal Ethical Committee of Göteborg (no. 293-01, 87-04, 17-05). For the iodo-[14C]-antipyrine (IAP) experiments dated pregnant Sprague—Dawley rats were purchased from Charles River Laboratories (Wilmington, MA, USA); experiments were approved by the Institutional Animal Care and Use Committee of the Hershey Medical Center/Pennsylvania State University.
Hypoxic Preconditioning
On PND6 rats were subjected to a PC interval of hypoxia. Animals were placed in a chamber perfused with a gas mixture of 8.0% oxygen in nitrogen for 3 h at 36°C. An additional group of rats, referred to as sham controls, were simultaneously exposed to normoxia (3 h, 36°C).
Hypoxic—Ischemic Insult
Twenty-four hours after completion of hypoxic PC, animals were subjected to HI. In brief, the pups were anesthetized with enflurane (3% for induction, 1.5% for maintenance) in a mixture of nitrous oxide and oxygen (1:1) and the left common carotid artery was ligated (6-0 suture) and cut. After the surgical procedure pups were returned to the dam for 1 h of recovery. Hypoxic—ischemia was induced by exposure to a humidified gas mixture (7.7% oxygen in nitrogen) at 36°C for 1 h 15 mins. The pups were allowed to adjust to the temperature and humidity of the chamber 10 mins before and 10 mins after hypoxia. After hypoxic exposure, the pups were returned to their dams. Owing to concerns that anesthesia during HI would sensitize the animal, the HI insult was reduced to 1 h in the Doppler flow experiments (see below). The reduction was made as a precaution to ensure survival for 1 week postinsult, for neuropathological scoring, and verification of differences in outcome between groups.
Neuropathological Analysis
On PND14, animals were deeply anesthetized by intraperitoneal injection of Pentothal sodium (50 mg/mL) and then intracardially perfusion-fixed with 0.9% NaCl followed by 5% buffered formaldehyde (Histofix, Histolab, Göteborg, Sweden). The brains were macroscopically scored for neuropathological outcome (Gustavsson et al, 2005).
Cerebral Blood Flow Measurement (IAP)
Cerebral blood flow was measured at the end of HI using a modification of the indicator fractionation technique, as previously described (Vannucci and Vannucci, 1997). Briefly, at the end of ischemia (90 mins) rat pups were injected subcutaneously with 5 μCi of iodo-[14C]-antipyrine (DuPont NEN, Boston, MA, USA). At 1 min after injection pups were decapitated and 10 μl of arterialized blood was solubilized overnight and then analyzed by liquid scintillation counting. The brain of each pup was removed and a sample (approximately 50 mg) of the ipsilateral hemisphere in the distribution of the middle cerebral artery was weighed, solubilized overnight, and analyzed by liquid scintillation counting. Cerebral blood flow was calculated from the concentration of the 14C-IAP tracer in the blood and brain tissue specimen at 1 min, assuming linearity in the input function from 0 to 1. The brain:blood partition coefficient (μ) was assumed to be equal to 0.94 (Lyons et al, 1987).
Cerebral Blood Flow Measurement (Laser Doppler Flowmetry)
The PC group (n = 18) was exposed to hypoxic PC and control rats to sham procedure (n = 14) on PND6. The following day, rats were subjected to HI but with some modification to allow CBF measurements. The carotid artery was ligated in anesthetized (enflurane; 3% for induction, 1.5% for maintenance) animals followed by a scalp incision to allow application of a blood flow probe directly on to the skull surface of the ipsilateral hemisphere (3 mm anterior to lambda, 3 mm lateral to the midline). Dental cement (Dentanol plus, Agntho's AB, Lidingö, Sweden) was used to attach the probe and as glue for wound closing. Administration of α-chloralose (VWR International AB, Stockholm, Sweden) intraperitoneally (6 mg/kg) was used for maintenance of anesthesia. At 30 mins after administration of α-chloralose, CBF measurements were initiated using laser Doppler flowmetry (Perimed PF5001, Perimed AB, Stockholm, Sweden) with the rat placed in a chamber kept at 36°C. Cerebral blood flow was measured during a 30 mins air preexposure period, followed by 1 h to 1 h 15 mins of hypoxia (7.7% oxygen in nitrogen) and during 30 mins of recovery. After completion of recording, the rat was allowed to wake up from anesthesia and was then returned to the dam. These rats were killed 1 week after HI and the brains were neuropathologically scored to confirm that PC provided protection in animals subjected to anesthesia during HI.
Gene Expression Analysis
The procedure of hypoxic PC was performed as previously described. Groups consisted of PC animals (n = 20) and sham controls (n = 20), drawn from 10 litters. Additional PC (n = 44) and sham controls (n = 36) from these litters were used for evaluation of neuroprotection, described below. For RNA isolation, pups were killed by decapitation at 0, 2, 8, and 24 h after the PC or the normoxic sham exposure. Five brains per group were collected at each time point. Brains were dissected and the cerebral cortex of both hemispheres was rapidly frozen on powdered dry ice. Total RNA was isolated according to the manufacturer's instructions (Qiagen, Hilden, Germany). The RNA was spectrophotometrically quantified at 260 nm and stored at −80°C.
Microarray analysis was performed at the NINDS consortium for microarray analysis (http://arrayconsortium.tgen.org/np2/public/microarrayResources.jsp). We used the Affymetrix 230 2.0 array, which comprises more than 31,000 probe sets, analyzing over 30,000 transcripts and variants from 28,000 well-substantiated rat genes. Further probe analysis was performed on Affymetrix. CEL files using the robust multi-array average algorithm (Bolstad et al, 2003), which performs three distinct operations: global background normalization, across array normalization, and log2 transformation of perfect match values (http://stat-www.berkeley.edu/users/bolstad/RMAExpress/RMAExpress.html). The robust multi-array average analysis, data management, statistical analysis, and gene ontology was performed using the web based GeneSifter™ software (http://login.genesifter.net/).
To verify results of the microarray analysis, real-time RT-PCR (LightCycler™, Roche Diagnostics, Mannheim, Germany) was used to examine mRNA expression for angiopoietin 2 (Angpt2), at 0 and 8 h, which was significantly regulated according to the microarray analysis. First strand cDNA was synthesized from the same total RNA used for the microarray preparations with the Superscript RNase H− reverse transcriptase kit (Life Technologies, Inc.) using random hexamer primers as previously described (Blomgren et al, 1999). Each 20 μl PCR contained 1/400 of the cDNA synthesis, 3 μmol/L glyceraldehyde-3-phosphate, or 4 μmol/L (Angpt2) MgCl2, 0.5 μmol/L forward and reverse primers and 2 μl Light-cycler Faststart DNA Master Cyber Green SYBR I (Roche Diagnostics). Oligonucleotide primers were designed for Angpt2 and glyceraldehyde-3-phosphate (CyberGene AB, Huddinge, Stockholm) with the following sequences and annealing temperatures/elongation times:
To verify the neuroprotective effect of hypoxic PC, littermates of animals used for the microarray analysis (PC n = 44; sham n = 36) were subjected to HI on PND7. Seven days post-insult animals were killed and the neuropathological score of brains were determined.
Cerebral Blood Flow Measurement and Nitric Oxide Synthase Inhibition
Animals of PND6 were either treated with the nonselective NOS inhibitor N-ω-nitro-
Additional experiments consisted of administration of
Measurement of Microvascular Density
Tissue samples were prepared from PC animals (n = 12) versus sham controls (n = 12) 24 h after hypoxic PC. For immunostaining, animals were deeply anesthetized by intraperitoneal injection of Pentothal sodium (50 mg/mL) and then intracardially perfusion-fixed with 0.9% NaCl followed by 5% buffered formaldehyde (Histofix, Histolab, Göteborg, Sweden). The brains were removed and postfixed overnight. Further, the brains were processed by graded dehydration using ethanol and xylene, embedded in paraffin and thereafter sectioned in 10 μm-coronal sections. Every 50th section was collected, yielding an average of nine sections per brain, and microvessels were visualized by immunolabeling against von Willebrand factor according to the following protocol. Antigen recovery was performed by boiling the sections in Target Retrieval Solution (Dako Cytomation, Glostrup, Denmark) for 10 mins and blocked with 4% goat serum in phosphate-buffered saline for 30 mins at room temperature. The sections were incubated with primary antibody (polyclonal rabbit anti-human von Willebrand factor, 1:400, Dako Cytomation) in blocking solution and the secondary biotin-labeled antibody (goatanti-rabbit, 1:250) for 1 h each at room temperature. Visualization was performed using Vectastain ABC Elite (Vector Laboratories) and 3,3-diaminobenzidine (DAB, 0.5 mg/mL) enhanced with ammonium nickel sulfate (15 mg/mL).
To evaluate changes in cortical microvasculature, we used a systematic random sampling procedure to apply a stereologic probe (area fraction fractionator) that estimates the fractional area and volume occupied by microvessels in cerebral cortex (Stereo Investigator 6, MicroBrightField Inc., Williston VT, USA). Cortical regions including white matter tracts, neocortex and archicortex were systematically sampled in one hemisphere at each level.
Statistics
The Mann—Whitney U-test was performed for nonparametric comparisons of brain damage and for microvascular volume fraction. Analysis of variance, followed by Fischer's PLSD post hoc analysis, was used for cerebral blood flow measurements. P-values <0.05 were considered statistically significant. Values are expressed as mean ± standard error of mean (s.e.m.). Statistical analyses were performed using Statview Version 5.0.1 (SAS Institute Inc., Cary, NC, USA).
The changes in gene expression over the 24 h experimental period (0, 2, 8, and 24 h) in the sham controls were analyzed using analysis of variance (with 0 h set as control) with Benjamini and Hochberg correction with false detection rate set at 0.05 and a threshold of 1.2-fold change for inclusion. In addition, a pairwise comparison of the 0 and 24 h time points in sham controls was carried out using a t-test and Benjamini and Hochberg false detection rate set at 0.05. Because there were significant changes in gene expression in sham controls over this 24 h period, the effect of PC gene expression was examined separately at each time point. Alterations of gene expression in response to PC was evaluated versus the time-matched sham controls at 0, 2, 8, and 24 h, t-test analysis with Benjamini and Hochberg correction (false detection rate: 0.05) and a threshold of 1.2-fold change for inclusion.
Results
Hypoxic Preconditioning Alters Cerebral Blood Flow During Hypoxia—Ischemia
It has previously been shown that on the induction of hypoxia after unilateral carotid ligation, the CBF of the ipsilateral hemisphere is reduced by 40% to 60% compared with the hemisphere contralateral to the ligation (Vannucci et al, 1988). Our measurements demonstrated that the decrease in CBF at the end of HI was reduced after hypoxic PC. Baseline CBF measured with IAP was 34.8 ± 4.0 ml/100g/min. At the end of HI CBF was reduced by 45.2% ± 3.5% in the PC group versus 62.0% ± 6.2% in controls (P < 0.01). These results were consistent with laser Doppler measurements demonstrating a decrease at the end of HI by 41% ± 5% in the PC group compared with 64% ± 4% in the control group (P < 0.01) (Figure 1). This latter group of animals was allowed to recover after HI and was later subjected to evaluation of neuropathology one week postinsult. Results confirmed the neuroprotective effect of hypoxic PC: the macroscopic brain injury score was 1.4 ± 0.3 in the PC group and 3.3 ± 0.2 in controls (P < 0.001).
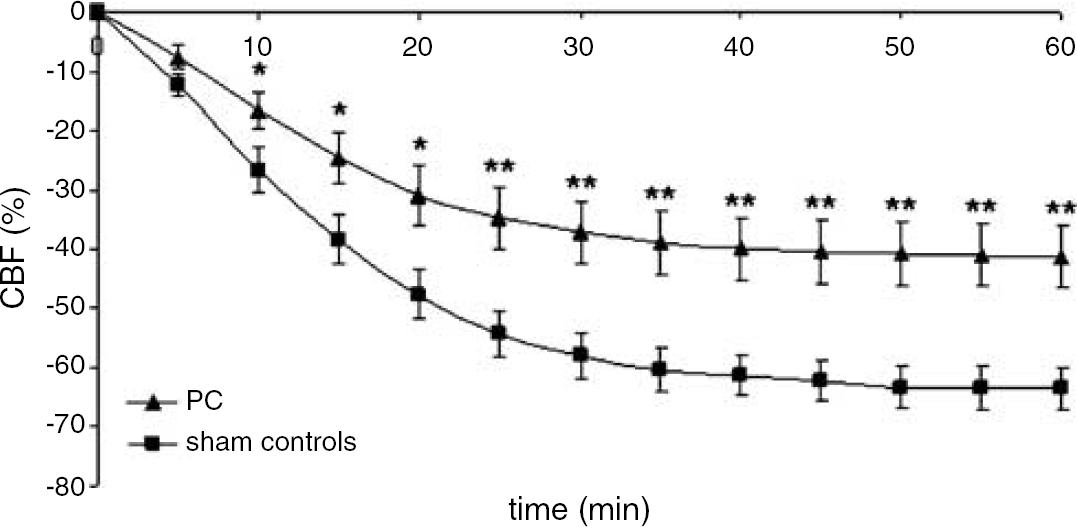
Laser Doppler measurements of cortical CBF (ipsilateral hemisphere) during HI in the PC and sham control groups. Values are expressed as percentage reduction in CBF compared with that measured during normoxic conditions before hypoxia, **P < 0.01 and *P < 0.05 for PC versus non-PC rats.
Changes in Global Expression of Vascular Related Genes after Hypoxic Preconditioning
Analysis of alterations in global gene expression revealed that 906 genes were significantly changed in PC versus sham controls at 0 h, 691 genes at 2 h, 389 genes at 8 h, and 114 genes at 24 h (t-test, Benjamini and Hochberg false detection rate 0.05, ≥1.2 fold change). Genes that may be related to regulation of angiogenesis and vasoregulation after PC were identified (Figure 2 and Table 1). The change in mRNA expression for Angpt2 detected with microarray at 0 and 8 h after PC was confirmed with RT-PCR (Figure 3). Results showed a significant upregulation at 0 h (P < 0.0001) and downregulation at 8 h (P < 0.01) compared with sham controls, and the fold change was similar when RT-PCR and microarray analyses were compared. To verify that PC was neuroprotective in this experiment, macroscopical evaluation of brains was carried out in littermates of those used for RNA isolation. These animals were subjected to PC or sham hypoxia on PND6 and HI on PND7; injury was evaluated on PND14. A significant reduction of hypoxic—ischemic brain injury was observed in animals exposed to PC hypoxia (0.8 ± 0.2, n = 44) in comparison with sham animals (2.7 ± 0.2, n = 36, P < 0.0001).
Vascular-related genes expressed in the immature brain after hypoxic preconditioning
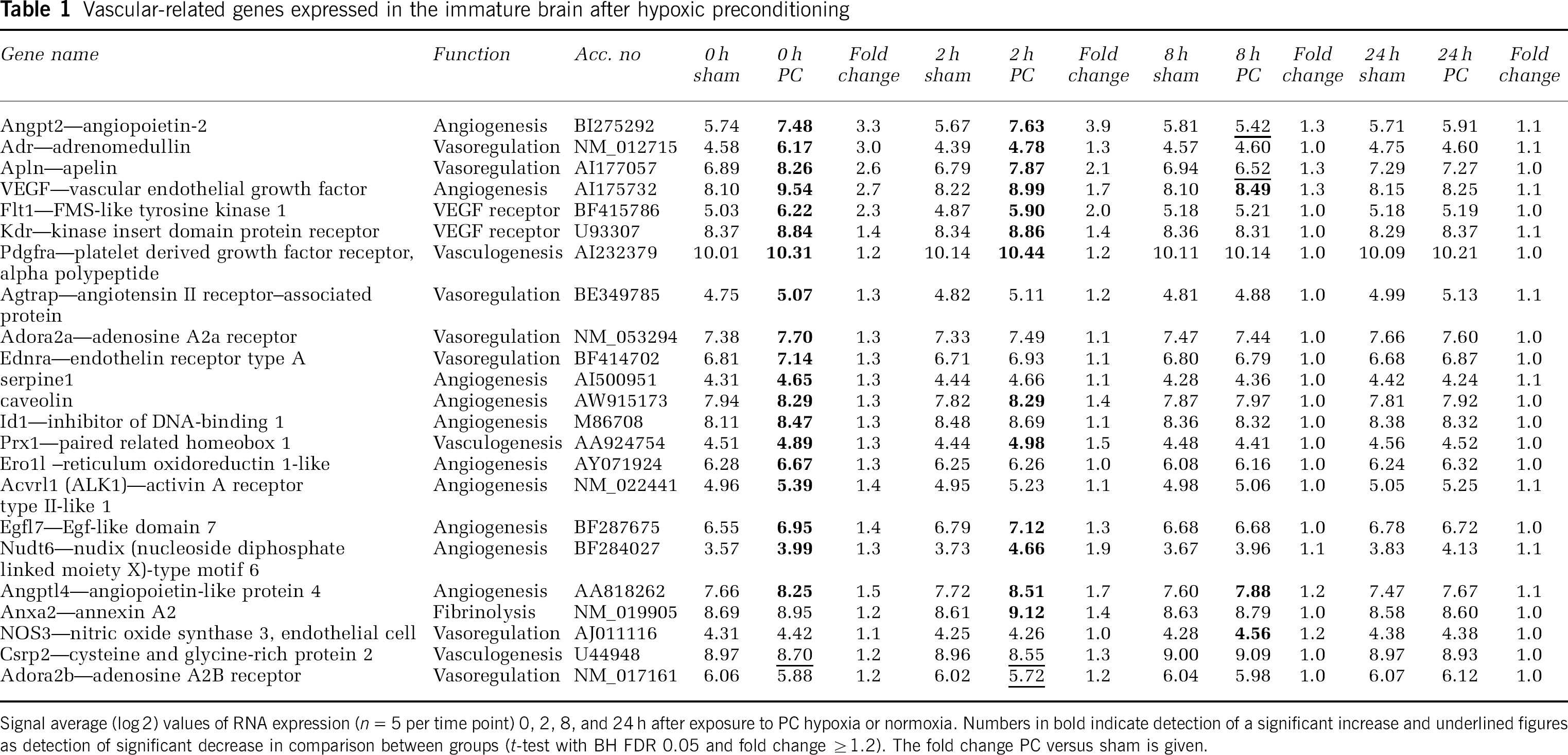
Signal average (log 2) values of RNA expression (n = 5 per time point) 0, 2, 8, and 24 h after exposure to PC hypoxia or normoxia. Numbers in B indicate detection of a significant increase and underlined figures as detection of significant decrease in comparison between groups (t-test with BH FDR 0.05 and fold change ≥1.2). The fold change PC versus sham is given.

Color-coded expression profiles of vascular-related genes at 0, 2, 8, and 24 h after exposure to PC hypoxia or normoxia (sham). Green indicates downregulation and red upregulation in PC versus time-matched controls. The color intensity reflects the difference in expression between PC and control for each gene (see Table 1 for comparison of expression among different genes).
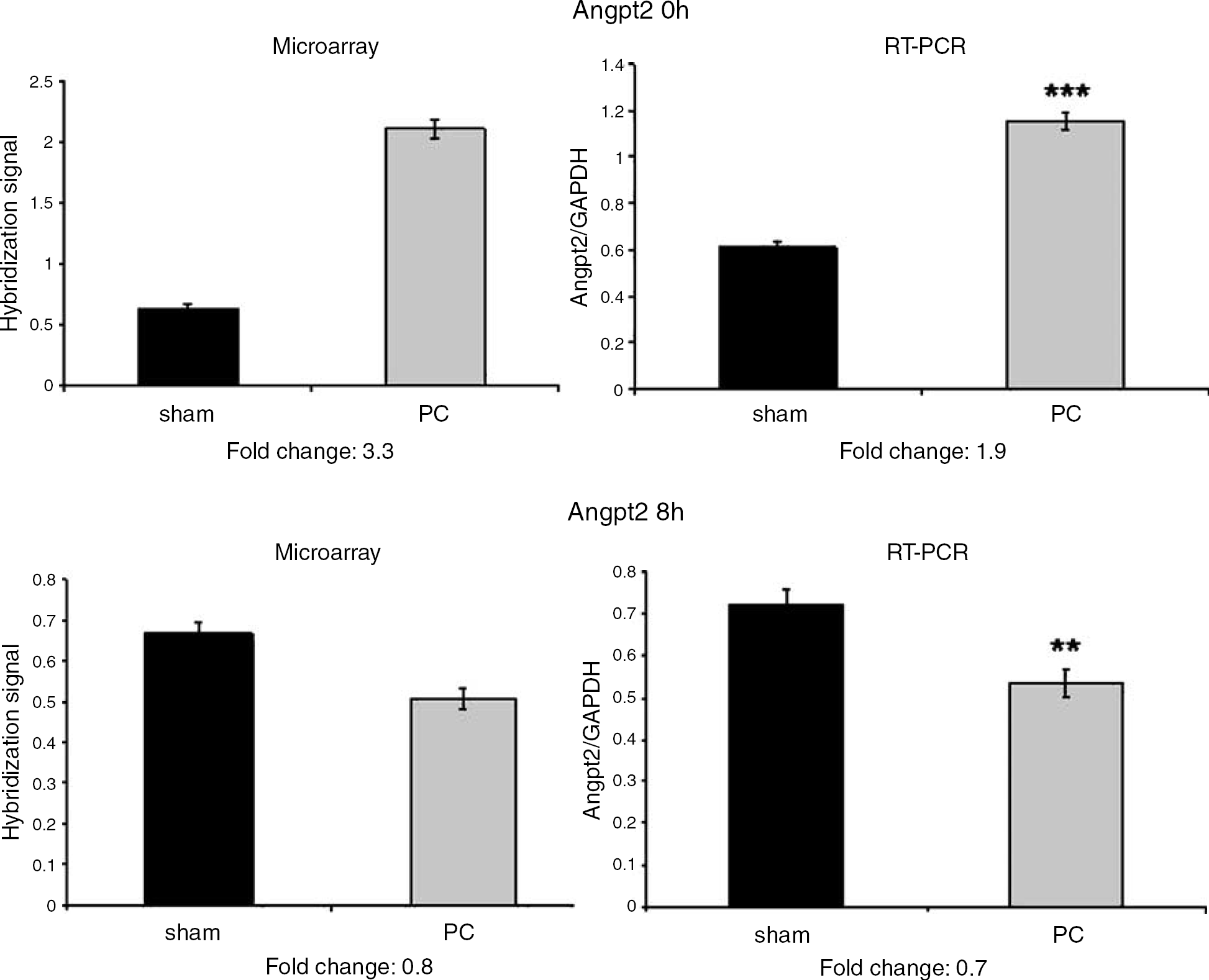
Expression of Angpt2 at 0 and 8 h using microarray analysis (Affymetrix 230 2.0) and RT-PCR. The early upregulation (0 h) and subsequent downregulation (8 h) of Angpt2 as detected with the microarray technique (see Figure 3) was confirmed by RT-PCR.
Vascular Response to Hypoxic Preconditioning was Independent of Nitric Oxide Production
Because previous findings indicated that the PC effect in the immature brain is NO-dependent (Gidday et al, 1999) and that eNOS (= NOS3) was significantly upregulated in response to hypoxic PC we next evaluated whether administration of a nonselective NOS-inhibitor (
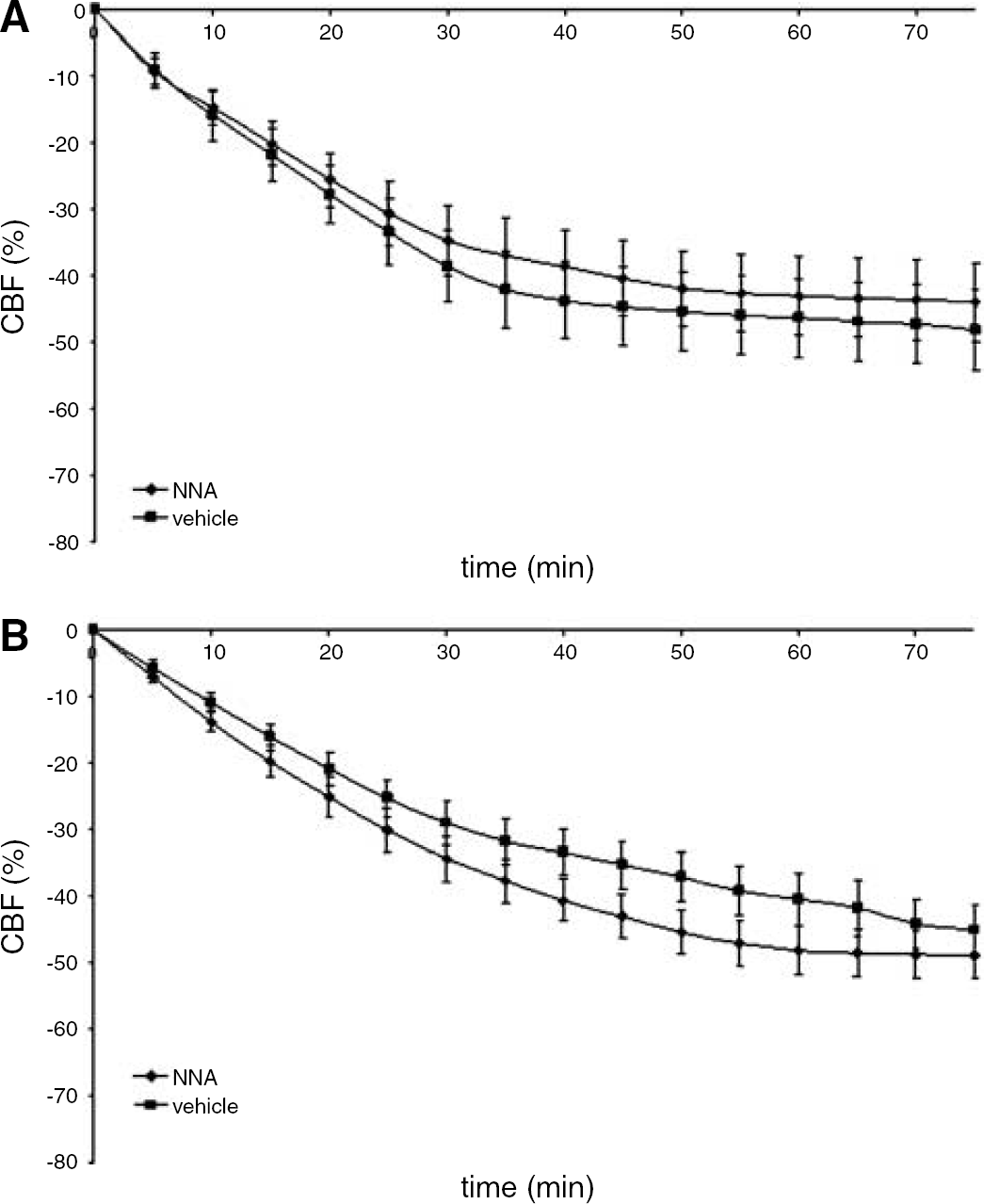
Laser Doppler measurement of cortical CBF (ipsilateral hemisphere) during HI in preconditioned animals treated
Structural Changes in the Cerebral Vascular bed After Hypoxic Preconditioning
Alterations in expression of genes related to angiogenesis after PC implies that modification of the cerebral vascular structure is likely to occur and that some of the improvement in CBF might be explained by these changes. Stereological analysis of microvascular volume fraction (the ratio of estimated capillary volume to the total volume) revealed a significant 18% difference (P < 0.05) between the PC groups (2.6% ± 0.1%) and controls (2.2% ± 0.1%) 24 h after PC (Figures 5 and 6).
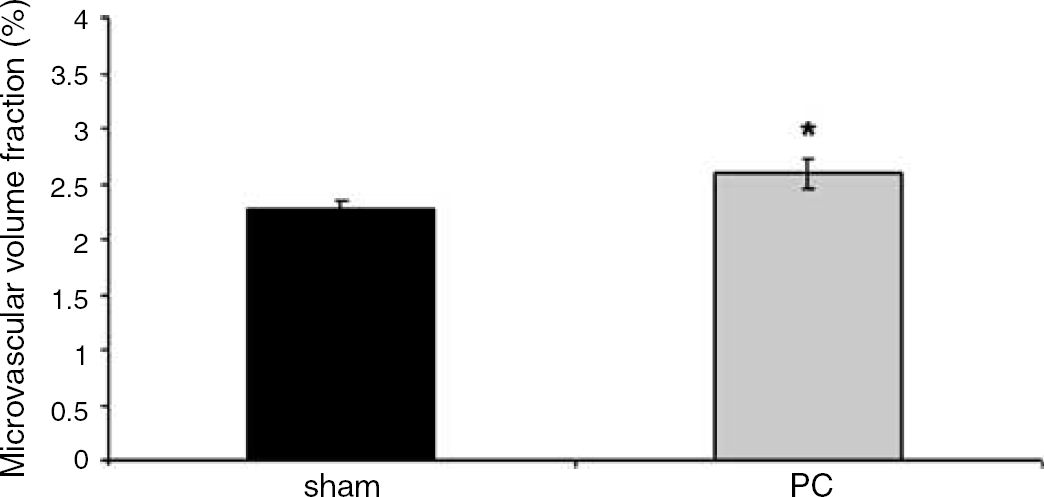
Microvascular volume fraction (ratio of estimated volume of capillaries and the total volume of cortical tissue analyzed) in PC treated animals and sham controls, *P < 0.05.
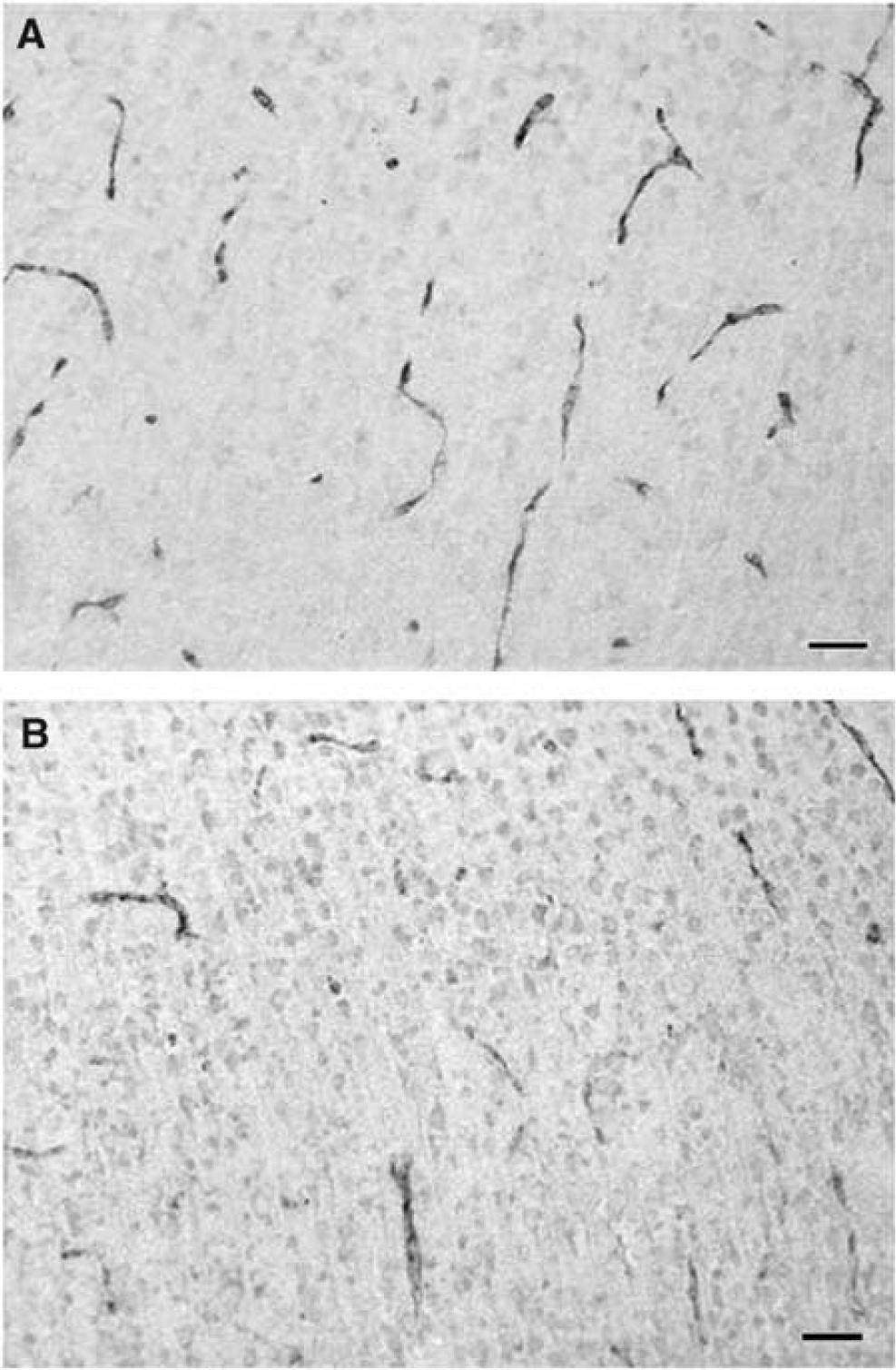
Microvessels in temporal cortex, immunolabeled against von Willebrand factor, 24 h after (
Discussion
Understanding the mechanisms responsible for hypoxic PC in the neonate is important because of the insights they may provide into endogenous neuroprotective pathways that could be targets for therapy. Our results highlight the probable role of previously unrecognized vascular mechanisms in this process. The finding that prior exposure to hypoxic PC attenuates the depth of ischemia associated with HI has not been reported previously, and this mechanism may account in part for the reduction in brain injury. Our results indicate that the relative preservation of blood flow is not dependent on the production of NO, but is accompanied by induction of numerous genes associated with vasoregulation and angiogenesis as well as a small but significant increase in vascular density 24 h post-PC exposure.
The detrimental cascade of HI injury in the immature brain is primarily related to insufficiency in the supply of oxygen and glucose resulting in failure in maintaining cellular metabolism and cellular integrity (Johnston et al, 2002). In the neonatal rodent model of HI, the distribution and extent of ischemic brain tissue injury is closely related to the regional reduction of CBF (Vannucci et al, 1988). Our results indicate that hypoxic PC affects the vascular response to HI, resulting in a less pronounced decrease of CBF (which may provide partial preservation of high energy phosphates during HI) and a subsequent reduction in brain damage. Such an interpretation is supported by a slower energy loss during HI after PC, although this finding also could be explained by increased tissue glycogen levels (Brucklacher et al, 2002). We demonstrated that the CBF value at the end of HI (as measured with the IAP technique) remained higher after PC than in non-PC sham animals and we confirmed this observation using the laser Doppler technique, which provided continuous data in the same animals over time. The laser Doppler data demonstrated that after PC the CBF was higher during the entire HI period. This finding was strengthened by follow-up neuropathological data in the same animals used for laser Doppler flow studies.
It has previously been shown that development of PC in the neonatal model involves production of NO, because a nonspecific NOS inhibitor administered directly before PC has been reported to eliminate the neuroprotective effect. As specific blockage of iNOS or nNOS had no effect on PC and it was hypothesized that PC protection may be mediated by eNOS (Gidday et al, 1999). On the basis of these earlier results and our current observation that the gene for eNOS is upregulated at 8 h after PC (Figure 2), we hypothesized that PC induces an increased eNOS-mediated vasodilation during the subsequent HI, which could explain the attenuated decrease of CBF during the insult after PC. However, we found that the same dosage of
It still remains to be clarified how hypoxic PC modifies the vascular bed and the CBF response to an ensuing HI insult. It is noteworthy that the small but significant change in vascular density that we observed in neonates occurred far more rapidly within 24 h than a similar response in adult models of chronic hypoxia. In the adult brain, exposure to chronic hypoxia stimulates angiogenesis and leads to an increase in capillary density after 1 week, resulting in decreased intercapillary distance and, consequently, to decreased diffusion distance (Lauro and LaManna, 1997). However, the number of newly formed vessels was incresed at the border of the infarction already 48 h after acute ischemia (Marti et al, 2000) and the angiogenic response to hypoxia has been shown to be remarkably fast in immature tissue in some situations (Nanka et al, 2006). This information in combination with the rapid induction of vascular genes (Table 1) support that the 18% increase in brain microvessel density we see at 24 h is real. It is likely that the angiogenic response is only the beginning of a more powerful regeneration of vessels that will continue during days and weeks of recovery after HI, and that may be more pronounced after PC. Such a scenario is supported by a recent study in the heart showing that hypoxic PC 24 h before a period of ischemia triggered rapid angiogenesis and improved contractile function over a period of weeks (Sasaki et al, 2002). The enhanced density may contribute to alterations in the CBF (Klein et al, 1986) improving tissue oxygenation during HI.
The availability of the rat genome microarray containing 30,000 transcripts and variants from 28,000 well-substantiated rat genes allowed us to monitor changes in substantially more genes than were reported by Sharp and Bernaudin (2004). Our results generally support the concept that HIF-1α is induced by PC (Bergeron et al, 2000) and that this transcription factor, in turn, upregulate multiple genes related to angiogenesis, trophic support, energy metabolism, and cell survival (Sharp and Bernaudin, 2004). However, we presently found that multiple vascular-related genes were induced after hypoxic PC and, interestingly, gene expression was already significantly altered at 0 h after a 3 h exposure to hypoxic PC. In addition to upregulation of vascular endothelial growth factor, adrenomedullin, and caveolin, which were reported by Bernaudin et al (2002) after PC in the neonatal rat, we found regulatory changes in many other vascular-related genes that have not been reported previously. Several of the genes that we found to be regulated after PC, for example, vascular endothelial growth factor and Angpt2, are mediators of vascular adaptation to chronic hypoxia in the adult setting (Pichiule and LaManna, 2002). However, even these genes may have protective functions in neurons as well as the vasculature, as has been reported for vascular endothelial growth factor (Kaya et al, 2005). Neuronal (Sugawara et al, 2001), astroglial, and microglial (Perez-Pinzon et al, 1999) elements have all been suggested to participate in the development of tissue resistance after PC (Perez-Pinzon et al, 1999).
Other genes related to angiogenesis are induced in these experiments include serpine1 (Pepper, 2001), caveolin (Griffoni et al, 2000), Id1 (Lyden et al, 1999), Ero1l (May et al, 2005), Acvrl1 (ALK1) (Seki et al, 2003), Egfl7 (Campagnolo et al, 2005), NUDT6 (Presta et al, 2005), and Angptl4 (Le Jan et al, 2003). In addition, Pdgra (Heldin and Westermark, 1999), Prx1 (Bergwerff et al, 1998), Crsp2 (Louis et al, 1997), and Anxa2 (Rescher and Gerke, 2004) were upregulated. Changes in the expression of vasoregulatory genes induced after PC include adrenomedullin (Lang et al, 1997), apelin (Lee et al, 2000), Agtrap (Daviet et al, 1999), Adora A2a and Adora A2b (Coney and Marshall, 1998), Ednra (Arai et al, 1990), and NOS3 (Griffith and Stuehr, 1995), which also could be important to explain the altered CBF response during HI. Hence, it was recently reported that pharmacological administration of adrenomedullin or transgenic overexpression of the adrenomedullin gene had a strong angiogenic effect after experimental stroke, which was associated with reduced brain injury and improved neuromotor function (Miyashita et al, 2006) in the adult brain. The role of the above vascular genes and their role for angiogenesis and neurogenesis in the setting of the vascular niche in immature CNS has to be addressed in future studies.
It is important to point out that extracerebral events could partly explain the difference in CBF response found after PC. Hence, hypoxic PC has been shown to improve pulmonary gas exchange in adult mice (Zhang et al, 2004) and hypoxic PC seems to enhance myocardial contractile function in adult rats (Sasaki et al, 2002), which certainly could increase cerebral perfusion pressure and affect the CBF response also in our immature model of PC. There is no information published on the effect of blood pressure, cardiac output, blood glucose, or blood gases in this PC model, which has to be addressed in the future.
Based on this study, which monitored changes in a nearly complete set of genes from the rat genome, we propose that PC also alters the expression of a large and complex network of vascular and angiogenic genes. It seems likely that these changes induce angiogenesis and expression of vasoregulatory mediators that improve vascular ability to maintain CBF during a subsequent HI challenge, thereby decreasing the brain's vulnerability. These vascular changes are likely to work in parallel with additional changes in gene expression or cellular metabolism within neurons and glia to provide the protective response.
Footnotes
Acknowledgements
The authors thank Georg Kuhn for assistance with stereological analysis and Anna-Lena Leverin and Eva Cambert for excellent technical support.