
Abstract
Ischemic preconditioning (IPC) induces neuroprotection to subsequent severe ischemia, but its effect on the cerebrovasculature has not been studied extensively. This study evaluated the effects of IPC on brain edema formation and endothelial cell damage that follows subsequent permanent focal cerebral ischemia in the rat. Transient (15 minute) middle cerebral artery occlusion (MCAO) was used for IPC. Three days after IPC or a sham operation, permanent MCAO was induced. Twenty-four hours after permanent MCAO, neurologic deficit, infarction volume, and water and ion content were evaluated. Six hours post-ischemia, blood–brain barrier (BBB) permeability was examined using [3H]-inulin. Water, ion contents, and BBB permeability were assessed in three zones (core, intermediate, and outer) depending on their relation to the MCA territory. Heat shock protein 70 (HSP70) was also examined as a potential marker of vascular injury. The model of IPC significantly reduced brain infarction and neurologic deficit. Compared with a sham operation, IPC also significantly attenuated brain edema formation in the intermediate (sham and IPC water contents: 5.99 ± 0.65 vs. 4.99 ± 0.81 g/g dry weight; P < 0.01) and outer zones (5.02 ± 0.48 vs. 4.37 ± 0.42 g/g dry weight; P < 0.01) of the ipsilateral hemisphere but not in the core zone. Blood–brain barrier disruption assessed by [3H]-inulin was significantly attenuated in the IPC group and the number of blood vessels that displayed HSP70 immunoreactivity was also reduced. Thus, IPC significantly attenuates ischemic brain edema formation, BBB disruption, and, as assessed by HSP70, vascular injury. Understanding the mechanisms involved in IPC may provide insight into methods for preserving cerebrovascular function during ischemia.
Endothelial cell dysfunction may play an important role in the pathophysiology of cerebral ischemia. Disruption of the endothelial cells that form the blood–brain barrier may lead to edema formation that can exacerbate brain injury and potentially lead to brain herniation (Betz and Dietrich, 1998). In addition, endothelial cell dysfunction leading to hemorrhagic conversion may be the leading limiting factor in tissue plasminogen activator reperfusion therapy for cerebral ischemia (NINDS t-PA Stroke Study Group, 1997; Jean et al., 1998). It is, therefore, important to develop mechanisms to limit endothelial cell injury after cerebral ischemia.
One mechanism that has been shown to protect parenchymal cells from cerebral ischemia is ischemic preconditioning (IPC). Thus, subjecting the brain to a short duration ischemic event can induce tolerance to subsequently larger ischemic insults (Kato et al., 1991; Kitagawa et al., 1990). Although originally demonstrated for global ischemia, short durations of focal ischemia also protect against damage after subsequent severe focal ischemia (Barone et al., 1998; Chen et al., 1996). There is evidence that IPC may occur in humans as well as animals (Alteri et al., 1998; Hakim, 1994; Weih et al., 1999). Although the concept of preconditioning has been widely studied, little attention has focused on the effects of preconditioning on blood–brain disruption and brain edema formation.
A 70 kDa heat shock protein (HSP70) is produced in the brain after a variety of brain injuries including ischemia, hypoxia–ischemia, epilepsy, and trauma (Brown et al., 1989; Ferriero et al., 1990; Nowak, 1985; Vass et al., 1989). There is a prominent HSP70 induction after focal cerebral ischemia where neurons, glia, and vascular cells may all up-regulate this protein (Kinouchi et al., 1993b; Li et al., 1992; Sharp et al., 1991). Heat shock protein 70 is a molecular chaperon that has been hypothesized, along with other heat shock proteins such as HSP32 and HSP27, to be a neuroprotective agent (Kato et al., 1994; Kirino et al., 1991; Panahian et al., 1999). However, it is also a marker of cell stress and cell injury and its production is down-regulated by cell protective agents (Abe et al., 1997; Gething and Sambrook, 1992; Gonzalez et al., 1989; Sloviter and Lowenstein, 1992). In the brain, laser-induced microvascular injury results in the induction of vascular HSP70 (Lindsberg et al., 1996). In addition, an in vitro study has shown that HSP70 can be produced by endothelial cells and that the expression of stress protein is a good marker of toxic damage (Wagner et al., 1999).
The current study examines brain edema formation and blood–brain barrier disruption after permanent middle cerebral artery occlusion (MCAO) in rats undergoing IPC or a sham operation. Vascular HSP70 was examined as a potential injury marker of cerebrovasculature.
MATERIALS AND METHODS
Animals and experimental protocols
The protocol for these animal studies was approved by the University of Michigan Committee on the Use and Care of Animals. Adult male Sprague–Dawley rats (Charles River Laboratories, Portage, MI, U.S.A.) weighing 275 to 350 g were used for all experiments. Rats were allowed free access to food and water before the experiment. Animals were anesthetized with pentobarbital (40 mg/kg, IP) before any surgery during which time body temperature was maintained at 37.5°C.
This study compared rats that underwent 15 minutes of transient MCAO (IPC) or a sham occlusion. After 3 days, the rats were used for 6 sets of experiments. In the first set, the rats underwent a further 24 hours of permanent MCAO before behavioral testing and assessment of infarction volume by 2,3,5-triphenyltetrazolium (TTC) staining. In the second set of experiments, the effect of IPC on brain water and ion contents 24 hours after permanent MCAO was determined. These rats were, again, also used for behavioral testing. In the third set, the effect of IPC on BBB permeability was examined by measuring blood to brain [3H]-inulin transport. In the fourth set, the effect of IPC on cerebral blood flow during subsequent MCAO was examined. In the fifth set, HSP70 was quantified by Western blotting analysis using a mouse anti-HSP70 monoclonal antibody (StressGen, Victoria, BC, Canada) in rats that had undergone IPC or a sham occlusion in combination with different durations of permanent MCAO. In the sixth, immunohistochemistry was performed to identify the cell types that expressed HSP70 at those different time points.
Middle cerebral artery occlusion
Transient (15 minute) occlusion of the MCA was achieved using the suture method of Zea Longa (Zea Longa and Weinstein, 1989). Briefly, the bifurcation of common carotid artery was exposed, the external carotid artery was ligated distally. A 3–0 monofilament nylon suture, its tip rounded by heating, was introduced into the internal carotid artery lumen through the stump of the external carotid artery and gentry advanced into the internal carotid artery 19 to 20 mm past the common carotid artery bifurcation to block the origin of MCA. After 15 minutes of MCAO, reperfusion was achieved by withdrawal of the suture from external carotid artery. For sham-occluded rats, the carotid arteries were exposed but no suture was inserted. After closing the skin incision, rats were allowed to recover from anesthesia, returned to their cages, and allowed to move and eat freely.
Three days after the transient MCA or sham occlusion, the rats were reanesthetized and a permanent MCA occlusion performed. Again the suture method of Zea Longa (Zea Longa and Weinstein, 1989) was used.
Morphometric measurement of infarct volume
Twenty-four hours after permanent MCAO, animals were reanesthetized with pentobarbital (40 mg/kg, IP) and right femoral artery was catheterized for blood sampling and measurement of pH, Po2, Pco2, blood glucose, and hematocrit. Animals were then decapitated and the brains rapidly removed and sectioned coronally at 2-mm intervals. All slices were incubated for 20 minutes in a 2% solution of TTC at 37°C and fixed by immersion in 2% paraformaldehyde solution. Using a computerized image analysis system (NIH image, version 1.61), the area of infarction of each section was measured. The total lesion volume was calculated by summing the infarct area in each section and multiplying by the distance between sections.
Measurement of brain water and ion concentration
All rats were decapitated at 24 hours post-MCAO. Brains were removed quickly from the skull, the hemisphere was divided, and the brain stem, hippocampus, and thalamus were discarded. The remaining telencephalic tissues ipsi- and contralateral to the MCAO were placed flat on a nonabsorbent surface and the portion inferior to the rhinal fissure was removed.
Three samples (ipsi- or contralateral) were taken from the remaining tissue using 5- and 7-mm cork borers (Dickinson and Betz, 1992; Martz et al., 1990). The core region was taken from the cortex and striatum underlying the MCA immediately distal to the region of occlusion (Fig. 1). The intermediate region was a ring of tissue surrounding the core zone, whereas the outer region consisted of the remainder of the cortical tissue. Brain samples were immediately weighed on an electronic analytical balance (model AE 100; Mettler Instrument, Highstown, NJ, U.S.A.) to obtain the wet weight. Brain samples then were dried in a gravity oven weight (Blue M. Electric, Blue Island, IL, U.S.A.) at 100°C for 24 hours to obtain the dry weight and water content determined as (Wet Weight - Dry Weight)/Dry Weight. Dehydrated samples were digested in 1 mL of 1 mol/L nitric acid for 1 week.
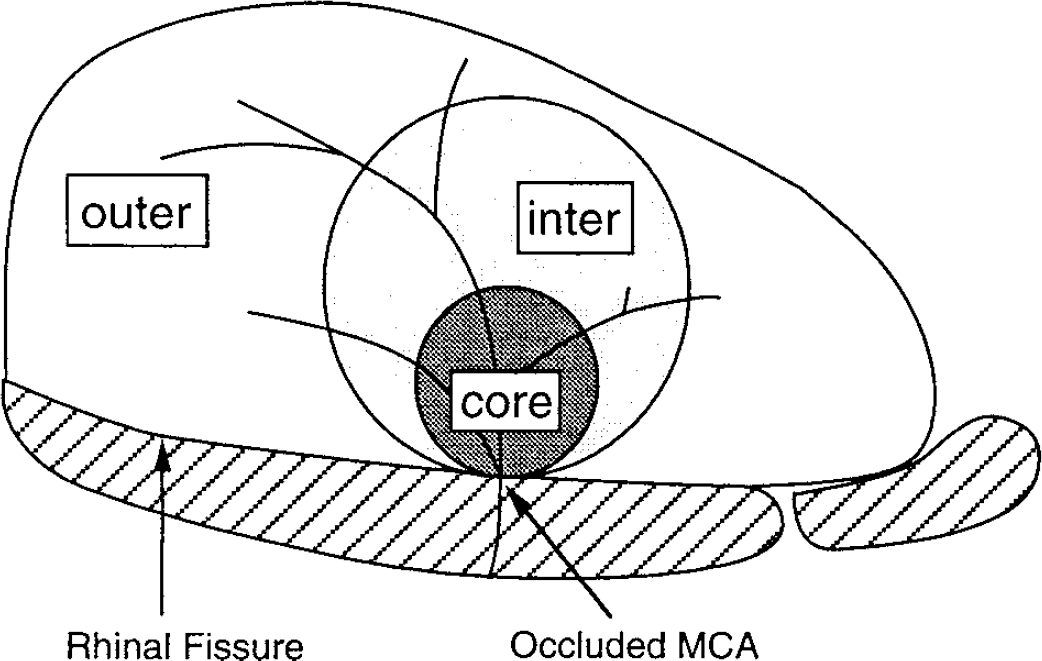
Punch scheme for cortical sampling. The ipsi- or contralateral cortex above the rhinal fissure was flattened and divided in core, intermediate (inter), and outer zone samples using 5 and 7 mm punches.
The sodium, potassium, and chloride contents of this solution were measured with the automatic flame photometer (model IL943, Instrumentation Laboratory) and a chloridometer (Haake Buchler Instruments). Ion content was expressed in milliequivalents per kilogram of dehydrated brain tissue (mEq/ kg dry wt).
Blood-brain barrier disruption
Six hours after permanent MCAO, rats were reanesthetized with pentobarbital and BBB integrity was examined. Blood–brain barrier disruption was assessed by measurement of the influx rate constant (Ki) for [3H]-inulin (New England Nuclear, Boston, MA, U.S.A.) using a method modified from Ohno et al. (1978), with [14C]-inulin (American Radiolabeled Chemicals, St. Louis, MO, U.S.A.) circulated for a short period of time, being used to correct for cerebral plasma volume. [3H]-inulin (50 μCi) was injected through a femoral vein 30 minutes before the end of the experiment, whereas [14C]-inulin (15 μCi), being used as a plasma volume marker, was given as a second injection 2 minutes before the end of experiment. The arterial concentration of [3H]-inulin was determined using a peristaltic pump (Gilson, Middleton, WI, U.S.A.) to create an artificial organ by continuous withdrawal of blood (0.05 mL/min) into a polyethylene catheter (PE 205). At the end of experiment, a terminal plasma sample was taken and the rat was killed by decapitation. Brains were rapidly removed and the cortices ipsi- and contralateral to the MCAO were sampled as described above. Samples were immediately weighed and digested in methylbenzethonium hydroxide. Scintillation fluid was added to the brain and plasma samples and radioactivity was counted using a Beckman 3801 liquid scintillation counter (Fullerton, CA, U.S.A.). The Ki was calculated from the formula:
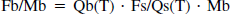
where Cbr is the counts per gram of brain, CPV is the plasma volume of the brain determined from the [14C]-inulin space, Ct is the terminal plasma concentration of [3H]-inulin, and ∞Cp dt is the integral of the [3H]-inulin plasma concentration (Cp) for the experiment. The latter was calculated from the radioisotope content of the continuously withdrawn arterial blood sample. The [3H]-inulin was purified using a G-25M Sephadex column (PD-10, Pharmacia, Piscataway, NJ, U.S.A.) before use.
Cerebral blood flow
One hour after permanent MCAO, rats were reanesthetized with pentobarbital and cerebral blood flow was determined. Cerebral blood flow was measured by the indicator fractionation technique (Van Uitert and Levy, 1978) using [14C]-N-isopropyl-p-iodoamphetamine ([14C]IMP; American Radiolabeled Chemicals; Williams et al., 1991). This method uses an intravenous bolus injection of a blood flow indicator followed by a constant rate of blood withdrawal through a femoral artery catheter to obtain the integral of the arterial isotope concentration. The withdrawal was started 5 seconds before intravenous injection of 5 μCi [14C]IMP. Two minutes later, the animal was killed by decapitation and blood withdrawal was stopped. The sample of withdrawn arterial blood was bleached with 30% H2O2 before addition of scintillation fluid and counting using a Beckman 3801 liquid scintillation counter. The brain was sampled as described above for the [3H]-inulin measurements and digested in methylbenzethonium hydroxide before counting.
Blood flow rates for the individual pieces of brain tissue were calculated using the following equation:
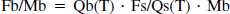
where Fb = the brain blood flow; Mb = the brain mass (in grams); Qb(T) = the quantity of indicator in the tissue at time T; Fs = the rate of blood withdrawal from t = 0 to t = T; Qs(T) = the quantity of indicator present in the withdrawal at time T. Cerebral blood flow is expressed as mL g−1 min−1.
Western blot analysis
Animals were anesthetized and decapitated at 0, 3, 6, and 24 hours after permanent MCAO. Brain was perfused with saline, and brain tissues were sampled as described above.
Brain tissues were immersed in 0.5 mL Western blot sample buffer (62.5 mmol/L Tris-HCL, pH 6.8, 2.3% sodium dodecyl sulfate, 10% glycerol, and 5% β-mercaptoethanol) and sonicated for 10 seconds. Fifty microliters of the sample solution was taken for protein assay (Bio-Rad, Hercules, CA, U.S.A.), whereas the rest was frozen at −20°C for later Western blotting. Western blot analysis was performed as described previously (Keep et al., 1997). Briefly, 50 μg protein was run on 7.5% polyacrylamide gels with a 4% stacking gel (SDS-PAGE) after 5 minutes of boiling at 95°C. The protein was transferred to hybond-C pure nitrocellulose membrane (Amersham International, Little Chalfont, Bucks, England). The membranes were blocked in 5% Carnation nonfat dry milk in TBST (150 mmol/L NaCl, 100 mmol/L Tris base, 0.1% Tween 20, pH 7.6) buffer for 1 hour at 37°C. After they were washed in TBST buffer 3 times, membranes were probed with 1:2000 dilution of the first antibody (mouse anti-rat HSP70, StressGen) for 1.5 hours at room temperature. The membranes were then washed with TBST buffer 3 times and immunoprobed again with 1:5000 dilution of the second antibody (peroxidase-conjugated goat anti-mouse IgG) (Bio-Rad) for 1 hour at room temperature. Finally, membranes were washed 3 times in TBST buffer, and the antigen-antibody complexes were visualized with the ECL chemiluminescence system (Amersham International) and exposed to a Kodak X-OMAT film. The relative densities of HSP70 protein bands were analyzed with a computerized image analysis system (NIH image, version 1.61).
Immunohistochemistry of HSP70
Rats underwent either 15 minutes of MCAO or a sham occlusion, as described above. After 3 days, the rats underwent a further 0, 3, 6, or 24 hours of permanent MCAO (n = 3 to 4 for each group) followed by transcardiac perfusion with 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (PBS; pH 7.4). Brains were removed, postfixed for 24 hours, and sectioned at 50 μm on a vibratome before processing for immunohistochemistry and cresyl violet staining.
For immunohistochemistry, sections were incubated in 1:10 goat serum for 30 minutes, rinsed, and incubated with a 1:500 dilution of the primary antibody (mouse anti-rat HSP70, StressGen).
Normal mouse IgG was used as negative control. After 3 washes in PBS, sections were incubated for 90 minutes with 1:800 dilution of biotinylated goat anti-mouse IgG (Chemicon, Temecula, CA, U.S.A.). After another three PBS washes, brain sections were incubated with avidin-biotinylated horseradish peroxidase (Vector Laboratories, Burlington, CA, U.S.A.) for 90 minutes. Brain sections then were rewashed three times in PBS and incubated with diaminobenzidine and hydrogen peroxide (Stable DAB, Research Genetics, Huntsville, AL, U.S.A). Sections then were washed in water for 5 minutes, dehydrated, and covered with a coverslip for microphotography.
Behavioral testing
Rats underwent either 15 minutes of MCAO or a sham occlusion. After 3 days, the rats underwent a further 24 hours of permanent MCAO and a neurologic examination was performed using a previously described methodology (Menzies et al., 1992). In brief, rats were scored as follows: 0 = no apparent deficits; 1 = contralateral forelimb flexion when suspended by the tail; 2 = decreased grip of the contralateral forelimb while tail pulled; 3 = spontaneous movement in all directions and contralateral circling only if pulled by tail; and 4 = spontaneous contralateral circling. Animals that had the features of the higher scores also showed all the features of the lower grades.
Statistical analysis
All parametric data in this study are presented as mean ± SD. Such data were analyzed with Student's t-test. The nonparametric behavior data was analyzed by means of Mann-Whitney U test. Statistical significance was accepted at P < 0.05.
RESULTS
There were no significant differences in blood gases, blood pH, blood glucose, and hematocrit among the groups. The combined mean physiologic variables are shown in Table 1.
Physiologic variables in each experimental group
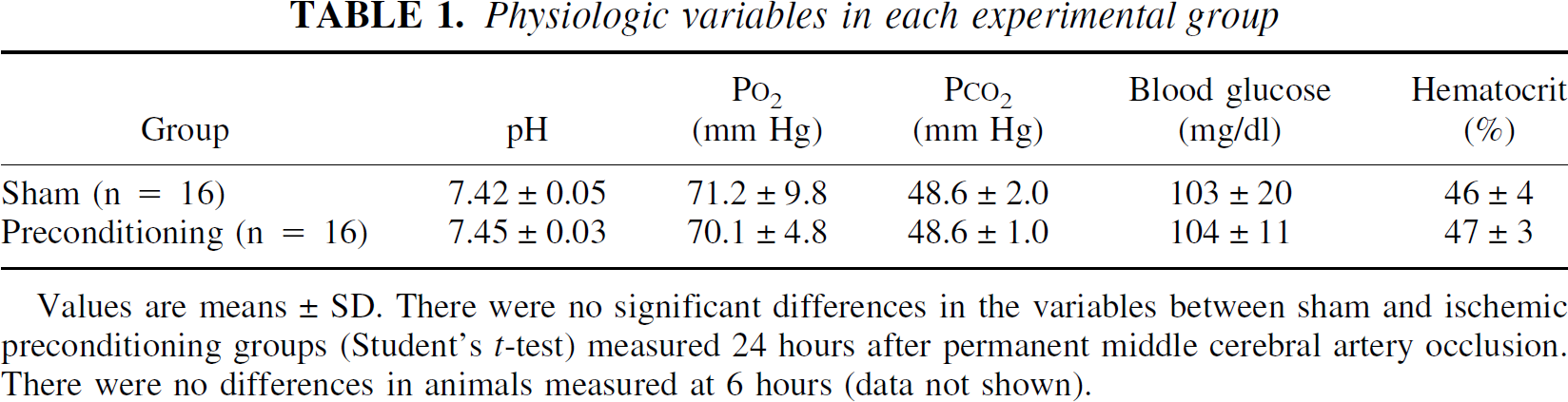
Values are means ± SD. There were no significant differences in the variables between sham and ischemic preconditioning groups (Student's t-test) measured 24 hours after permanent middle cerebral artery occlusion. There were no differences in animals measured at 6 hours (data not shown).
Infarction volume and behavioral testing
Fifteen minutes of MCAO, the preconditioning insult, did not result in a TTC-demarcated lesion three days later. The effect of this IPC on infarct volume after 24 hours of permanent MCAO is shown in Fig. 2. Ischemic preconditioning resulted in a significant 41% reduction in total infarction volume compared with sham control (P < 0.01). This protection occurred in cortex and basal ganglia. In addition, twenty-four hours after permanent MCAO, the IPC group had significantly (P < 0.05) less motor deficit than sham-operated rats (Table 2; mean scores 1.2 and 2.0, respectively).
Neurologic scores in each experimental group
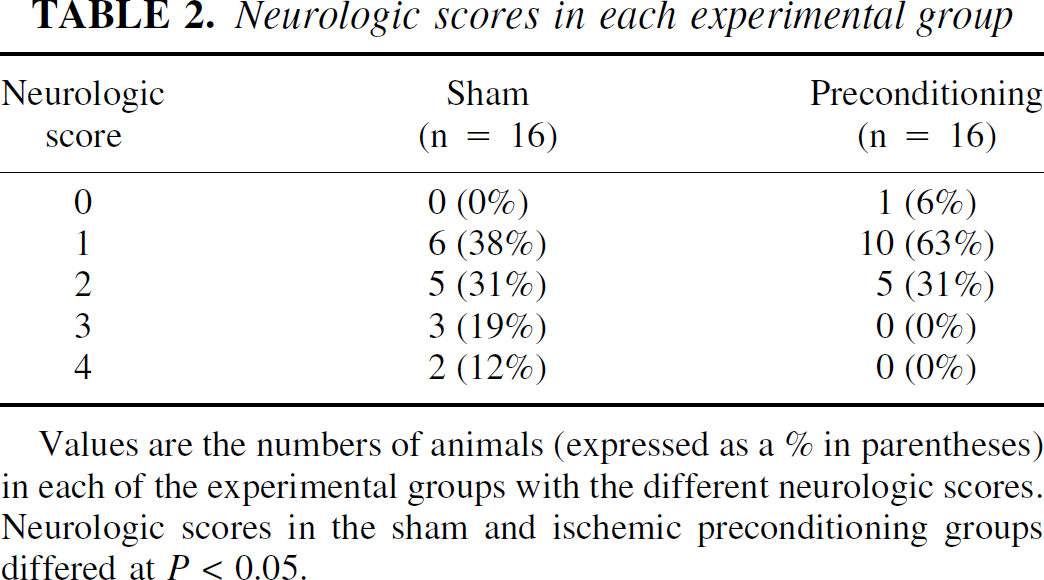
Values are the numbers of animals (expressed as a = in parentheses) in each of the experimental groups with the different neurologic scores. Neurologic scores in the sham and ischemic preconditioning groups differed at P < 0.05.
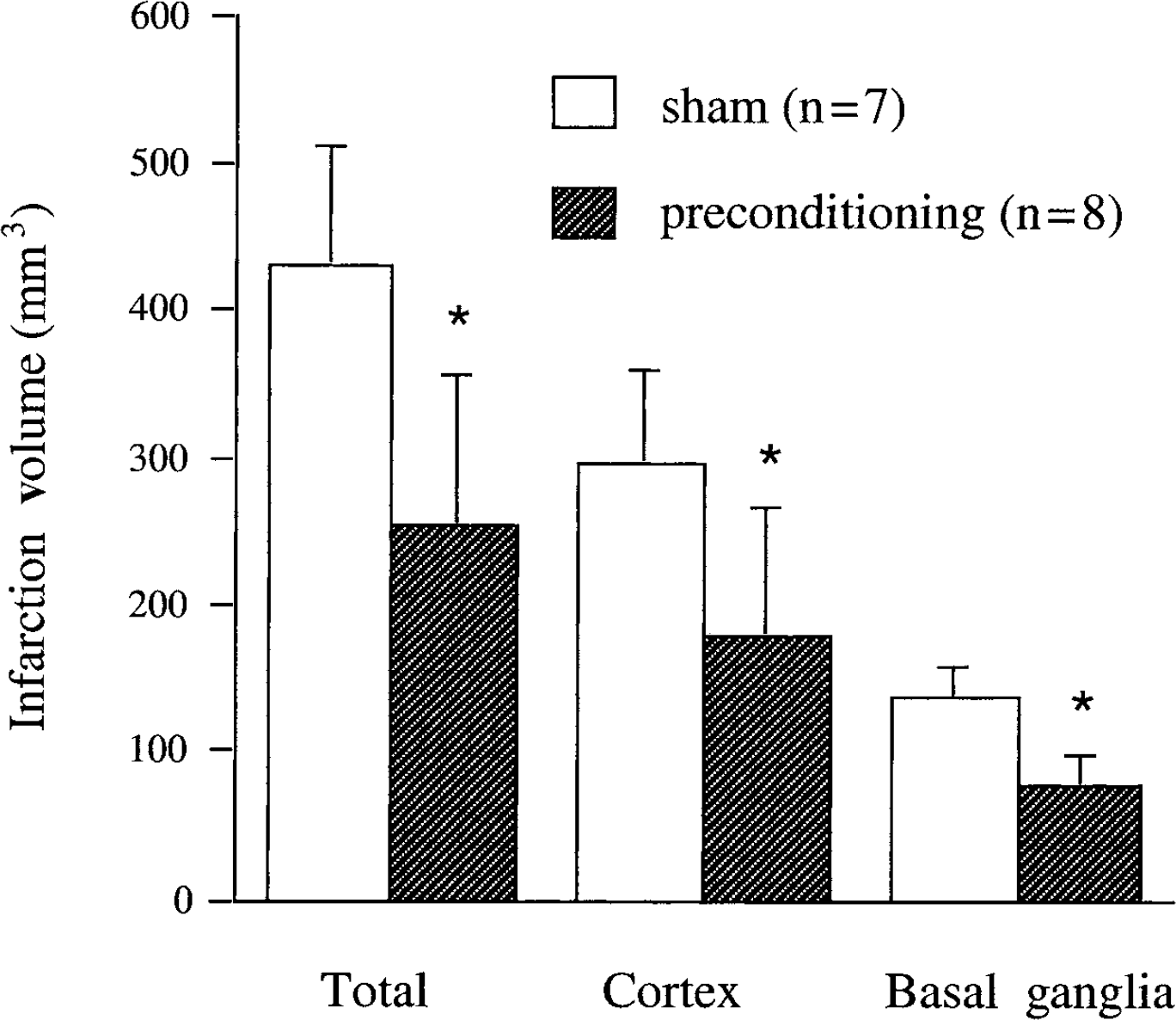
Infarct volume 24 hours after permanent middle cerebral artery occlusion (MCAO). Animals received 15 minutes of ischemic preconditioning (n = 7) or a sham operation (n = 8) 3 days before permanent MCAO. Values are expressed as mean ± SD. *P < 0.01 vs. sham.
Brain edema formation
Sham-operated rats that underwent 24 hours of permanent MCAO 3 days later had a pronounced increase in brain tissue water content in core, intermediate, and outer zone samples (an increase of 2.77 ± 0.85, 2.27 ± 0.66, and 1.02 ± 0.42 g/g dry weight, respectively) compared with the contralateral side (Fig. 3). In comparison, animals that received IPC had significantly less edema formation in their intermediate and outer zone samples (an increase of 1.31 ± 0.8 and 0.55 ± 0.35 g/g dry weight, respectively, compared with contralateral), although the edema in the core zone (2.50 ± 1.17 g/g dry weight) was not significantly different from shams. The reduction in brain edema formation with IPC in the intermediate and outer zones was associated with reduced sodium and chloride accumulation (Fig. 4A and 4C) and a reduction in potassium loss (Fig. 4B).
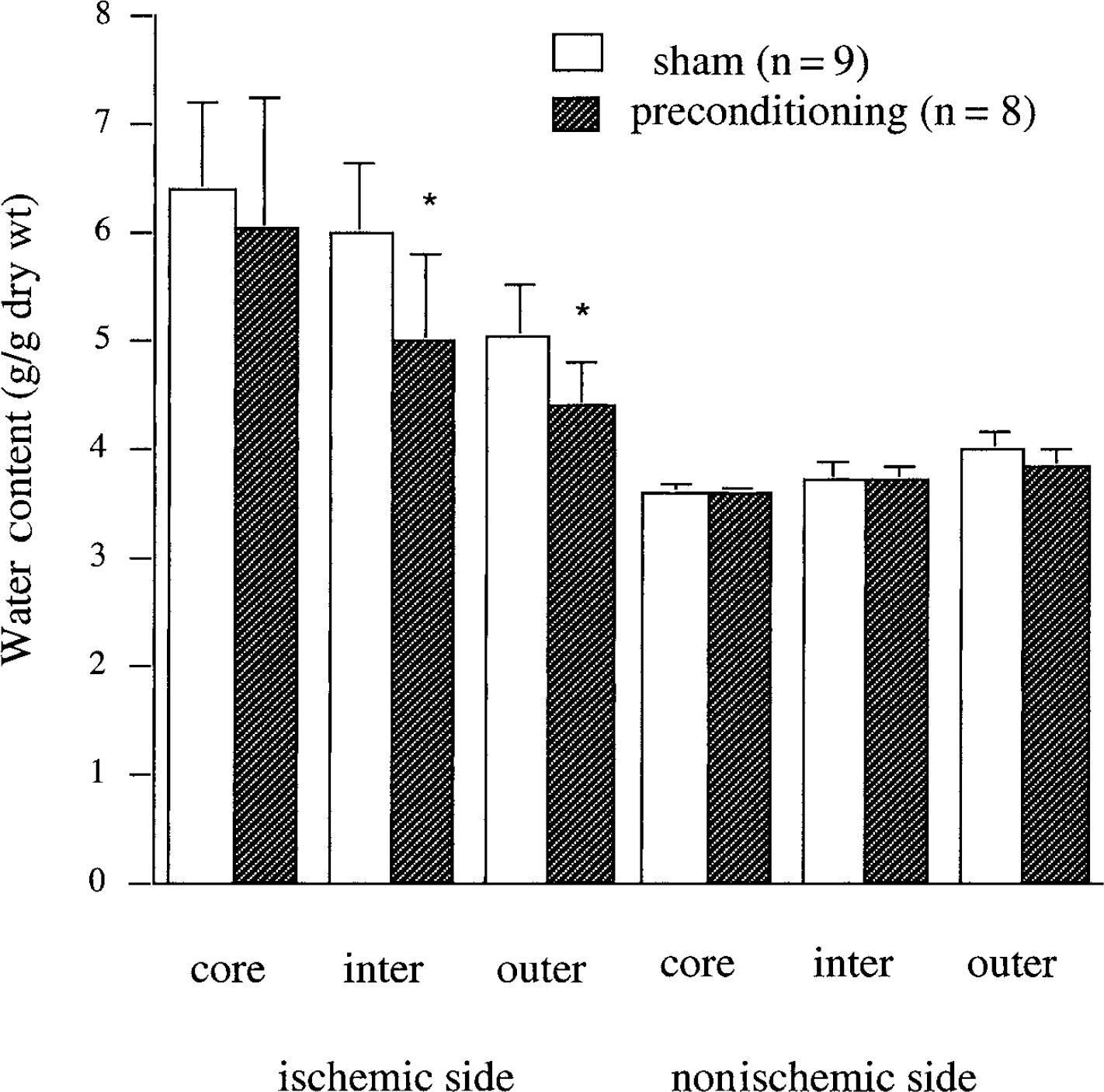
Brain water content 24 hours after permanent middle cerebral artery occlusion (MCAO). Measurements are from the core, intermediate (inter), and outer zones of the ischemic and nonischemic hemispheres. Rats received 15 minutes ischemic preconditioning (n = 8) or a sham operation (n = 9) 3 days before MCAO. Values are expressed as mean ± SD. *P < 0.01 vs. sham.
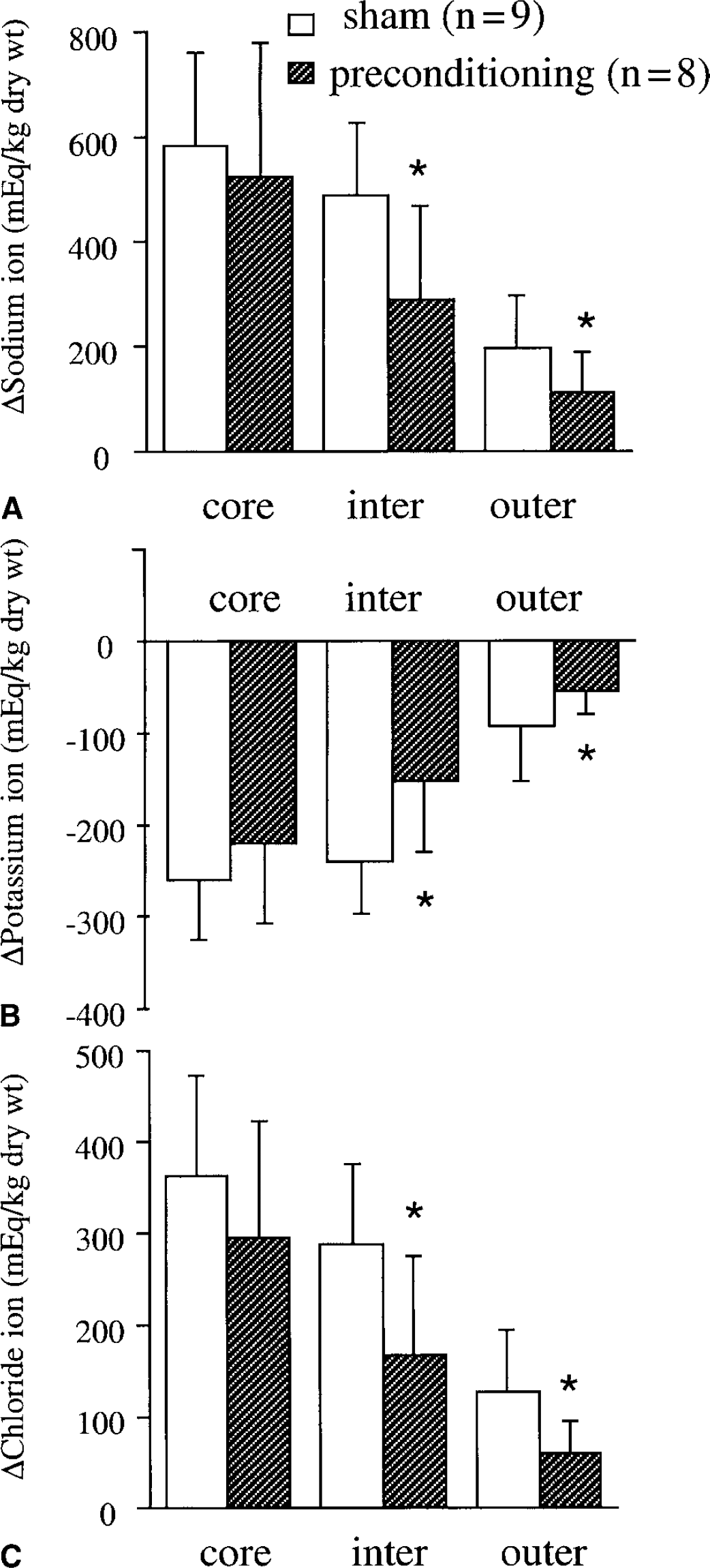
Changes in brain sodium
Blood–brain barrier permeability
Rats that had previously undergone a sham occlusion had marked BBB disruption after 6 hours of subsequent permanent MCAO. Thus, the influx rate constant (Ki) for [3H]-inulin was increased by 190%, 79%, and 43% in the core, intermediate, and outer zones compared with contralateral (Fig. 5). This BBB disruption was markedly attenuated in the IPC group (Fig. 5), where, for example, there was only a 75% increase in Ki in the core sample. There were no significant differences between the sham and IPC groups in the Ki of the contralateral hemisphere or the cerebellum.
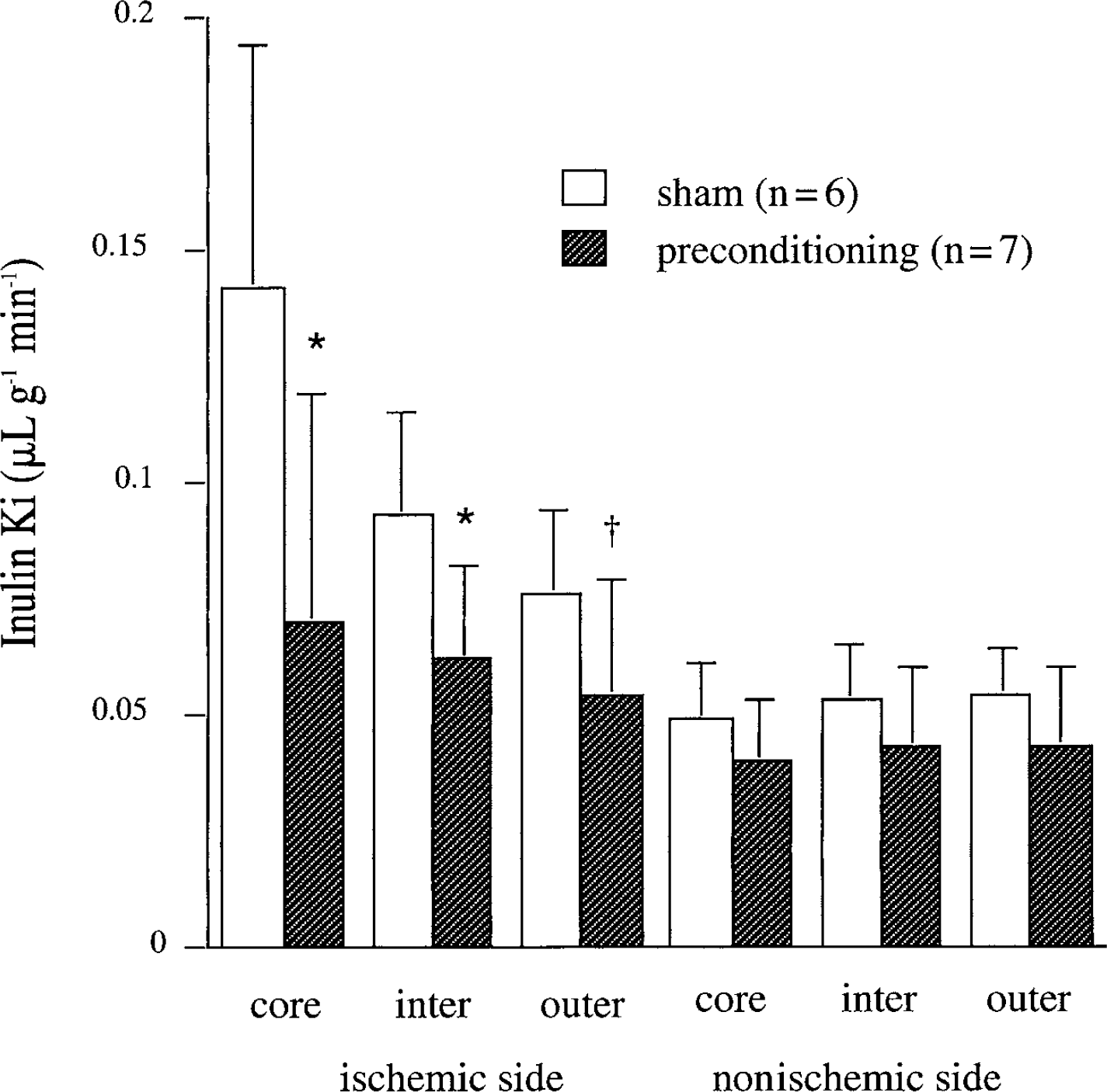
The influx rate constants (Ki) for [3H]-inulin in core, intermediate (inter), and outer zones ipsi- and contralateral to the site of middle cerebral artery occlusion (MCAO). Rats had undergone 15 minutes of ischemic preconditioning (n = 7) or a sham operation (n = 6) 3 days before 6 hours of permanent MCAO. Values are means ± SD. *P < 0.05 vs. sham. †P < 0.05 (one-tailed) vs. sham.
Cerebral blood flow
Both sham- and IPC-treated groups had a similar decline in cerebral blood flow upon permanent MCAO three days later in core, intermediate, and outer zones (Table 3). Similarly, contralateral blood flows were not different between the two groups.
Cerebral blood flow after 1 hour of permanent MCA
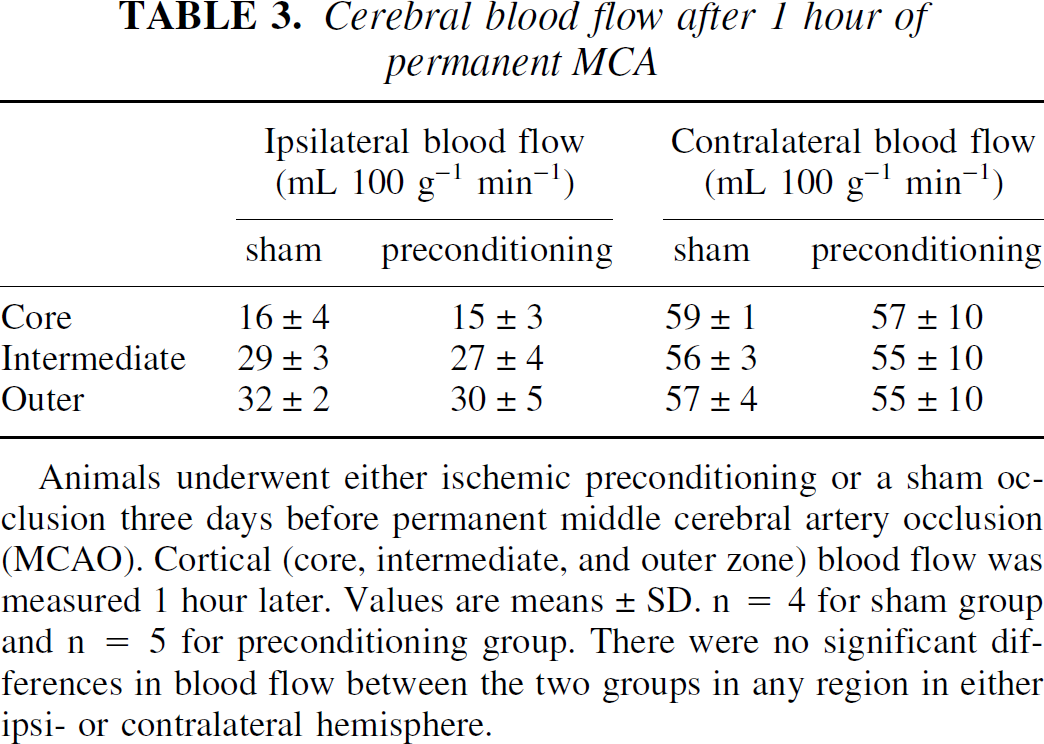
Animals underwent either ischemic preconditioning or a sham occlusion three days before permanent middle cerebral artery occlusion (MCAO). Cortical (core, intermediate, and outer zone) blood flow was measured 1 hour later. Values are means±SD. n = 4 for sham group and n = 5 for preconditioning group. There were no significant differences in blood flow between the two groups in any region in either ipsi- or contralateral hemisphere.
Heat shock protein 70 immunohistochemistry
Three days after 15 minutes of focal ischemia (IPC), a large number of ipsilateral cortical neurons were HSP70 immunoreactive (HSPir) in contrast with sham-operated rats (Fig. 6A and 6B). This was reflected in HSP70 Western blots on the ipsilateral cortical tissue from the two groups of rats (Fig. 6E). Ischemic preconditioning also induced HSPir in neurons in the striatum in contrast with sham-operated rats (Fig. 6C and 6D). In contrast with the parenchymal effects, HSP70ir blood vessels were not found after three days with IPC alone, nor were they found in rats that underwent sham occlusion (Fig. 6A and 6B).
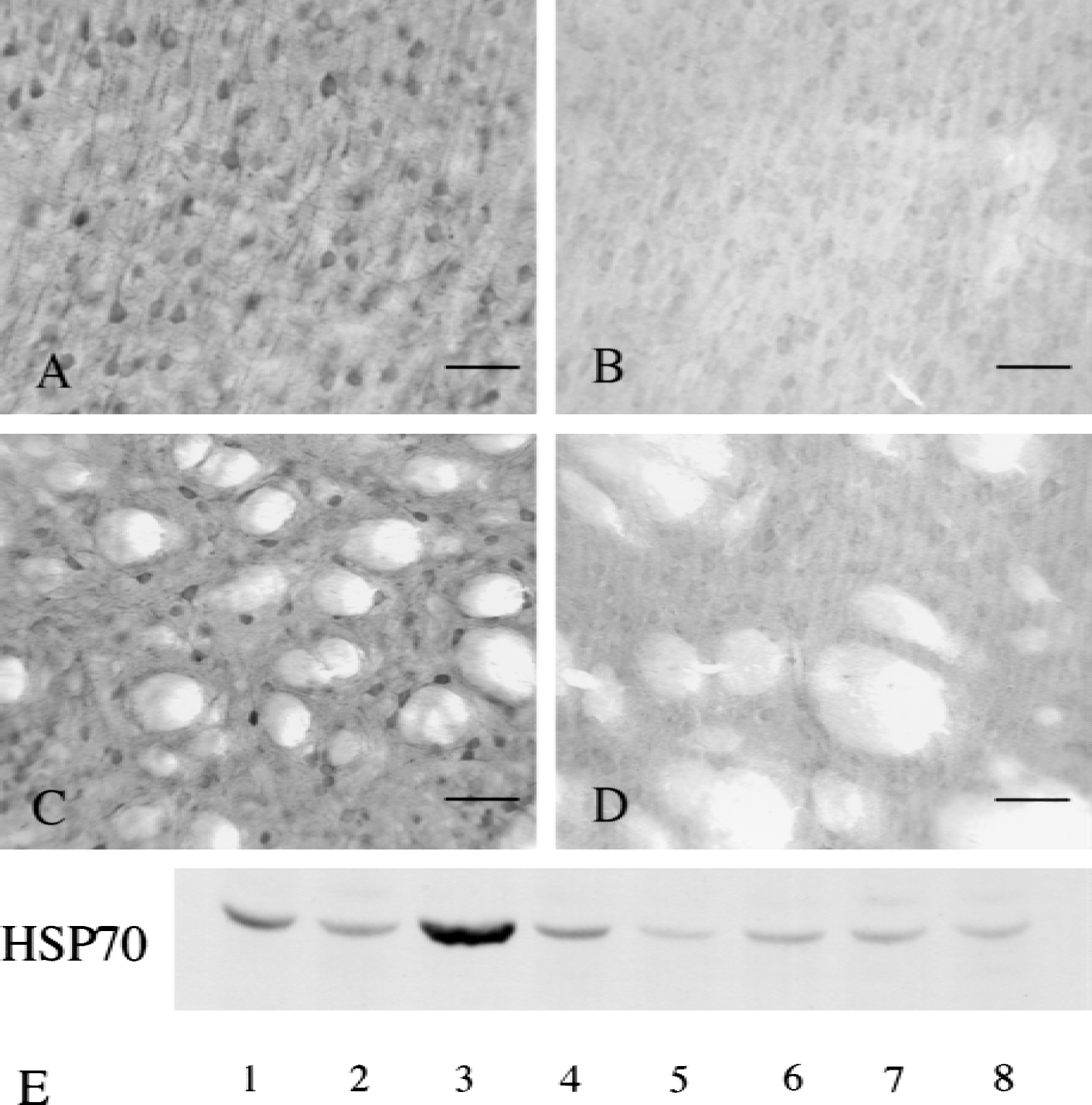
Cerebral heat shock protein 70 (HSP70) immunohistochemistry 3 days after 15 minutes of ischemic preconditioning (core cortex
In rats that received the previous sham occlusion, permanent MCAO resulted in the induction of HSP70 immunoreactivity in blood vessels at 3, 6, and 24 hours (for example, Fig. 7B in the core and Fig. 7D in the intermediate zone). The number of HSP70ir vessels in both ipsilateral cortex and basal ganglia remained approximately constant over the first 24 hours of ischemia (Fig. 8A and 8B). In comparison with sham-operated animals, rats that had undergone IPC 3 days before the onset of the permanent ischemia had far fewer HSP70ir blood vessels in the cerebral cortex 3, 6, and 24 hours after the onset of the permanent MCAO (Figs. 7A, 7C, and 8A). Ischemic preconditioned rats also had significantly less HSP70ir blood vessels than sham-operated rats in the basal ganglia at 3 hours after the onset of permanent ischemia (Fig. 8B).
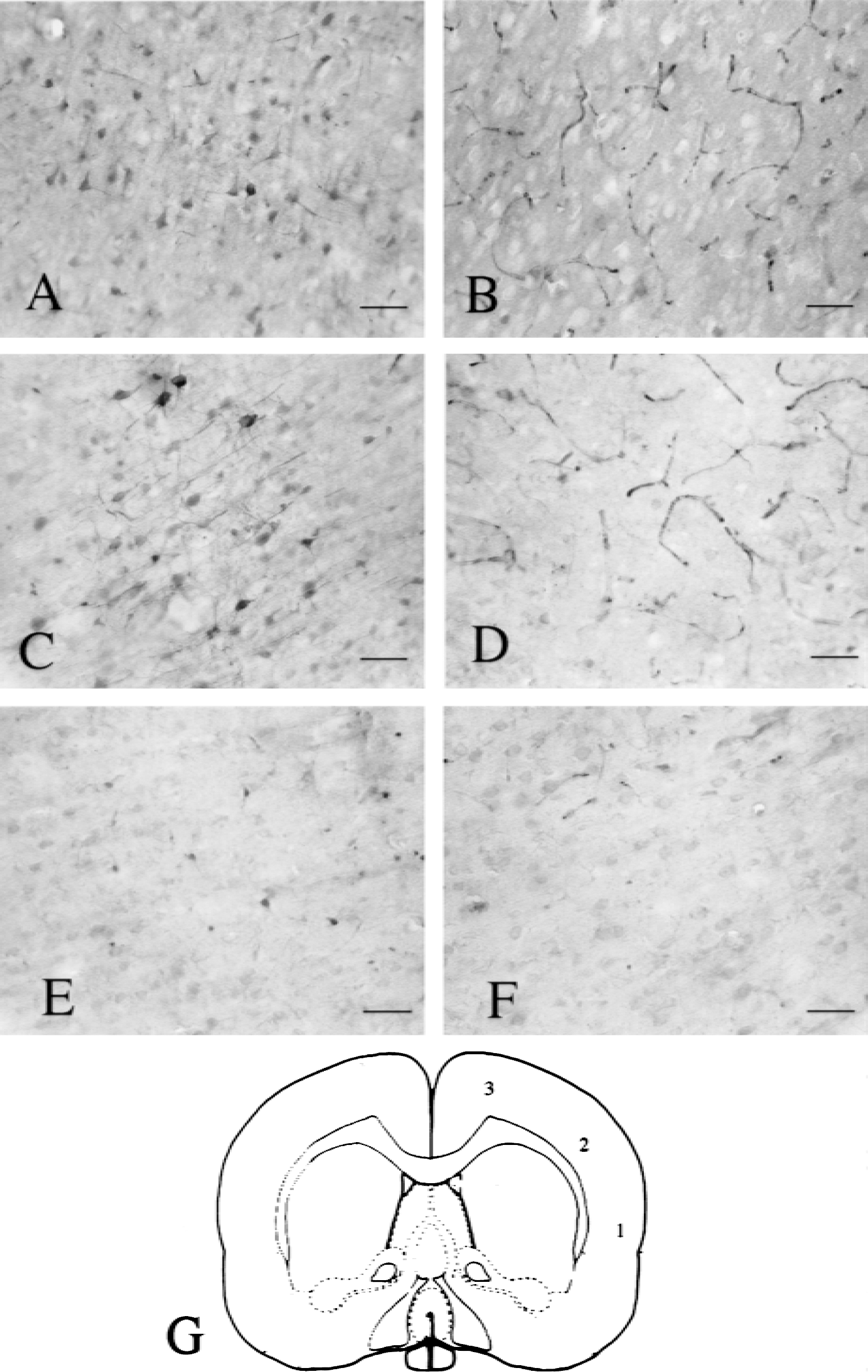
Heat shock protein 70 (HSP70) immunohistochemistry 6 hours after permanent middle cerebral artery occlusion (MCAO) in rats that had either ischemic preconditioning
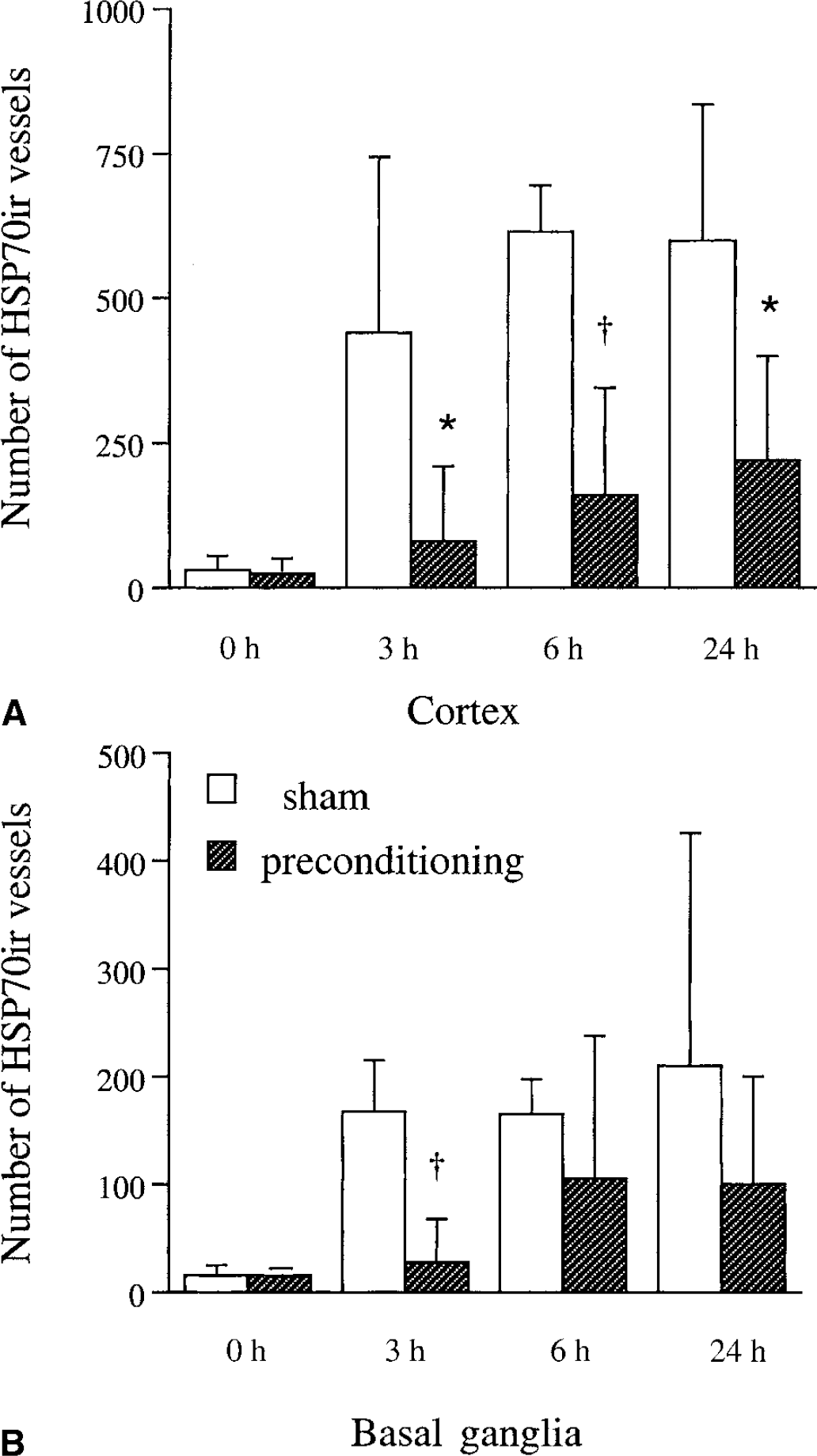
The number of heat shock protein 70 (HSP70) immunoreactive blood vessels 0, 3, 6, and 24 hours after permanent middle cerebral artery occlusion (MCAO) in rats that had undergone ischemic preconditioning or a sham operation. The HSP70ir vessel was measured in the cortex
At 6 and 24 hours, there was a tendency for a reduction with IPC but it did not reach significance. The distribution of HSP70ir blood vessels 6 hours after permanent MCAO in IPC and sham-treated rats is shown in Fig. 9C and 9D. In rats that had previously undergone a sham operation, the distribution of HSP70ir blood vessels was similar to that of the TTC-demarcated lesion found at 24 hours. In contrast, in IPC-treated rats, HSP70ir blood vessels were restricted to a very small area of the cortex and striatum (Fig. 9C).
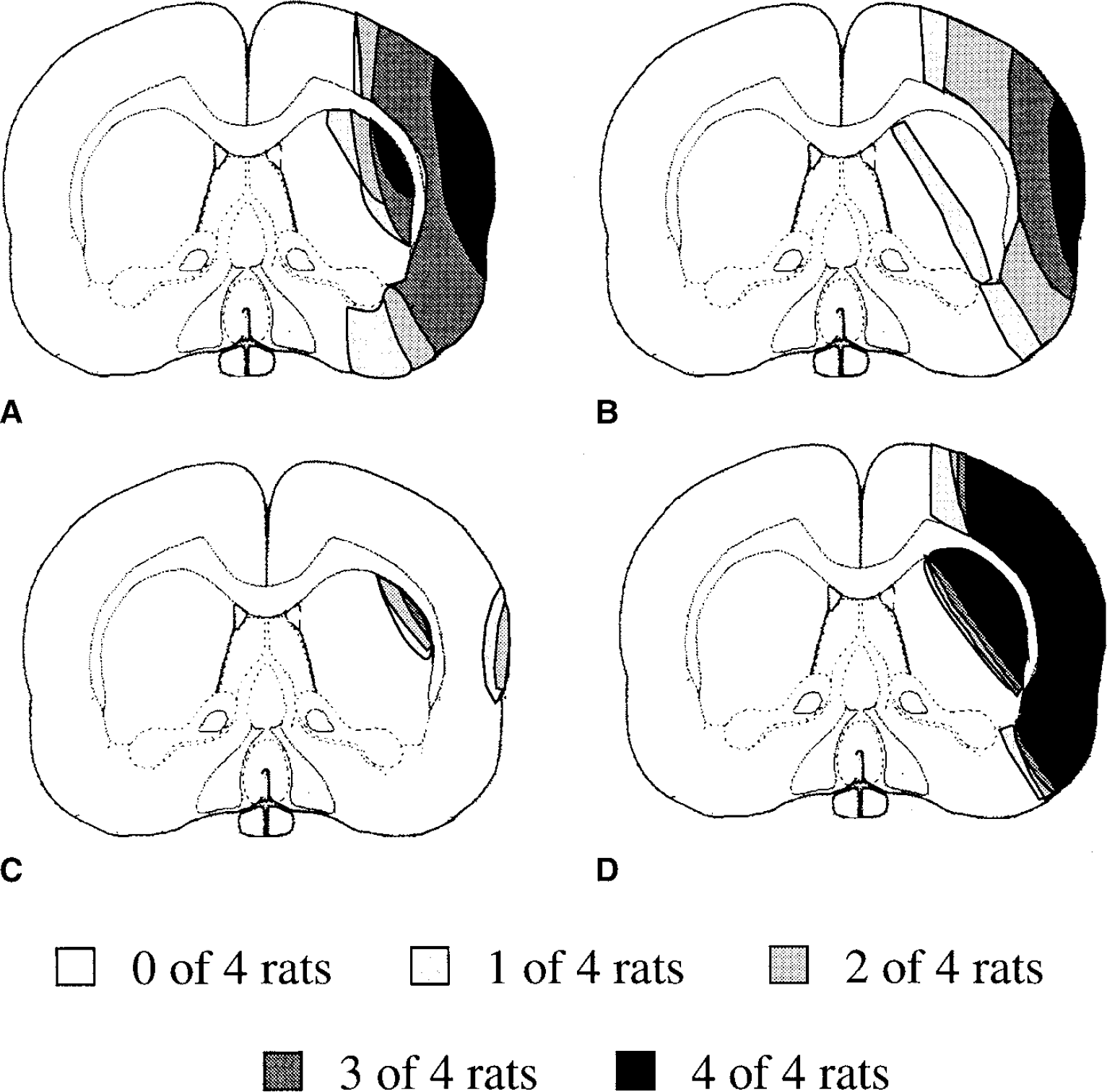
Distribution of HSPir 6 hours after permanent middle cerebral artery occlusion (MCAO) in rats that had previously undergone either ischemic preconditioning
In contrast with vascular HSP70ir, 6 hours after permanent MCAO the distribution of parenchymal cell HSP70ir was not markedly different between IPC and sham-treated rats (Fig. 9A and 9B) with the exception of the striatum, where very little parenchymal cell HSP70ir was seen in sham-treated rats, presumably because of the extensive parenchymal cell damage in this region.
DISCUSSION
The current study demonstrates that ischemic preconditioning, along with reducing infarct volume and motor deficit, significantly attenuates brain edema formation and BBB disruption after permanent MCAO. These results indicate that IPC protects the cerebrovasculature, a hypothesis also supported by a reduction in the number of HSP70ir blood vessels during permanent ischemia in IPC-treated rats.
Fifteen minutes of MCAO was used for ischemic preconditioning. Preliminary data indicated that this was an insult insufficient to induce an infarct. It was, however, sufficient to induce parenchymal cell HSP70 three days later. This response was somewhat variable (Fig. 6) probably as a result of differences in the degree of ischemia induced during the IPC insult. Imaging techniques that would allow measurements of blood flow during the initial temporary ischemia and brain injury during the subsequent permanent ischemia would enable a better correlation between the extent of the first insult and the degree of cerebrovascular protection.
Ischemic preconditioning attenuated brain edema formation in the intermediate and outer zones, but not in core zone. Thus, it appears that IPC may not be able to reduce edema formation in areas with low flows. Brain edema formation during permanent focal cerebral ischemia involves the movement of water and ions (primarily Na+ and Cl−) from blood to brain. Edema formation has both vasogenic (because of BBB disruption) and cytotoxic components (Betz, 1996).
The authors, therefore, examined whether BBB disruption was inhibited in IPC rats and found a marked reduction in disruption 6 hours after permanent MCAO indicating that IPC has a protective effect on the endothelial cells that form the BBB. This may contribute to the reduced edema formation in IPC-treated rats and may also contribute to reduced parenchymal cell injury by limiting the entry of potential harmful compounds from blood to brain. Interestingly, protection against BBB disruption was found in all three zones examined including the core. This may reflect the relative importance of cytotoxic and vasogenic edema formation in the different zones although it could also reflect the difference in time point for the 2 measurements (6 hours for BBB permeability and 24 hours for edema) if IPC only delays BBB disruption in the core. Protection of the cerebrovasculature in the core of the infarct with IPC, as evinced by reduced BBB disruption, is different from the parenchymal effects of IPC that appear to be limited to the penumbra (Barone et al., 1998; Chen et al., 1996).
Further evidence for a protective effect of IPC on the cerebrovasculature comes from HSP70 immunohistochemistry. Heat shock protein 70 has been thought of both as a neuroprotective agent (Kirino et al., 1991) and as a marker of cell stress (Gonzalez et al., 1989; Wagner et al., 1999) including endothelial injury (Lindsberg et al., 1996). The reduction in the number of HSP70ir blood vessels during the permanent MCAO by previous IPC suggests reduced cerebrovascular injury or stress. The profound reduction with IPC in the area of brain with such HSP70ir blood vessels (Fig. 9) also suggests that the cerebrovascular effects of IPC are not purely mediated by a reduction in infarct volume.
Most studies of BBB permeability during permanent focal ischemia conclude that there is no significant disruption until approximately 4 to 6 hours after the onset of ischemia (Betz and Dietrich, 1998). In the current study, there were HSP70ir blood vessels 3 hours after the onset of ischemia. This suggests that HSP70 may be produced in the cerebrovasculature before BBB disruption. Active transport at the BBB during ischemia also is inhibited before disruption (Keep et al., 1999).
Endothelial production of HSP70 thus may be an early sign of endothelial stress. Identifying such early vascular changes is important given that BBB disruption and resultant hemorrhagic transformation currently limits the use of plasminogen activator reperfusion therapy.
A number of mechanisms have been proposed to induce cerebrovascular damage during cerebral ischemia including energy failure, inflammation, free radicals, and arachidonic acid production (Betz, 1996). The mechanism by which IPC may protect against such damage remains to be elucidated. In common with other studies (Matsushima and Hakim, 1995; Chen et al., 1997; Barone et al., 1998), the current results indicate that the protective effects of IPC are not mediated by a change in cerebral blood flow during the subsequent ischemic event. Many substances are produced after IPC through induction of gene expression (Chen and Simon, 1997); these include interleukin-1 receptor antagonist and free radical scavengers (Barone et al., 1998; Kato et al., 1995; Ohtsuki et al., 1996; Toyoda et al., 1997) that might reduce inflammation- and free radical–induced damage. Although these substances are involved in the parenchymal cell protective effect, they may also contribute to the cerebrovascular protective effect. Because of the multiple potential links between parenchymal cells and the cerebrovasculature, it is difficult to isolate whether parenchymal cell protection may contribute to cerebrovascular protection or cerebrovascular protection may contribute to parenchymal protection. Notably, however, neuronal injury alone (induced by excitatory amino acids) does not appear to be sufficient to cause cerebrovascular HSP70 induction (Dutcher et al., 1998; Gass et al., 1995; Gonzalez et al., 1989), indicating that the ischemia-induced cerebrovascular HSP70 up-regulation modulated by IPC may be a direct effect on the vasculature. In addition, as noted above, although the effects of IPC on lesion volume are primarily penumbral (Barone et al., 1998; Chen et al., 1996), this study indicates that there is a reduction in BBB disruption even within the core of ischemic territory.
As mentioned above, HSP70 itself has been suggested to be involved in the neuroprotective effect of IPC (Kirino et al., 1991), and a number of studies, including this study, have shown that IPC induces the production of HSP70 in neurons (Chen et al., 1996; Chen and Simon, 1997). Prolonged cerebral ischemia up-regulates HSP70 in endothelial cells in normal rats and, to a reduced extent, IPC-treated rats, as shown in the current study and previous studies (Kinouchi et al., 1993a; Lindsberg et al., 1996). The current study does not address the question of whether such production of HSP70 by the vasculature is protective. Heat shock protein 70 is preferentially induced in injured but still viable cells (Nowak, 1991; Sloviter and Lowenstein, 1992), and there is evidence that the induction of HSP70 in endothelial cell protects it from injuries such as those induced by ischemia–reperfusion (Chen et al., 1997).
In conclusion, ischemic preconditioning can attenuate brain edema formation and cerebrovascular injury induced by permanent MCAO. Determining the mechanisms by which IPC preserves endothelial cell function and reduces edema formation during ischemia may be an important strategy for brain protection.