
Abstract
A brief period of sublethal cerebral ischemia, followed by several days of recovery, renders the brain resistant to a subsequent lethal ischemic insult, a phenomenon termed ischemic preconditioning or tolerance. Ischemic tolerance was established in the rat two-vessel occlusion model of ischemia, induced by occlusion of both carotid arteries in combination with hypotension. Ischemic preconditioning (3 minutes) provided maximal neuroprotection when induced 2 days prior to a lethal ischemic insult of 9-minute duration. Neuroprotection persisted for at least 8 weeks. Since neurotransmission has been implicated in ischemic cell death, the effect of ischemic preconditioning on tyrosine phosphorylation of proteins and on the levels of glutamate receptor subunits in hippocampus and neocortex was studied. Regional levels of tyrosine phosphorylation of proteins in general and the N-methyl-
If the brain is subjected to a transient period of sublethal global or focal ischemia (Kitagawa et al., 1990), hypoxia (Gorgias et al., 1996), seizures (Sasahira et al., 1995), or spreading depression (Kawahara et al., 1995; Kobayashi et al., 1995), it becomes resistant to damage induced by a subsequent period of lethal stress. Ischemic preconditioning has been demonstrated in the gerbil (Kato et al., 1991; Kitagawa et al., 1991) and rat (Liu et al., 1992) brain but has also been observed in other tissues such as the heart (Baxter et al., 1994; Kuzuya et al., 1993; Lawson and Downey, 1993). In the nonconditioned brain, transient cerebral ischemia causes selective neuronal damage particularly to the hippocampal CA1 region in the gerbil (Ito et al., 1975), rat (Pulsinelli et al., 1982), and human (Petito et al., 1987). The degeneration of the pyramidal neurons in CA1 develops slowly during reperfusion following ischemia and is evidenced by light microscopy at 2 to 3 days of recovery (Kirino, 1982; Pulsinelli et al., 1982). In these ischemia models, an episode of preconditioning ischemia of 2 to 5 minutes in duration and with an interval of 2 to 3 days between the sublethal and lethal ischemic insults provides a maximal neuroprotective effect.
The cellular mechanism of tolerance induction is still poorly understood. Activation of adenosine receptor-coupled ATP-dependent potassium channels (Heurteaux et al., 1995) or N-methyl-
Signal transduction in the brain is severely affected by cerebral ischemia (Wieloch and Kamme, 1998), also affecting tyrosine phosphorylation of cellular proteins (Hu and Wieloch, 1994). Since cell signaling is tightly linked to cell survival (Anderson, 1997), a study on the effect of ischemic preconditioning on cellular signal transduction is warranted.
The aim of this study was to establish the model of ischemic tolerance in the two-vessel occlusion model of cerebral ischemia (Smith et al., 1984) and then to study the effect of ischemic preconditioning on cell signaling after the second ischemic insult. For that purpose, we investigated the tyrosine phosphorylation of proteins as a measure of activation of cell signaling, focusing in particular on the phosphorylation of the NMDA receptors. These results were presented previously in abstract form (Wieloch and Shamloo, 1996).
MATERIALS AND METHODS
Experimental groups
The experimental paradigm for the histopathologic studies is indicated in Fig. 1A and that for the western analysis in Fig. 1B. For histology, 13 groups of animals were assigned to either 1 or 8 weeks of recovery. Eight groups of animals were included in the western analysis, in which the changes in signal transduction and glutamate receptor levels after preconditioned ischemia were studied in the synaptosomal fraction. Three groups of animals were used to verify the signal transduction changes observed in the synaptosomal fraction by western blotting in the whole CA1 and neocortex (see Table 1). Nonconditioned animals were subjected to sham surgery followed by 2 days of recovery and then 6, 9, or 15 minutes of ischemia. Preconditioned rats were subjected to 3 minutes of ischemia and 2, 4, 7, or 14 days of recovery before being subjected to 6, 9, or 15 minutes of ischemia. In both nonconditioned and preconditioned groups, a recovery time of 0, 30 min, 24 hours, 1 week, or 8 weeks was allowed prior to killing. Animals in the sham-operated and 3-minute ischemia groups were allowed 2 days and 1 week of recovery, respectively.
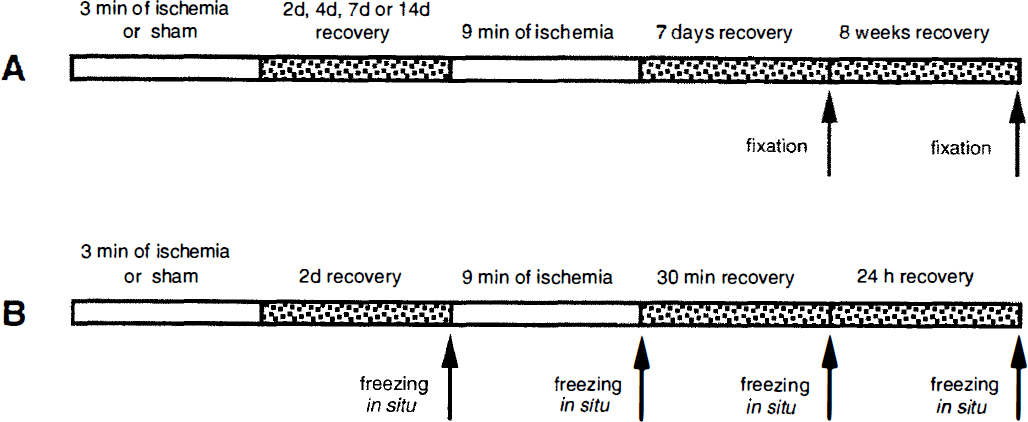
The experimental protocols for the histopathologic study
The number of animals in the different groups of the study
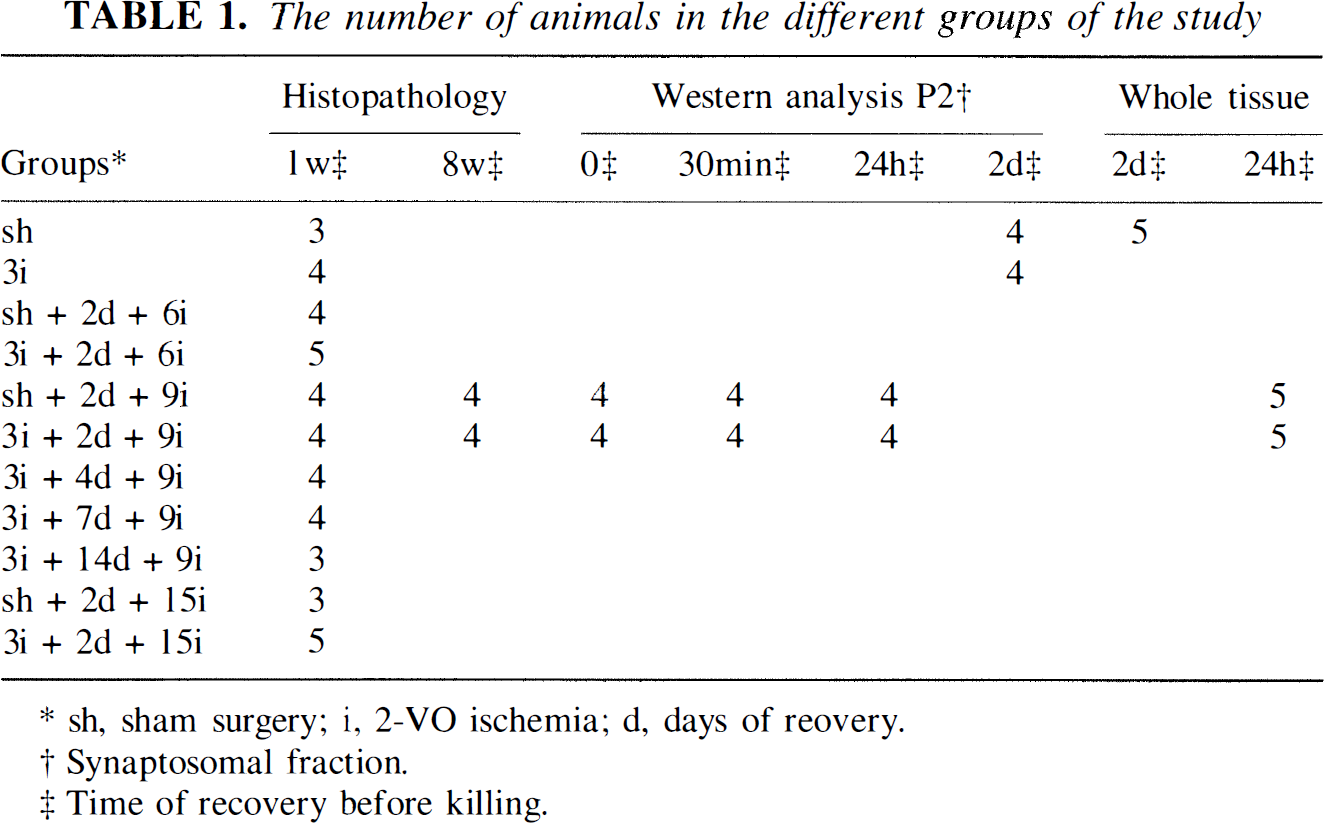
sh, sham surgery; i, 2-VO ischemia; d, days of reovery.
Synaptosomal fraction.
Time of recovery before killing.
Surgical procedures
All animal experiments were approved by the ethical committee at Lund University. Male Wistar rats, weighing 300 to 340 g (Møllegaard's Breeding Center, Copenhagen, Denmark), were used. The animals were fasted overnight with access to water before surgery. We employed the two-vessel occlusion model of global cerebral ischemia in our experiments (Smith et al., 1984). Anesthesia was induced with 3% halothane in N2O/O2 (70:30). The animals were intubated and connected to a respirator, and the halothane concentration reduced to 1.2 to 1.5%. A tail artery and a tail vein were cannulated for blood pressure recordings, blood sampling, and drug infusions. To allow rapid withdrawal of blood for induction of ischemia, a soft Silastic catheter was inserted into the inferior caval vein through the right jugular vein. The common carotid arteries were isolated and encircled with loose ligatures. Electrodes for recording of bipolar EEG were inserted into the temporal muscles, and EEG activity was recorded before, during, and after ischemia. After surgery, the halothane was decreased to 0.3 to 0.5%, and the ventilation and O2 supply were adjusted to give an arterial P
Animals for histopathologic analyses were reanesthetized with 3.5% halothane, tracheotomized, artificially ventilated at either 7 days or 8 weeks of recovery, and perfusion-fixed with 0.9% NaCl followed by phosphate-buffered 4% formaldehyde. The brains were allowed to postfix in situ for 1 hour and were then removed, dehydrated, and embedded in paraffin. The animals to be used for western analysis, destined to be killed immediately at the end of ischemia (0 minutes) or after 30 minutes of recirculation, remained on halothane anesthesia, and the brain was frozen in situ with liquid nitrogen. The animals killed at 24 hours of recovery after lethal ischemia and 2 days of recovery after sublethal ischemia or sham surgery were reanesthetized in a similar way as described above, and the brains were frozen in situ with liquid nitrogen. The neocortex, the CA1 region, and the CA3/dentate gyrus were dissected out at −17°C and stored at −80°C until analysis. The animals used for western analysis of whole-tissue homogenate were treated in similar way.
Postischemic temperature measurements
After the second ischemic episode, the animals were maintained in a 12: 12-hour light/dark cycle, with the lights off from 6 p.m. to 6 am. Rectal core temperature was measured with a digital thermometer (Type CTD 85, Ellab, Copenhagen, Denmark) at 8
Histopathologic procedures
The paraffin-embedded brains were sectioned at 5 µm and stained with a combination of celestine blue and acid fuchsin (Auer et al., 1984). Brain damage was quantified at a coronal level ~3.8 to 4.0 mm caudal to bregma by visual counting (Auer et al., 1984) of intact violet-stained neurons in a blinded manner. The number of undamaged neurons from three defined fields, 400 µm in length, of the CA1 subsector of the dorsal hippocampus (Nellgard and Wieloch, 1992), the medial, the middle, and the lateral CA1 (at the border to CA3), was calculated in both hemispheres. The results are presented as undamaged neurons as percentage of the sham-operated group value. Damage in temporal neocortex, CA3, and hilus of dentate gyrus is presented as number of damaged, acidophilic neurons. The damage in striatum was quantified at a magnification of ×400 by counting the number of undamaged neurons in two defined microscopic fields, 400 µm in diameter, in the dorsal and the lateral striatum in both hemispheres. The results are presented as cell loss in as percentage of the sham values.
Subcellular fractionation
Frozen tissue was homogenized with a Teflon-glass homogenizer in 1:10 (mg/µL) homogenization buffer (HB) consisting of 50 mmol/L 3-[N-morpholino]propanesulfonic acid (pH 7.6), 2 mmol/L dithiothreitol, 0.1 mmol/L sodium orthovanadate, 3 mmol/L ethyleneglycol bis(aminoethylether)tetraacetate, 20 µg/mL leupeptin, 5 µg/mL aprotinin, 10 µg/mL pepstatin, and 0.32 mol/L sucrose, and spun at 800 g for 10 minutes at 4°C. The supernatant was spun at 9,200 g for 15 minutes at 4°C in a SE12 rotor (Sorval). The pellet, the synaptosomal fraction (P2), was suspended in homogenization buffer containing 0.1% Triton X-100 (HBT). The protein concentration was determined (Lowry et al., 1951) before freezing the samples at −80°C for later analysis.
Protein preparation from whole tissue
Frozen tissue was homogenized by sonication with a ultrasonic homogenizer (Cole-Palmer Instruments) at an output setting of 30 for 2 × 10 seconds in 1:10 (mg/µL) HB on ice. The resulting homogenate was suspended in 1:10 (mg/µL) HB with 0.2% Triton X-100 (HBT) for 2 × 10 seconds by sonication, and the protein concentration was determined using BioRad DC protein assay kit (500-0116) before freezing the samples at −80°C for later analysis.
Electrophoresis and immunoblotting
Sodium dodecyl sulfate (8%) polyacrylamide gel electrophoresis (SDS-PAGE) was carried out (Laemmli, 1970). The samples (50 µg of the subcellular fraction protein and 100 µg of whole-tissue protein) were mixed with a 5 × SDS sample buffer of 0.3 mol/L Tris-HCl (pH 6.8), 25% β-mercaptoethanol, 12% SDS, 25 mmol/L ethylenediaminetetraacetic acid, 20% glycerol, and 0.1% bromphenol blue, boiled for 3 min. One sample from each experimental group was applied on the same gel and subjected to SDS-PAGE at a constant current of 20 mA (stacking gel) and 30 mA (separating gel). The proteins on the gel were electrotransferred onto polyvinylidene fluoride (PVDF) membrane (Bio-Rad transblot; pore size, 0.2 mm). After electrophoresis (Towbin et al., 1979) at a constant voltage of 14 V overnight, the PVDF membrane was washed three times with Tris-buffered saline containing 0.1% Tween 20 (TBST; pH 7.4) and then preincubated with a blocking solution of 3% bovine serum albumin in TBST for 1 hour at room temperature. The PVDF membrane was incubated at 4°C overnight with a primary antibody solution containing a rat polyclonal antibody against phosphotyrosine (Affinity Nottingham, U.K.), NMDA R2A (anti-NR2A; Chemicon, U.S.A.), NMDA R2B (anti-NR2B; Chemicon), NMDA R1 (anti-NR1; Chemicon), α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid (AMPA) receptor subunit 1 (anti-GluR1; Chemicon), and AMPA receptor subunit 2/3 (anti-GluR2/3; Chemicon). After incubation with the primary antibody, the PVDF membrane was washed three times with TBST at room temperature and then incubated with secondary antibody conjugated with horseradish peroxides for 1 hour at 4°C. Finally, the immunoreactivity of the synaptosomal fraction and whole-tissue proteins was visualized using Amersham ECL (Amersham, U.K.) and ECL+ (Amersham) western blotting systems, respectively.
The PVDF membrane containing samples from the synaptosomal fraction was exposed to a Kodak X-Omat film at various times to obtain a range of optical densities that was linear with regard to the concentration of the proteins to be analyzed. The films subjected to intermediate exposure times were selected for quantification, which was conducted by computerized image analysis using the program Image (Dr. Wayne Rasband, NIH, Bethesda, MD, U.S.A.). In one series of experiments, samples from whole-hippocampal tissue were blotted on to the PVDF membrane, which was quantified with a DIANA-II CCD camera system (Raytest Isotopenmessgerate GmbH, Straubenhardt, Germany) using the TINA 2.0 program (Raytest Isotopenmessgeräte GmbH). This analysis system provides gray values that are linear in relation to the concentration of the proteins applied to the gel.
Statistical analysis
Data are presented as mean ± SD values or mean values and the 99% confidence interval (CI) of the data (99% CI: lower confidence limit to upper confidence limit). The neuronal cell loss was assessed by analysis of variance followed by Scheffé's test (P < 0.05) or by the two-tailed unpaired Student's t test (P < 0.05, P < 0.01, P < 0.001). The significance of the changes in tyrosine phosphorylation of proteins over time was assessed by comparing the 99% CIs (P <0.01) with sham values or by paired Student's t test (P < 0.05) with Bonferroni correction at particular times of recovery.
RESULTS
Histopathologic outcome after ischemic preconditioning
The rectal and skull temperatures in all experimental animals were kept at 37 ± 0.5°C during ischemia. The postischemic rectal temperature in preconditioned and nonconditioned animals was measured for 2 months after the second ischemic episode and is presented in Fig. 2. There was no significant difference between the preconditioned and nonconditioned groups at any time point. Also, there was no significant difference in blood P
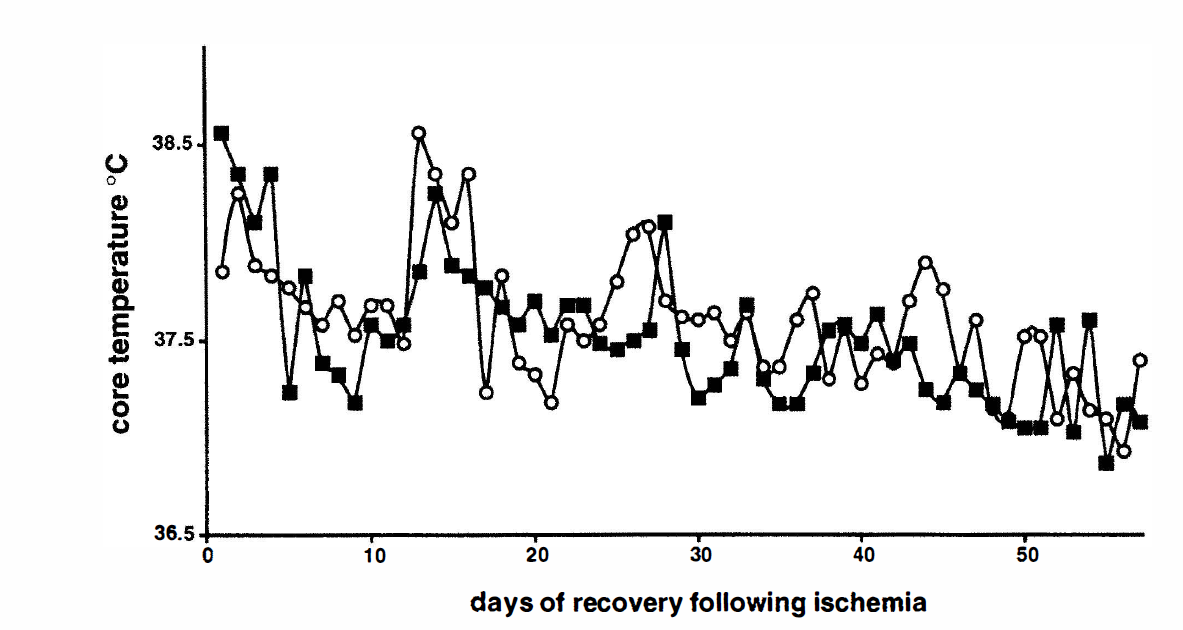
The mean rectal temperature during 8 weeks of recovery following 9 minutes of ischemia. The filled squares represent rats preconditioned with 3 minutes of ischemia followed by 2 days of recovery and subsequently 9 minutes of ischemia. The open circles represent values from sham-operated animals with 2 days of recovery and then 9 minutes of ischemia. There was no statistically significant difference between preconditioned and nonconditioned animals.
After a series of preliminary experiments, we chose a 3-minute ischemic episode as the preconditioning insult, which provided optimal protection against secondary ischemic insults of variable duration when assessed at 7 days after the second ischemia. Three minutes of ischemia alone did not cause damage to the CA1 neurons. When sham-operated animals (Fig. 3A) were exposed to 6 minutes of ischemia after a 2-day recovery period, 60 ± 14% (Scheffé's test, P < 0.05) neuronal cell loss was observed (Fig. 4A). In the preconditioned group, no damage was induced by a second ischemic event of 6-minute duration. Increasing the lethal ischemia time to 15 minutes (Fig. 4B) augmented neuronal cell loss in nonconditioned animals to 84 ± 10% (P < 0.05). A 3-minute preconditioning ischemia mitigated neuronal cell loss to 30 ± 17% (P < 0.05); however, no complete protection was attained. Preconditioning also provided a robust protection in the CA3 region, neocortex, and hilus of the dentate gyrus but not striatum (Table 2).
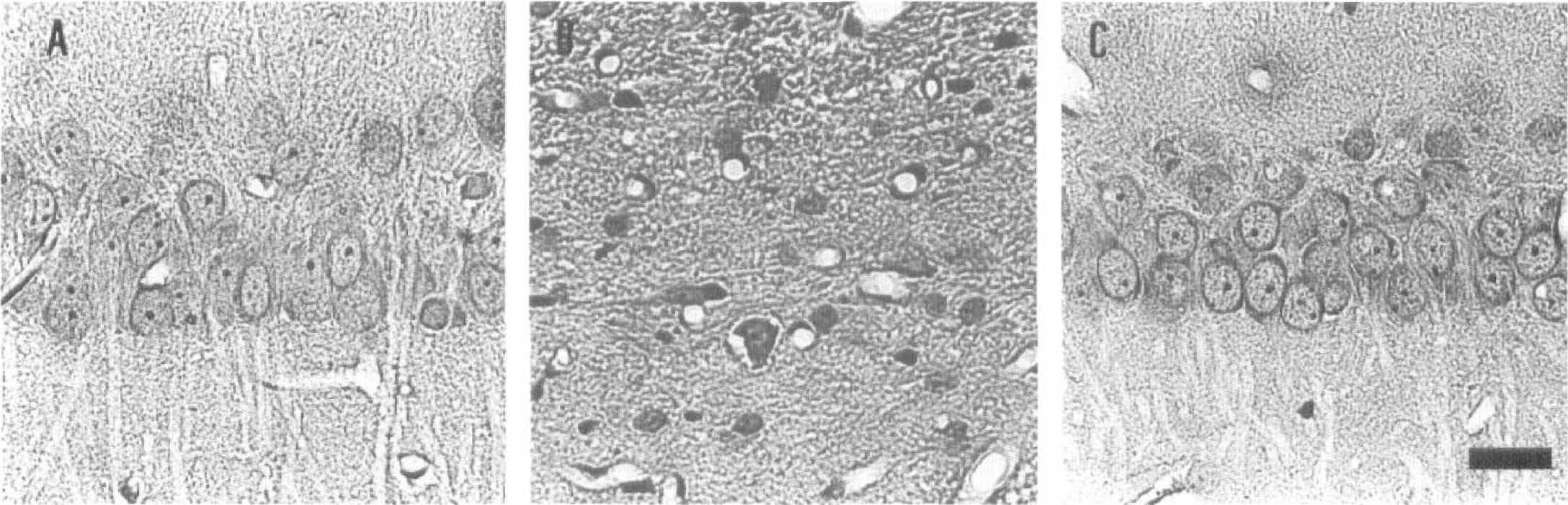
Photomicrographs of 5-µm-thick sections of the rat brain at the level of the middle CA1 region of the dorsal hippocampus from sham-operated animals
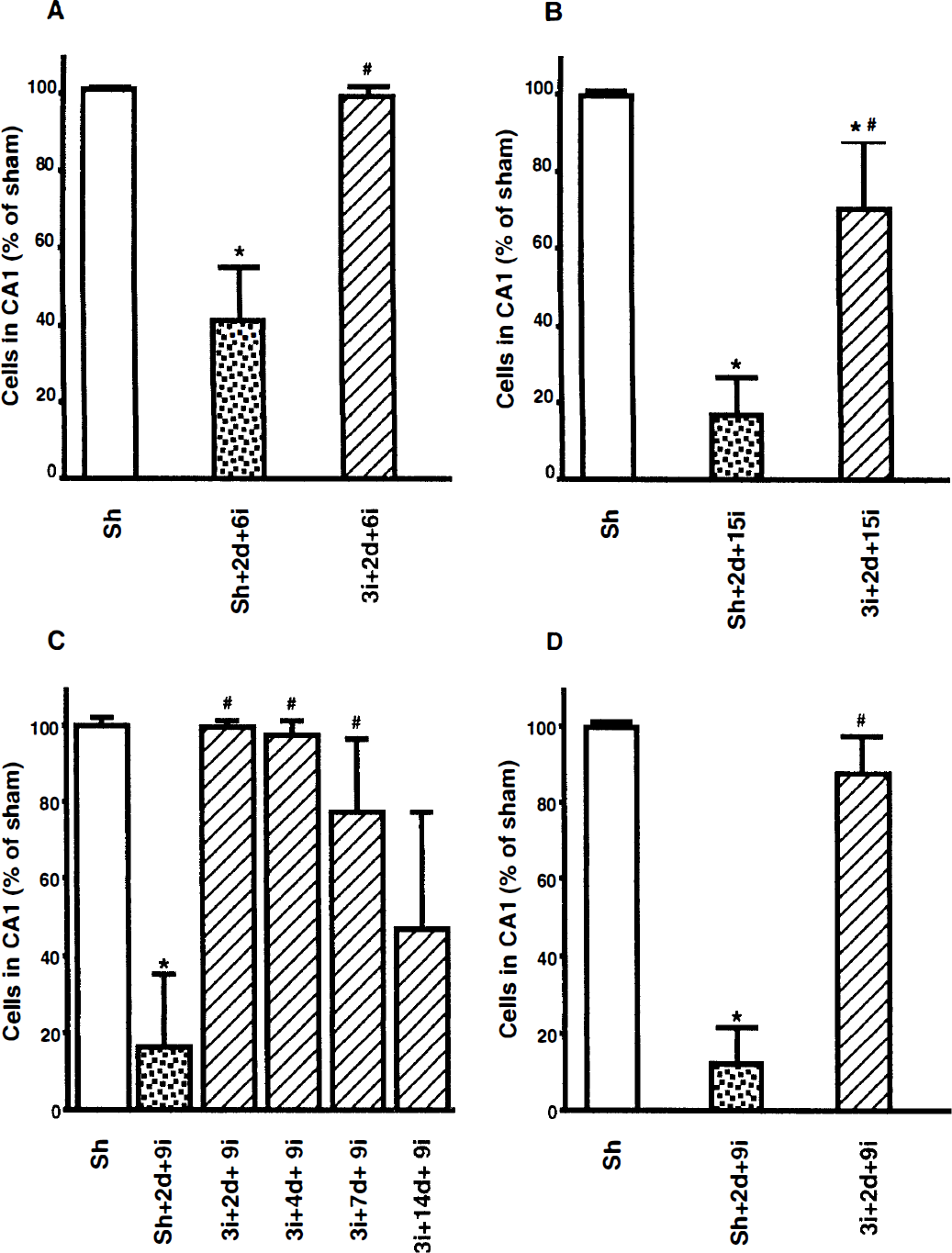
Number of neurons in the CA1 region of the rat hippocampus, as percentage of values of the sham-operated group (Sh). Animals were sham-operated or preconditioned with 3 minutes of ischemia (3i) followed by 2 days of recovery (2d) and subsequently subjected to either 6
Neuronal damage in hippocampal CA3 region, the hilus of dentate gyrus, striatum and neocortex at 1 week recovery after 15 minutes of ischemia with or without 3 minutes ischemic preconditioning

The values are mean ± SD. Results are presented as number of damaged, acidophilic neurons. Statistical significant differences between preconditioned and nonconditioned animals are indicated as †P < 0.05; ‡P < 0.001, unpaired Student's t-test.
The values are mean ± SD. Results are presented as cell loss in percent of the sham group. The differences between preconditioned and nonconditioned animals was statistically evaluated by the unpaired Student's t-test.
After ischemia of 9 minutes, 84 ± 18% (P < 0.05) neuronal cell loss was found in the CA1 region when assessed at 7 days of recovery (Fig. 4C) and 88 ± 9% (P < 0.05) at 8 weeks of recovery (Figs. 3B and 4D). Three minutes of preconditioning ischemia yielded an almost complete protection against damage induced by 9 minutes of ischemia, when initiated at 2 days of recovery following the preconditioning ischemia (Fig. 4C). The 9-minute ischemia was therefore chosen as the standard second ischemic insult. At an interischemic interval of 4 days, protection by preconditioning ischemia remained essentially unchanged. With 7 days of recovery between the sublethal and lethal ischemic insults, neuronal cell loss increased to 23 ± 19%. When the time interval increased to 14 days, 54 ± 30% neuronal cell loss was seen. With 8 weeks of recovery following 9 minutes of ischemia, preceded by 3 minutes of preconditioning ischemia with an interischemic interval of 2 days, 12 ± 9% fewer neurons were found in the CA1 region when compared with sham-operated animals (Figs. 3C and 4D).
Changes in level of tyrosine-phosphorylated proteins in synaptosomal fraction
The changes in the level of tyrosine-phosphorylated (PTyr) proteins were studied in the synaptosomal fraction of rat hippocampus and neocortex with or without preconditioning after 9 minutes of ischemia, followed by either 30 minutes or 24 hours of reperfusion. When the SDS polyacrylamide gel was stained for total protein content, no difference in protein staining among the experimental groups in synaptosomal fraction was noted (Fig. 5). Figure 6 shows an autoradiogram of a PVDF membrane immunostained with an antibody directed to phosphorylated tyrosine residues of proteins in the synaptosomal fraction of CA1 region of the hippocampus. The protein levels on the immunoblot were calculated and are presented as percentage of that in sham-operated animals.
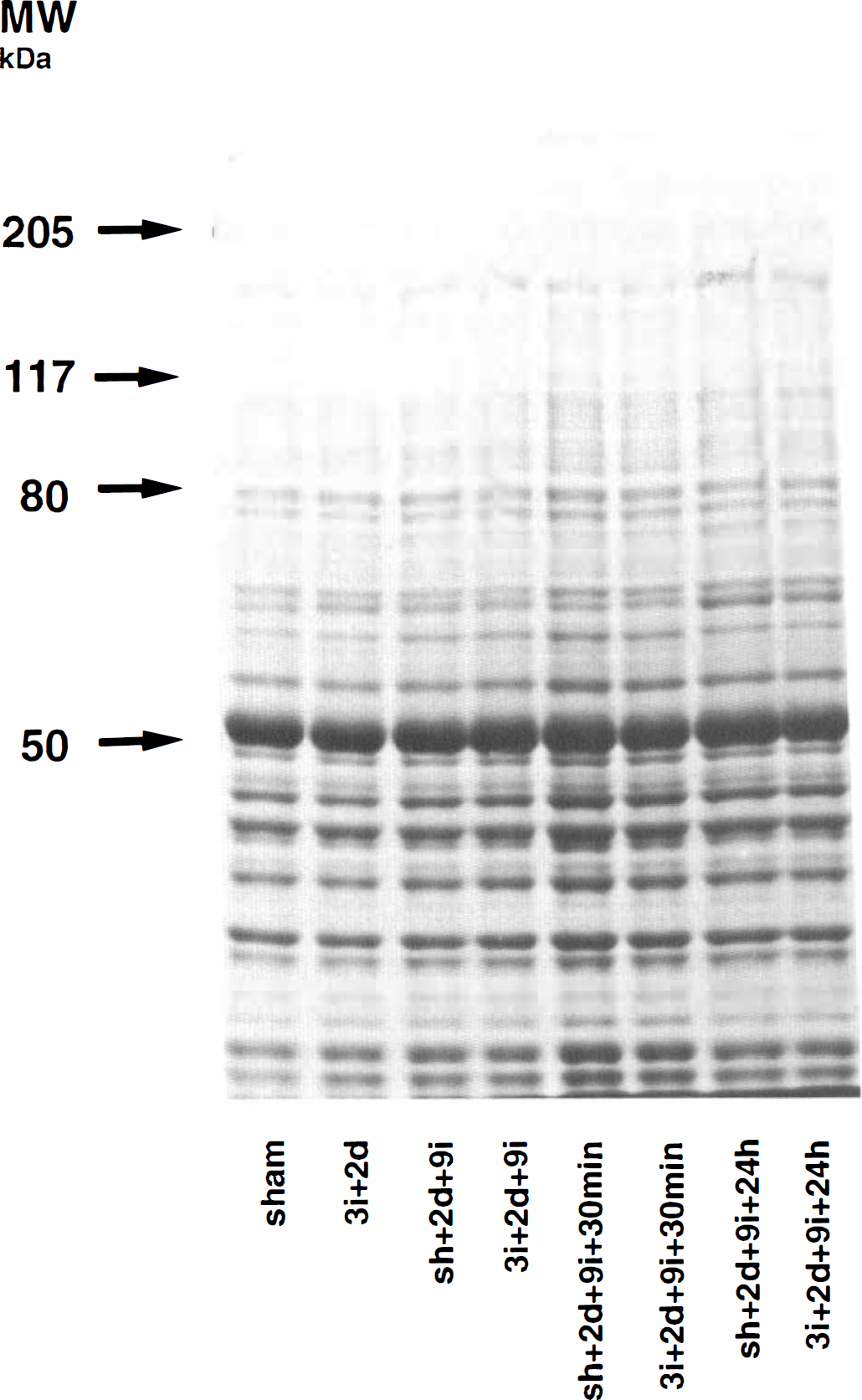
An 8% SDS polyacrylamide gel from crude synaptosomal fraction of rat neocortex. The gel was stained with Commassie Blue. Similar amounts of protein (50 µg) were loaded in each slot. Molecular weight standards are indicated by arrows.
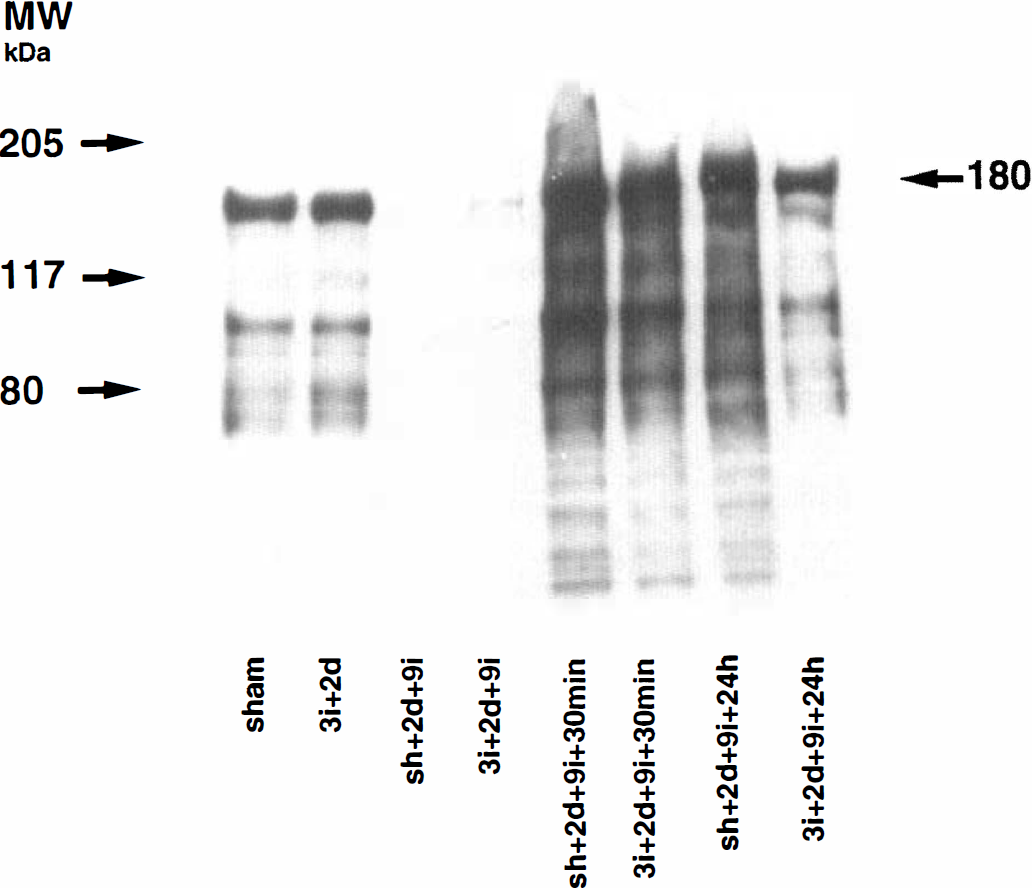
An immunoblot of the protein bands in the crude synaptosomal fraction from the CA1 region of sham-operated rats (Sh) or rats exposed to 3 minutes of ischemia and 2 days of recovery (3i + 2d) or rats either sham-operated or preconditioned with 3 minutes of ischemia followed by 2 days of recovery and 9 minutes of subsequent ischemia with 0, 30 minutes, or 24 hours of recovery.
Figure 7A shows the levels of the PTyr proteins in the synaptosomal fraction of the CA1 region of hippocampus. The levels of PTyr proteins at 2 days after 3 minutes of ischemia were unchanged when compared with control levels. At the end of 9 minutes of ischemia, PTyr immunoreactivity decreased to 21% (99% CI: 5 to 34; P < 0.01) and 26% (99% CI: 11 to 37; P < 0.01) of the sham levels in nonconditioned and preconditioned brains, respectively. Also, at 30 minutes of recirculation, the levels of PTyr proteins in synaptosomal fraction were increased to 168% (99% CI: 135 to 193; P < 0.01) and 130% (99% CI: 65 to 180) of the sham levels in nonconditioned and preconditioned groups, respectively. In contrast, by 24 hours of recirculation, the levels of PTyr proteins increased to 160% (99% CI: 121 to 189; P < 0.01) of sham levels in nonconditioned brains, whereas in preconditioned brains, the immunoreactivity remained at 104% (99% CI: 53 to 143) of the sham group. At this time of recirculation, the total levels of PTyr proteins were 55% higher (paired Student's t test with Bonferroni correction, P < 0.05) in nonconditioned than preconditioned animals. There were no significant differences between nonconditioned and preconditioned animals at the end of ischemia or after 30 minutes of recirculation.
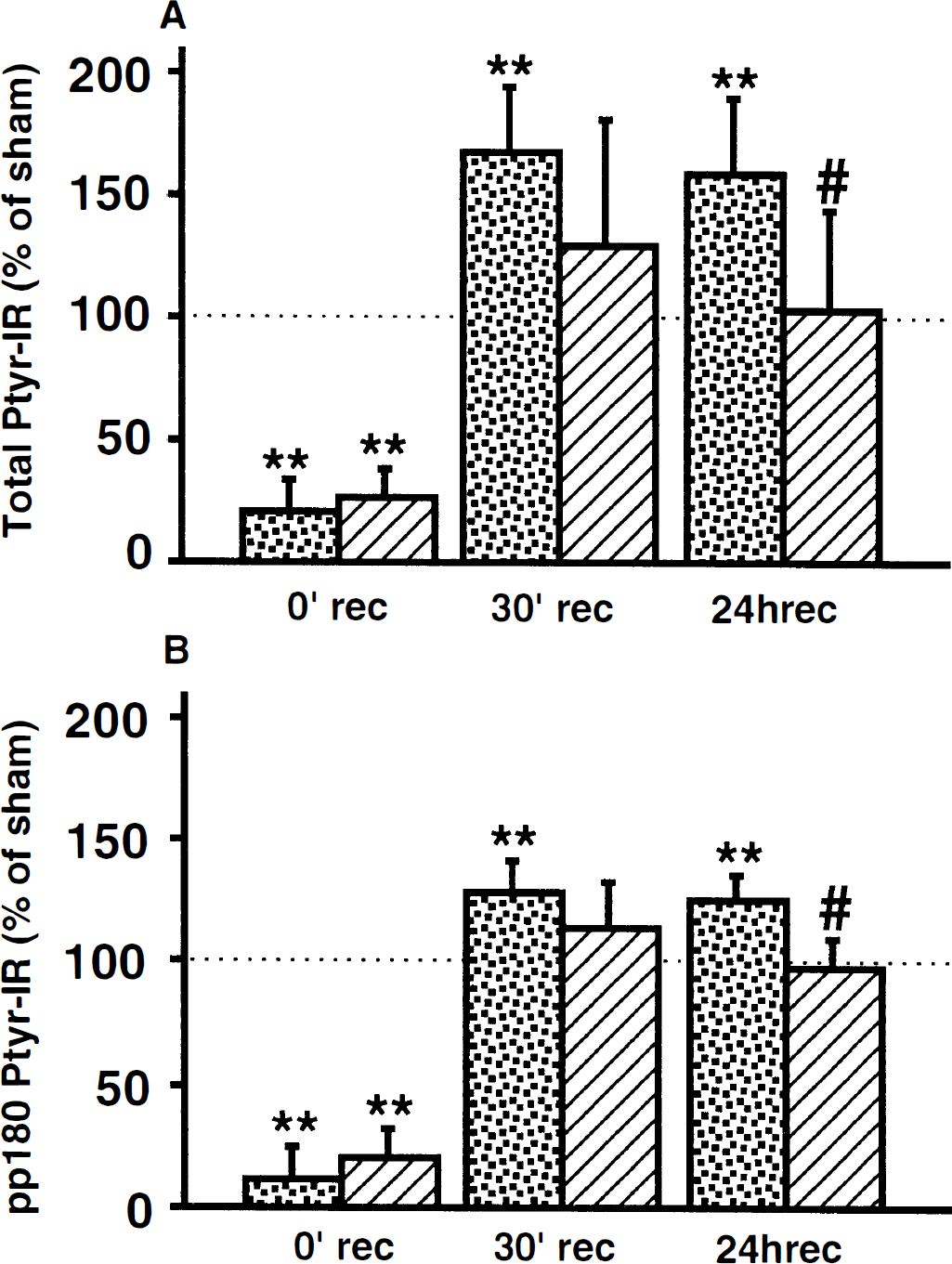
Changes in the tyrosine phosphorylation of proteins in the crude synaptosomal fraction from the CA1 region.
A similar pattern of phosphorylation was seen in the levels of the PTyr protein pp180 (Fig. 7B). Three minutes of ischemia followed by 2 days of recovery did not significantly alter the tyrosine phosphorylation of pp180 in the synaptosomal fraction. At the end of 9-minute ischemia, the protein was almost completely dephosphorylated. During the recirculation period, the pp180 band was rephosphorylated in both preconditioned and nonconditioned brains. At 30 minutes of recirculation, the PTyr levels of pp180 were increased to 129% (99% CI: 111 to 142; P < 0.01) and 113% (99% CI: 89 to 132) of the sham values in nonconditioned and preconditioned brains, respectively. Conversely, at 24 hours of recirculation, PTyr immunoreactivity levels significantly increased to 126% (99% CI: 114 to 136; P < 0.01) of sham levels in nonconditioned brains, whereas in preconditioned brains, the level was 97% (99% CI: 82 to 109) of the sham value. At this time of recirculation, the tyrosine phosphorylation of pp180 in nonconditioned brains was ~30% higher than in the preconditioned group (paired Student's t test with Bonferroni correction, P < 0.05). There were no significant differences between nonconditioned and preconditioned animals at the end of ischemia or after 30 minutes of recirculation.
Figure 8A shows the levels of the PTyr proteins in synaptosomal fraction of the rat neocortex as a percentage of sham-operated animal values. A similar pattern of phosphorylation as seen in the CA1 region was observed. At the end of 9 minutes of ischemia, the phosphorylation of the proteins decreased to 36% (99% CI: 30 to 42; P < 0.01) and 35% (99% CI: 23 to 43; P < 0.01) of sham value, at 30 minutes of recovery it increased to 151% (99% CI: 75 to 208) and 133% (99% CI: 99 to 159), and at 24 hours of recovery to 149% (99% CI: 130 to 163; P < 0.01) and 96% (99% CI: 66 to 119) of the sham group in nonconditioned and preconditioned groups, respectively. The level of protein tyrosine phosphorylation in nonconditioned animals at 24 hours of recovery was 50% higher than in preconditioned ones (paired Student's t test with Bonferroni correction, P < 0.05). There were no significant differences between nonconditioned and preconditioned animals at the end of ischemia or at 30 minutes of recirculation.
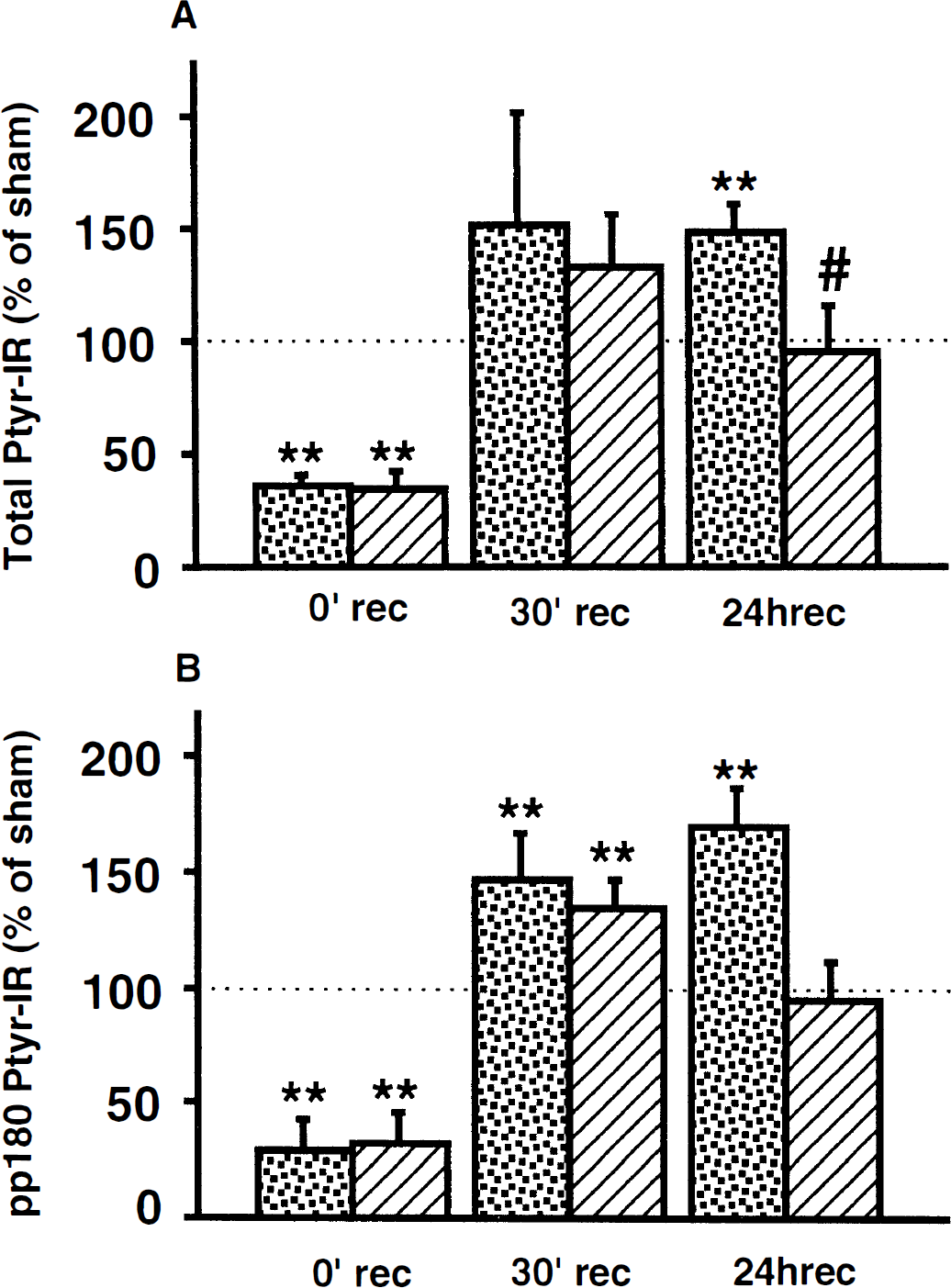
Changes in the tyrosine phosphorylation of proteins in the crude synaptosomal fraction from rat neocortex.
Figure 8B shows the levels of pp180 in neocortex as a percentage of the values of sham-operated animals. At the end of 9-minute ischemia, the phosphorylation decreased to ~30% (99% CI: 7 to 48; P < 0.01) of sham levels in both groups. During the recirculation period, rephosphorylation occurred in both preconditioned and nonconditioned brains. At 30 minutes of recirculation, the levels of PTyr pp180 increased to 147% (99% CI: 117 to 170; P < 0.01) and 134% (99% CI: 116 to 148; P < 0.01) compared with the sham value in nonconditioned and preconditioned brains, respectively. Similarly as in the hippocampus, the PTyr levels increased at 24 hours of recirculation to 170% (99% CI: 145 to 189; P < 0.01) of sham levels in nonconditioned animals, whereas in the preconditioned brains, the levels decreased to the sham value 94% (99% CI: 68 to 114). At this time of recirculation, the tyrosine phosphorylation of pp180 in nonconditioned brains was ~75% higher than in the preconditioned group. There was no significant difference between nonconditioned and preconditioned animals at the end of ischemia or at 30 minutes or 24 hours of recirculation.
To assess whether the changes in tyrosine phosphorylation at 24 hours after ischemia were due to redistribution of phosphorylated proteins, we assessed the levels of PTyr proteins in total tissue homogenates of cortex and CA1. Figure 9 shows an autoradiogram of PTyr proteins in the whole-tissue homogenate of the CA1 region of the hippocampus subjected to 9 minutes of ischemia with or without preconditioning.
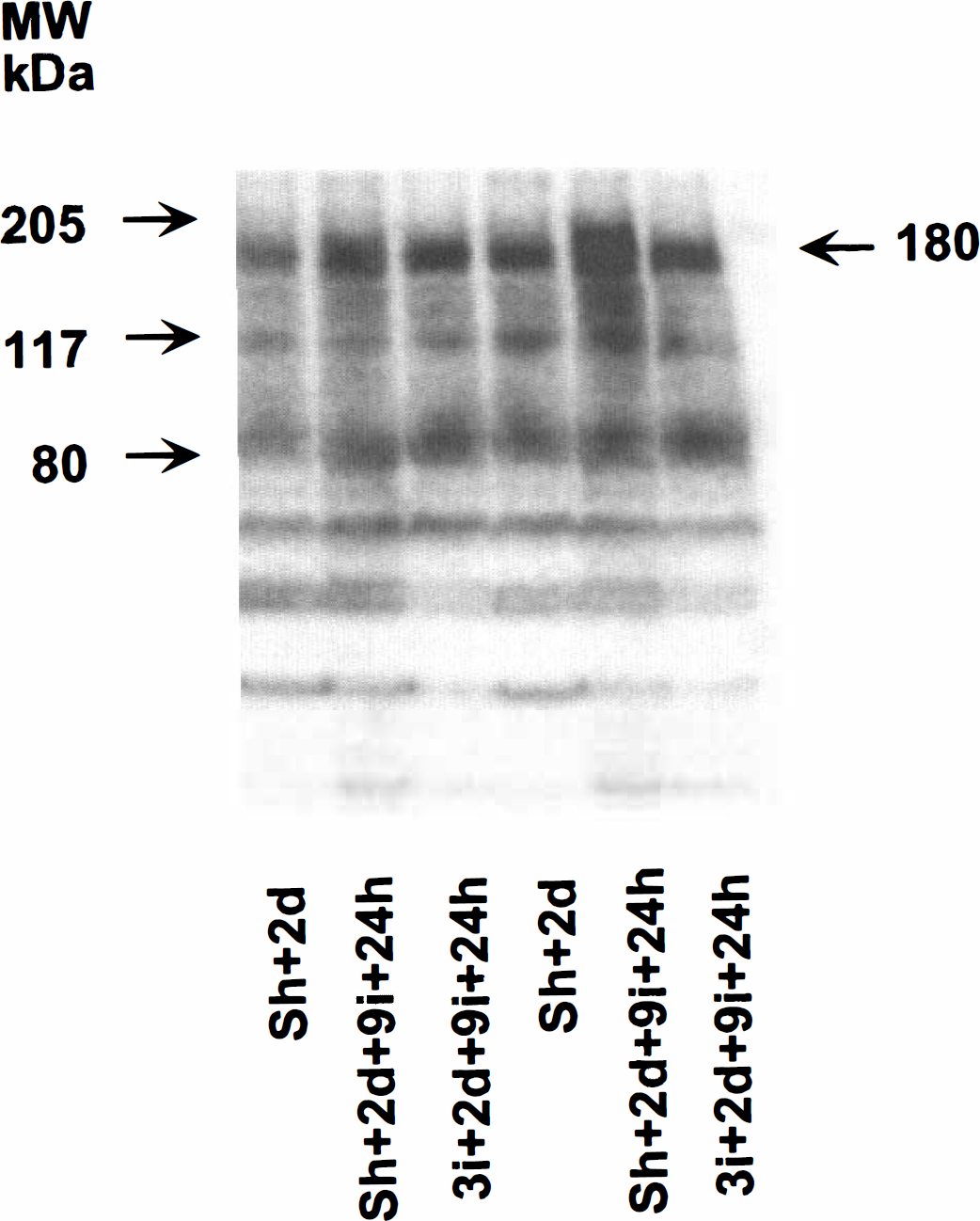
An immunoblot of the whole-tissue homogenate from the CA1 region of sham-operated rats with 2 days of recovery (Sh + 2d) and rats either sham-operated or preconditioned with 3 minutes of ischemia followed by 2 days of recovery and 9 minutes of subsequent ischemia with 24 hours of recovery.
Figure 10A shows the calculated levels of total PTyr protein in whole-tissue homogenate of rat CA1. The levels are expressed as gray values (arbitrary units; AU). The total levels of PTyr proteins increased at 24 hours after nonconditioned ischemia by 79% (Scheffé's test, P < 0.05) compared with the sham-operated group. The level of PTyr proteins in preconditioned animals was not significantly different from that in the sham group but was 70% (P < 0.05) lower than in the nonconditioned ones.
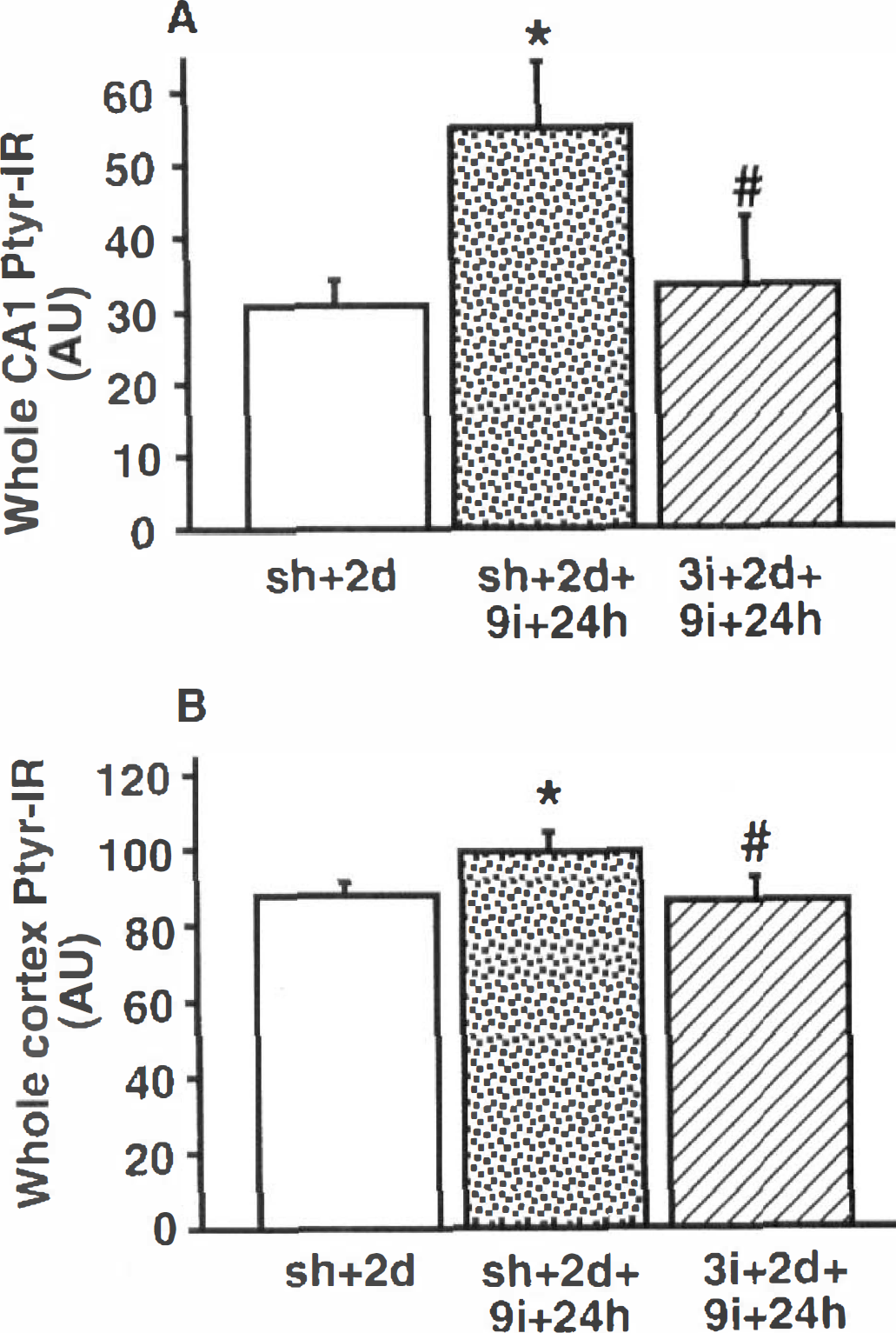
Changes in the tyrosine phosphorylation of proteins in the whole-tissue homogenate from rat CA1 and neonates.
Figure 10B shows the calculated levels of the total PTyr proteins in whole-tissue homogenate of the rat neocortex expressed as gray values (arbitrary units; AU). The total PTyr level at 24 hours of reperfusion after nonconditioned ischemia was 12% (P < 0.05) higher than in the sham-operated group. At the same time point of recovery, the PTyr levels in preconditioned animals were 3 and 9%. (P < 0.05) lower than in the sham and nonconditioned groups, respectively.
Changes in AMPA and NMDA receptor subunits
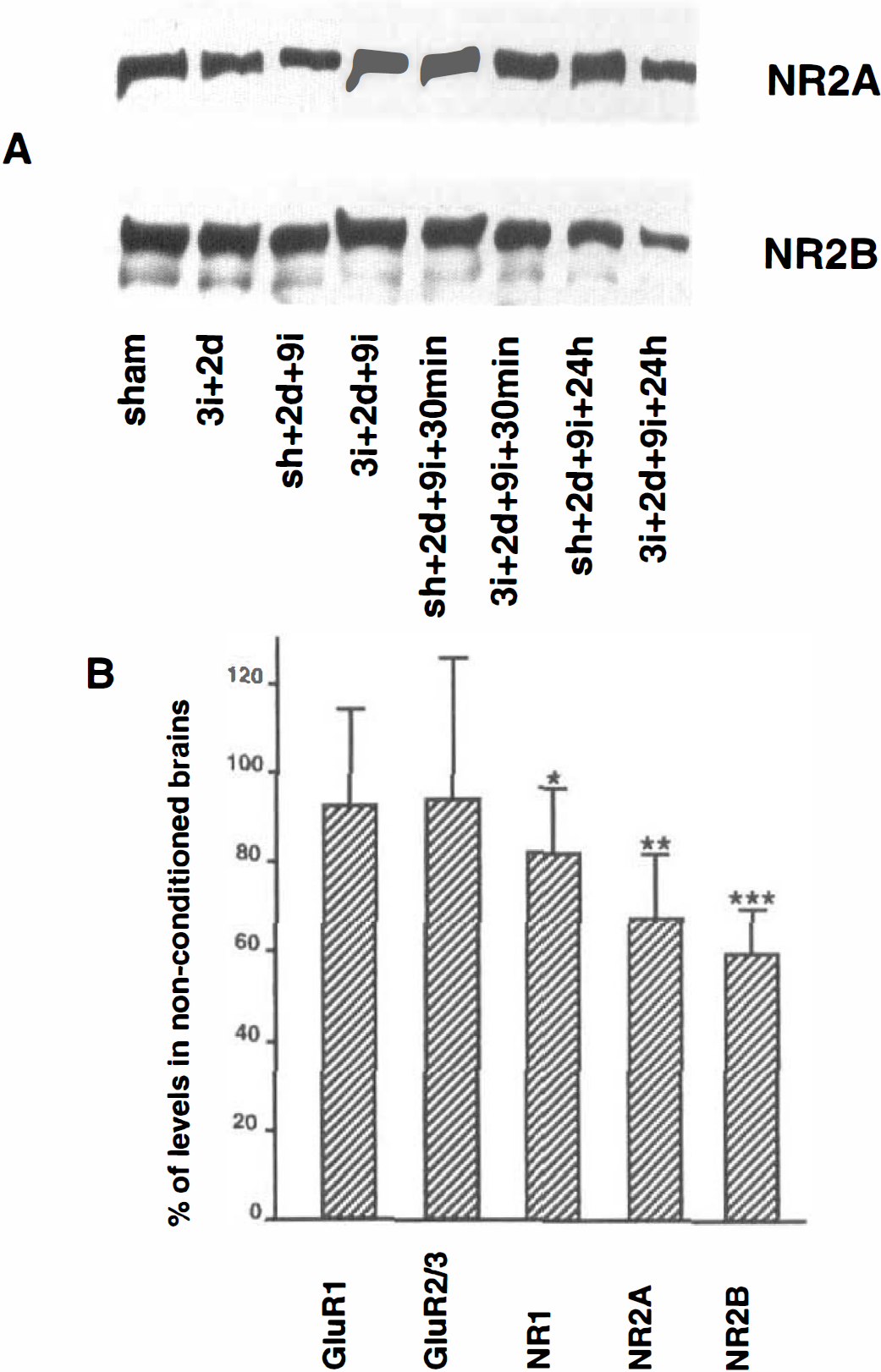
The levels of the glutamate receptor subunits were measured to investigate whether this could account for the decrease in tyrosine phosphorylation of the pp180 protein at 24 hours of recovery. The levels of the AMPA and NMDA receptor subunits were determined on western blots using antibodies directed to GluR1, GluR2/3, NR1, NR2A, and NR2B. Figure 11A shows the immunoblots of NR2A and NR2B in the synaptosomal fraction from neocortex, and the levels of the different subunits are displayed in Fig. 11B. Preconditioning did not influence these values. There was no difference in the levels of GluR1 and GluR2/3 in the synaptosomal fraction of neocortex at 24 hours of recovery in neocortex. In contrast, a significant decrease in NR1 immunoreactivity by 18 ± 14% (unpaired Student's t test, P < 0.05) was noted in neocortex of the preconditioned animals. Also, the levels of NR2A and NR2B decreased in neocortex by 33 ± 14% (P < 0.01) and 41 ± 10% (P < 0.001), respectively, at 24 hours of recirculation compared with the nonconditioned brains. In the hippocampal CA1 region, preconditioning did not influence the levels of NR2A and NR2B at 24 hours of recovery. Also, the levels of NR2A and −2B in the whole homogenate of neocortex were assessed. A small decrease in the levels of NR2A and −2B was found in neocortex of preconditioned animals but was not statistically significant (data not shown).
DISCUSSION
Ischemic tolerance
The results demonstrate that exposure of the brain to 3 minutes of cerebral ischemia in the two-vessel occlusion model of global transient ischemia provides powerful protection against ischemic neuronal damage caused by a subsequent 6 to 15 minute period of ischemia. At the preconditioning times studied, protection is evident in all hippocampal subregions and neocortex. These results are in agreement with earlier data obtained in the rat (Liu et al., 1992) and the gerbil (Kato et al., 1991; Kitagawa et al., 1990). We also demonstrate that the protection is maintained for 8 weeks and that there is no effect of preconditioning on postischemic body temperature that could possibly contribute to the neuroprotection observed in our study (Coimbra et al., 1996; Corbett and Crooks, 1997).
In our model we found that 1 to 2 minutes of ischemia is insufficient to induce tolerance, but 3 minutes provides protection. Evidently, induction of ischemic tolerance requires energy failure sufficient to cause collapse of ionic gradients across the plasma membrane (Abe and Nowak, 1996; Kristian et al., 1994). However, energy failure per se is not a prerequisite for induction of tolerance as cortical spreading depression, associated with similar ion fluxes as seen during ischemia (Hansen, 1985) but with minimal changes in ATP levels (Lauritzen et al., 1990), also induces tolerance (Kobayashi et al., 1995). This suggests that the calcium influx into cells that occurs during ischemia may activate the induction of tolerance. This hypothesis is supported by the observation that blockade of the NMDA receptors (Kato et al., 1992) as well as adenosine receptor and potassium channel blockers mitigate the neuroprotective effect induced by tolerance (Heurteaux et al., 1995). As receptor activation stimulates protein kinase cascades and affects calcium ion homeostasis, cell signaling in general and the glutamate and calcium ion homeostasis in particular seem to be involved in events that induce tolerance (Wieloch et al., 1997).
Protein tyrosine phosphorylation and ischemic tolerance
The protein kinases are enzymes that phosphorylate proteins, a reaction reversed by protein phosphatases. The changes in tyrosine phosphorylation of proteins observed during ischemia therefore reflect changes in the relative activation of protein kinases and phosphatases and are indicators of the cell signaling in the brain after ischemia. Two days after the preconditioning ischemia, tyrosine phosphorylation of synaptosomal proteins, particularly the NR2 subunit, was not different from that in sham-operated animals. Therefore, the preconditioning does not appear to affect the total levels of tyrosine phosphorylation of proteins at the recovery time when a neuroprotective effect is obtained, i.e., 2 days of recovery.
During ischemia the ATP levels decrease; therefore, tyrosine phosphorylation ceases and the phosphatase action dominates, leading to dephosphorylation of the proteins. The levels of PTyr proteins decrease at the end of the second ischemic insult and are not different between the preconditioned and nonconditioned brains. Apparently, preconditioning does not affect the extent of protein dephosphorylation during the second ischemia.
The tyrosine phosphorylation recovers rapidly during early reperfusion and is markedly enhanced (Hu and Wieloch, 1994), persisting until 24 to 48 hours of reperfusion (Wieloch et al., 1997; Takagi et al., 1997). During the early recovery, cell signaling is markedly disturbed due to transmitter release during ischemia and the breakdown of ion homeostasis, leading to changes in the phosphorylation state of proteins (Wieloch et al., 1996). At 30 minutes of recovery, though, tyrosine phosphorylation of proteins is not notably affected by preconditioning. The rephosphorylation of the protein apparently occurs with similar rates in preconditioned and nonconditioned brains, and the balance between protein tyrosine kinase and phosphatase activities therefore seems to be unaffected by preconditioning in the early recovery period (30 minutes).
At 24 hours of recovery, clear effects of preconditioning on protein phosphorylation are observed. In nonconditioned brains, tyrosine phosphorylation of proteins is elevated in both neocortex and hippocampus. In particular, the NR2 subunit phosphorylation is increased (Takagi et al., 1997). This enhancement is mitigated by preconditioning, and the phosphorylation state of the proteins, particularly the NR2 subunit, is essentially normal at this time of recovery. In the neocortex, the levels of NR2 phosphorylation decrease to below sham levels. This is most probably due to a selective decrease in the NR2A and NR2B subunits in neocortex (Takagi et al., 1997). The decrease in the levels of the NMDA receptor subunits by preconditioning ischemia is seen in neocortex but not hippocampus where tyrosine phosphorylation recovers to control levels. The changes are selective for the NMDA receptor, as the AMPA receptor subunit GluR1 and GluR2/3 levels are not affected. There is also a slight decrease in the NR1 subunit levels.
The selective decrease in NMDA receptor protein levels in neocortex does not appear to be due to a decreased protein synthesis. After global ischemia, protein synthesis is persistently depressed in vulnerable cells and is transiently depressed in resistant cells such as CA3 neurons (Bergstedt et al., 1993; Widmann et al., 1991). After preconditioning ischemia, protein synthesis is also transiently depressed in vulnerable neurons but recovers after 6 hours. In preconditioned brains, the recovery of protein synthesis following the second ischemia is facilitated; i.e., it recovers even faster than after the short preconditioning ischemia (Kato et al., 1995; Nakagomi et al., 1993). This suggests that the NMDA receptor complex is either transcriptionally down-regulated or selectively degraded by a proteolytic process in neocortex, activated by preconditioning ischemia. As there was no equivalent decrease in the hippocampus, there is no obvious correlation between the down-regulation of NMDA receptors and tolerance. The absence of a difference in the levels of NR2A and NR2B in whole homogenates suggests that the down-regulation occurs at synaptic membranes. Recently, it was reported that the levels of NR2A/B in hippocampus decrease by ~50% at 24 hours after 15 minutes of four-vessel occlusion ischemia (Zhang et al., 1997). The consequences of the decrease in the NMDA receptor complex in neocortex are not clear. However, the decrease may cause impairment of cortical functions such as working memory (Corbett and Crooks, 1997).
In contrast to degradation of NMDA receptor in neocortex, the normalization of tyrosine phosphorylation of proteins in the postischemic phase by preconditioning ischemia correlates with cell survival. The pattern of tyrosine phosphorylation is normalized at 24 hours, suggesting that preconditioning enhances the recovery of cell signaling in the postischemic phase. This could be accomplished by an inhibition of protein kinases or enhancement of phosphatases. However, the effect of preconditioning is a general normalization of tyrosine phosphorylation and suggests that the effect is upstream from the kinases and phosphatases, i.e., at the second messenger or receptor level. The protein kinases are among other things regulated by calcium ions, which suggests that processes regulating calcium homeostasis would be affected by ischemic preconditioning (Ohta et al., 1996).
Recently it was shown that long-term potentiation or anoxic potentiation was prevented in hippocampal slices from preconditioned brains (Kawai et al., 1997), Anoxic long-term potentiation has been proposed to be due to increased phosphorylation of proteins regulating NMDA receptor function (Hammond et al., 1994). This is in line with our data suggesting that preconditioning normalizes or decreases postischemic NMDA receptor phosphorylation.
CONCLUSION
In summary, our results show that ischemic preconditioning enhances a normalization of tyrosine phosphorylation of proteins in general and the NMDA receptor subunits in particular, following the second ischemic insult. We therefore propose that preconditioning will protect neurons against the deleterious second ischemic insult by expressing proteins that prevent the activation of a persistent and detrimental cell-signaling cascade.
Footnotes
Acknowledgments
The authors thank Prof. Tracy McIntosh for valuable discussion, and Profs. Anders Holtsberg and Ulf Strömberg at Lund University for valuable help concerning the statistical analysis.