
Abstract
A prominent cognitive impairment after traumatic brain injury (TBI) is hippocampal-dependent memory loss. Although the histopathologic changes in the brain are well documented after TBI, the underlying biochemical mechanisms that contribute to memory loss have yet to be thoroughly delineated. Thus, we determined if calcium/calmodulin-dependent protein kinases (CaMKs), known to be necessary for the formation of hippocampal-dependent memories, are regulated after TBI. Sprague—Dawley rats underwent moderate parasagittal fluid-percussion brain injury on the right side of the parietal cortex. The ipsilateral hippocampus and parietal cortex were Western blotted for phosphorylated, activated α-calcium/calmodulin-dependent protein kinase II (α-CaMKII), CaMKIV, and CaMKI. α-Calcium/calmodulin-dependent protein kinase II was activated in membrane subcellular fractions from the hippocampus and parietal cortex 30 mins after TBI. CaMKI and CaMKIV were activated in a more delayed manner, increasing in phosphorylation 1 h after TBI. The increase in activated α-CaMKII in membrane fractions was accompanied by a decrease in cytosolic total α-CaMKII, suggesting redistribution to the membrane. Using confocal microscopy, we observed that α-CaMKII was activated within hippocampal neurons of the dentate gyrus, CA3, and CA1 regions. Two downstream substrates of α-CaMKII, the AMPA-type glutamate receptor GluR1, and cytoplasmic polyadenylation element-binding protein, concomitantly increased in phosphorylation in the hippocampus and cortex 1 h after TBI. These results demonstrate that several of the biochemical cascades that subserve memory formation are activated unselectively in neurons after TBI. As memory formation requires activation of CaMKII signaling pathways at specific neuronal synapses, unselective activation of CaMKII signaling in all synapses may disrupt the machinery for memory formation, resulting in memory loss after TBI.
Introduction
Traumatic brain injury (TBI) is a significant health concern, affecting 1.4 million people in the United States annually at a cost of $56 billion (Langlois et al, 2004). More than five million people are coping with disabilities from TBI. Underlying these disabilities are both focal and diffuse brain pathologies that result from the initial trauma and subsequent progressive secondary injury. Contusion formation, neuronal death, and diffuse axonal tract damage are the typical pathologies seen in TBI victims (Bigler et al, 2002; MacKenzie et al, 2002; Serra-Grabulosa et al, 2005; Zhang et al, 2005). These pathologies continue to progress from hours to days after the initial TBI and all are potential therapeutic targets. Unfortunately, there are no pharmacological therapies available to people suffering from TBI (Narayan et al, 2002).
The majority of TBI incidents are clinically classified as mild insults producing amnesia lasting less than 24h (Kraus and Nourjah, 1988). However, 50% of the people who sustain mild TBI exhibit cognitive impairments lasting 1 year after the head injury (Parker and Rosenblum, 1996). The most common cognitive impairments are learning and memory deficits. The hippocampus, which is absolutely critical for the formation of declarative memories, is particularly vulnerable to damage during TBI and in vivo magnetic resonance imaging has revealed a high prevalence of hippocampal atrophy among both moderate and severe TBI patients (Tate and Bigler, 2000; Tomaiuolo et al, 2004; Ariza et al, 2005; Hopkins et al, 2005). This most likely contributes to the significant percentage of TBI patients reporting long-term hippocampal-dependent, declarative memory impairments (Mathias and Mansfield, 2005; Serra-Grabulosa et al, 2005).
Hippocampal-dependent learning requires the activation of AMPA and NMDA-type glutamate receptors, which influx calcium into the postsynaptic neuron. Intracellular calcium activates multiple protein kinases that subserve memory formation (Selcher et al, 2002). These include α-Calcium/calmodulin-dependent protein kinase II (α-CaMKII), calcium/calmodulin-dependent protein kinase I (CaMKI), calcium/calmodulin-dependent protein kinase IV (CaMKIV), p44/p42 mitogen-activated protein kinase (MAPK), and protein kinase C (Atkins et al, 1998; Giese et al, 1998; Weeber et al, 2000; Kang et al, 2001; Schmitt et al, 2005).
Although a great deal is known about the signaling pathways regulated in the hippocampus during memory formation, how these pathways are affected during head injury is less well understood. Not surprisingly, these pathways are also important for the formation of hippocampal-dependent memories. These studies have found that p44/p42 MAPK and c-jun N-terminal protein kinase are activated in the hippocampus after TBI (Dash et al, 2002; Mori et al, 2002; Otani et al, 2002; Carbonell and Mandell, 2003; Raghupathi et al, 2003; Griesbach et al, 2004a; Lu et al, 2005). Correspondingly, the transcription factor cAMP-responsive element binding protein (CREB), a downstream substrate of MAPK, is phosphorylated after TBI in the hippocampus (Dash et al, 1995; Carbonell and Mandell, 2003; Griesbach et al, 2004a; Hu et al, 2004). However, global changes in calcium signaling through glutamate excitotoxicity and potassium depolarization waves suggest that many calcium-dependent protein kinases are likely to be regulated during TBI (Takahashi et al, 1981; Katayama et al, 1990; Fineman et al, 1993; Nilsson et al, 1993; Osteen et al, 2004). Using a model of TBI that elicits hippocampal-dependent memory impairments, fluid-percussion brain injury (FPI) (Lyeth et al, 1990; Bramlett et al, 1997a; Carbonell et al, 1998; Sanders et al, 1999; Griesbach et al, 2004b), we determined if calcium/calmodulin-dependent protein kinases known to be critical for the formation of hippocampal-dependent memories, such as α-CaMKII, CaMKIV, and CaMKI, are affected by TBI.
Materials and methods
Traumatic Brain Injury Model
All procedures were in compliance with the NIH Guide for the Care and of Laboratory Animals and approved by the University of Miami Animal Care and Use Committee. Traumatic brain injury was induced in male Sprague—Dawley rats (270 to 320 g, Charles River Laboratories, Raleigh, NC, USA) using the parasagittal FPI device overlying the right, ipsilateral parietal cortex (Lotocki et al, 2004). All possible measures were taken to reduce animal suffering and the numbers of animals used in this study. Rats were anesthetized with 3% halothane, 70% N2O, and a balance of 30% O2, then intubated endotracheally and mechanically ventilated using a Harvard rodent ventilator (Harvard Apparatus, Holliston, MA, USA) with 1.5% halothane, 70% N2O, and 30% O2. Pancuronium bromide (0.5 mg/kg) was intravenously administered to immobilize the rats and facilitate mechanical ventilation. The femoral artery and vein were cannulated to deliver drugs and monitor arterial blood pressure, blood gas, and serum glucose. Arterial blood gases were measured 15 mins before TBI and up to 4 h after TBI. Rectal and temporalis muscle thermistors were used to maintain a core brain temperature of 37°C using self-adjusting feedback warming lamps. To produce TBI, the animals were placed in a stereotaxic frame and a 4.8 mm craniotomy (3.8 mm posterior to bregma, 2.5 mm lateral to the midline) was created to anchor a modified plastic 18 gauge syringe hub (8 mm length, PrecisionGlide needle, Becton Dickinson, Franklin Lakes, NJ, USA) over the exposed dura of the right parietal cortex. At 24 h after the craniotomy, the animals were anesthetized, intubated, and then placed over the FPI device. The device was connected to the plastic syringe hub and a moderate pressure pulse of 1.8 to 2.2 atmospheres was delivered at bregma level −3.8 mm. Sham-operated animals received all of the surgical steps, but did not undergo the FPI insult. If mortality or lung edema resulted from the FPI, animals were excluded from analysis (n = 2 of 82 animals).
Western Blots
Animals were anesthetized, decapitated, and the right, injured parietal cortex encompassing the area of injury and the ipsilateral hippocampus were rapidly dissected on ice and frozen in liquid nitrogen within 1 min after removal of the brain. Animals were killed at 15 mins (n = 7 TBI animals, n = 4 sham animals), 1 h (n = 7 TBI animals, n = 4 sham animals), 4 h (n = 7), and 24 h (n = 7 TBI animals, n = 4 sham animals) for Western blot analysis. However, the changes in phospho-α-CaMKII threonine 286 (pCaMKII Thr286) after FPI are highly labile. Thus, for pCaMKII Western blot analysis, animals were anesthetized and the brains were frozen in situ with liquid nitrogen to preserve the phosphorylation status of CaMKII. Animals were killed at 30 min, 4 h, and 24 h after FPI and compared with sham animals (n = 6 for each group). The cortices and hippocampi were dissected at −12°C in a glove box and then fractionated.
We were interested in specifically studying the biochemical changes that occur at the synapse; thus, the tissues were fractionated to yield a membrane subcellular preparation containing synaptosomes as previously described with minor modifications (Hu et al, 1999; Liu et al, 2004). The tissue was homogenized using a Dounce homogenizer (35 strokes, 4°C) in 1.5 ml of lysis buffer: 15 mmol/L HEPES pH 7.6, 0.25 mmol/L sucrose, 1 mmol/L MgCl2, 2.5 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT, 1.25 μg/ml pepstatin, 25 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mmol/L benzamidine, 0.5 mmol/L PMSF, 0.1 mmol/L Na3VO4, 50 mmol/L NaF, 2 mmol/L Na4P2O7, and 1 × phosphatase inhibitor cocktail set I (Calbiochem, La Jolla, CA, USA). For pCaMKII, because tissue was a limiting factor, the samples were centrifuged only once (10,000g, 10 mins 4°C) to generate a crude membrane pellet containing synaptosomes and nuclei, which was resuspended in lysis buffer with 10% TX-100, and a supernatant that was primarily cytosol that was used for Western blotting. For phospho-CaMKI threonine 177 (pCaMKI Thr177) and phospho-GluR1 serine 831 (pGluR1 Ser831), which are not as labile after FPI, the total, nonfractionated homogenates were centrifuged (800g, 10 mins 4°C) to remove the pellet containing primarily nuclei. The supernatants were centrifuged again (10,000g, 10 mins 4°C) and the membrane pellet containing synaptosomes was resuspended in lysis buffer with 10% TX-100 for use in Western blotting. Each subcellular fraction was assayed for total protein using the Coomassie Plus assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Samples were boiled with 1 × sample buffer for 7 to 9 mins at 95°C. Equal amounts of protein (30 μg/lane) from each fraction were electrophoresed (12.5% SDS-PAGE). The membrane subcellular fractions containing synaptosomes and nuclei were Western-blotted for pCaMKII (1:1000, Cell Signaling Technology, Beverly, MA, USA) and α-CaMKII (1:2000, Cell Signaling Technology). The more purified synaptosome subcellular fractions were Western-blotted for pCaMKI (1:1000, generous gift of TR Soderling, Oregon Health and Sciences University, Portland, OR, USA and N Nozaki, Kanagawa Dental College, Yokosuka, Kanagawa, Japan), CaMKI (1:1000, Affinity BioReagents, Golden, CO, USA), pGluR1 (1:1000, Upstate Cell Signaling Solutions, Charlottesville, VA, USA), and GluR1 (1:5000, Upstate Cell Signaling Solutions). Nonfractionated total homogenates were Western blotted for phospho-CPEB threonine 171 (pCPEB Thr171, 1:20 of hybridoma supernatant, generous gift of TR Soderling and N Nozaki), CPEB (1:20 of hybridoma supernatant, TR Soderling and N Nozaki), phospho-CaMKIV threonine 196 (pCaMKIV Thr196, 1:1000, generous gift of TR Soderling and N Nozaki), CaMKIV (1:1000, Transduction Laboratories, San Jose, CA, USA), and α-CaMKII (1:2000, Cell Signaling Technology). Total protein for each fraction was normalized by Western blotting for β-tubulin (1:5000, Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA, USA). Epitopes were visualized with HRP-conjugated secondary antibodies (1:1000 to 1:5000, Cell Signaling Technology) using the Phototope HRP Western blot detection system (Cell Signaling Technology) and developed on film (Phenix x-ray film BX, Phenix Research Products, Hayward, CA, USA). Films were developed to be in a linear range and densitized using LabWorks software (Ultra-Violet Products, Upland, CA, USA). Levels of phospho-protein immunoreactivity (e.g., pCaMKII) were normalized to total protein immunoreactivity (e.g., α-CaMKII), and then to β-tubulin immunoreactivity.
Confocal Microscopy
Double-label fluorescence confocal microscopy was performed as previously described (Hu et al, 2004). For confocal microscopy analysis, animals were killed at 30min, 4h, and 24 h after FPI and compared with sham animals (n = 3 to 4 for each group). Animals were anesthetized with 3% halothane and perfused for 30 mins with ice-cold 4% phosphate-buffered paraformaldehyde while maintaining the animals' breathing with a respirator. The brains were removed and sectioned (50 μm thick) with a Leica vibratome (Leica Microsystems, Inc., Exton, PA, USA). Sections were washed twice in Tris-buffered saline (TBS) for 5 mins at room temperature (RT), washed in 0.01 mol/L citrate buffer pH 6.0 for 5 mins at RT, and then heated for 10 mins in a 100°C water bath to improve antigenicity. The sections were cooled for 10min, washed with 0.2% TX-100 in TBS for 20min, and blocked for 1 h at RT in 3% bovine serum albumin (BSA) and 0.1% TX-100 in TBS. Sections were incubated overnight at 4°Cin 1% BSA and 0.1% TX-100 in TBS with the following primary antibodies: pCaMKII Thr286 (1:400, Cell Signaling Technology) and α-CaMKII (1:400, Cell Signaling Technology). To ensure specificity of the antibodies, controls lacking the primary antibody were included (Hu et al, 1995b; Hu and Wieloch, 1995; Tang et al, 2004). Sham and TBI sections were immunostained in parallel. Sections were washed twice in 0.1% TX-100 in TBS and then incubated with secondary antibody (fluorescein-labeled anti-rabbit, 1:200, Molecular Probes, Carlsbad, CA, USA) and propridium iodide (1:300, Molecular Probes) for 1 h at RT. Sections were rinsed twice in 0.1% TX-100 in TBS, mounted, and coverslipped with Vectashield mounting medium (Vector Laboratories Inc., Burlingame, CA, USA). Sections were analyzed with a Zeiss laser scanning confocal microscope (Zeiss, Inc., Thornwood, NY, USA). Sham and TBI sections were imaged using identical microscopy settings.
Statistical Analysis
Data presented are mean ± standard deviation. All means are represented as the fold increase above sham animal levels. Statistical analyses are one-way ANOVAs with post hoc Tukey HSD t-tests. *Denotes P < 0.05, **P < 0.01, and ***P < 0.001.
Results
To determine if calcium/calmodulin-dependent protein kinases (CaMKs) were regulated after TBI, we Western blotted subcellular fractions from the ipsilateral, injured hippocampus and parietal cortex of animals subjected either to moderate parasagittal FPI or sham surgery. Using antibodies to autophosphorylated, activated α-CaMKII Thr286 (pCaMKII), we observed a significant increase in pCaMKII 30 mins after TBI (4.1 ± 2.0 fold increase above sham levels, n = 6) in membrane subcellular fractions from the hippocampus (Figure 1). At 4 and 24 h after TBI, pCaMKII levels were still elevated nonsignificantly above sham levels (1.8 ± 0.7, n = 6 and 2.571.5, n = 6, respectively). In the parietal cortex, pCaMKII increased in membrane subcellular fractions at 30 mins (2.9 ± 2.5, n = 6), 4h (1.6 ± 0.7, n = 6), and 24h (2.7 ± 2.7, n = 6) after TBI; however, this was more variable than the changes seen in the hippocampus. In both the hippocampus and parietal cortex, the increases in pCaMKII were accompanied by an increase in total α-CaMKII levels in membrane subcellular fractions, with a loss of α-CaMKII immunoreactivity in the cytosolic fraction (Figure 2). These results suggest that as α-CaMKII underwent autophosphorylation after TBI, it redistributed from the cytosol to the membrane.
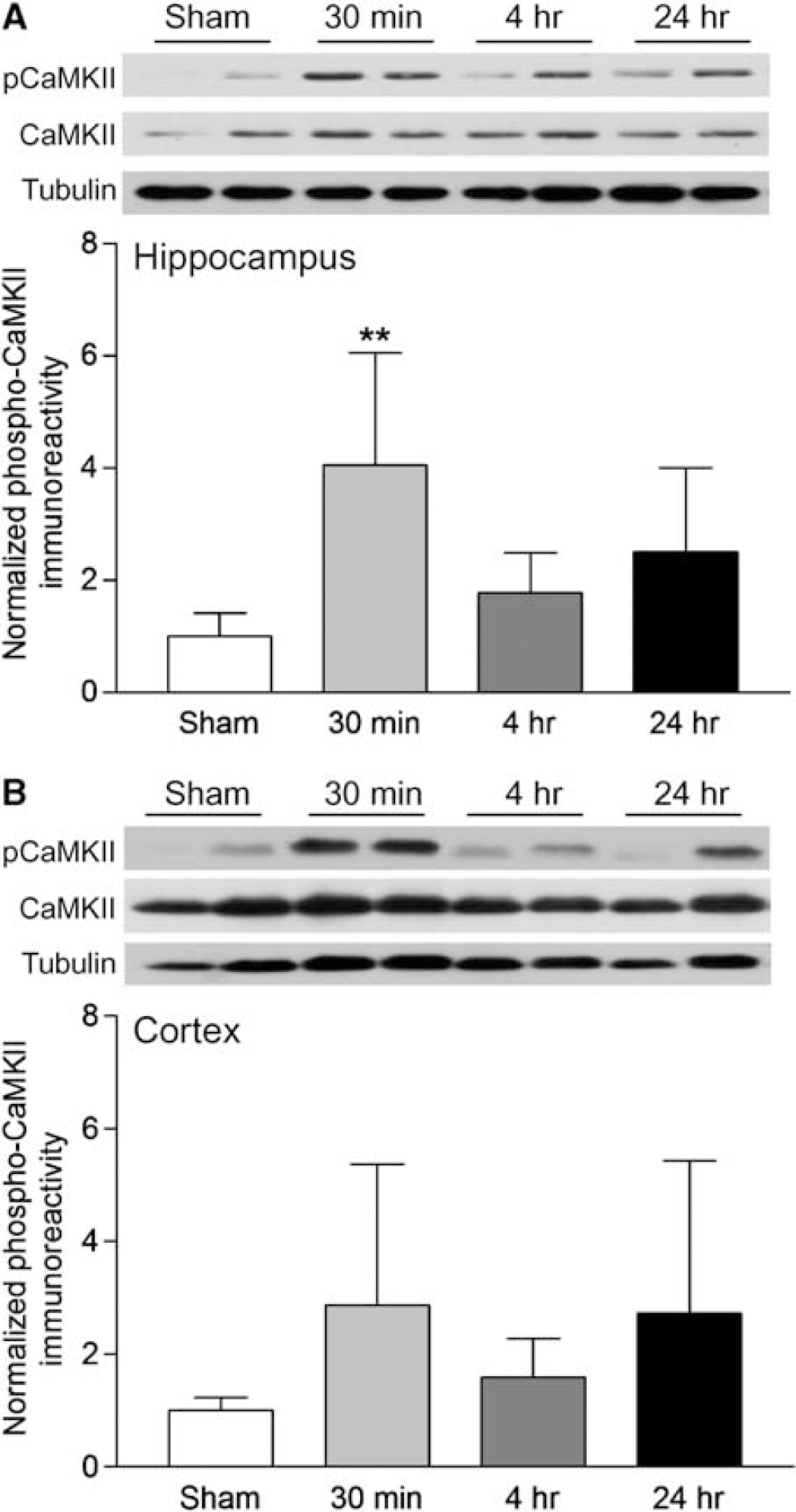
Activation of α-CaMKII after TBI. (
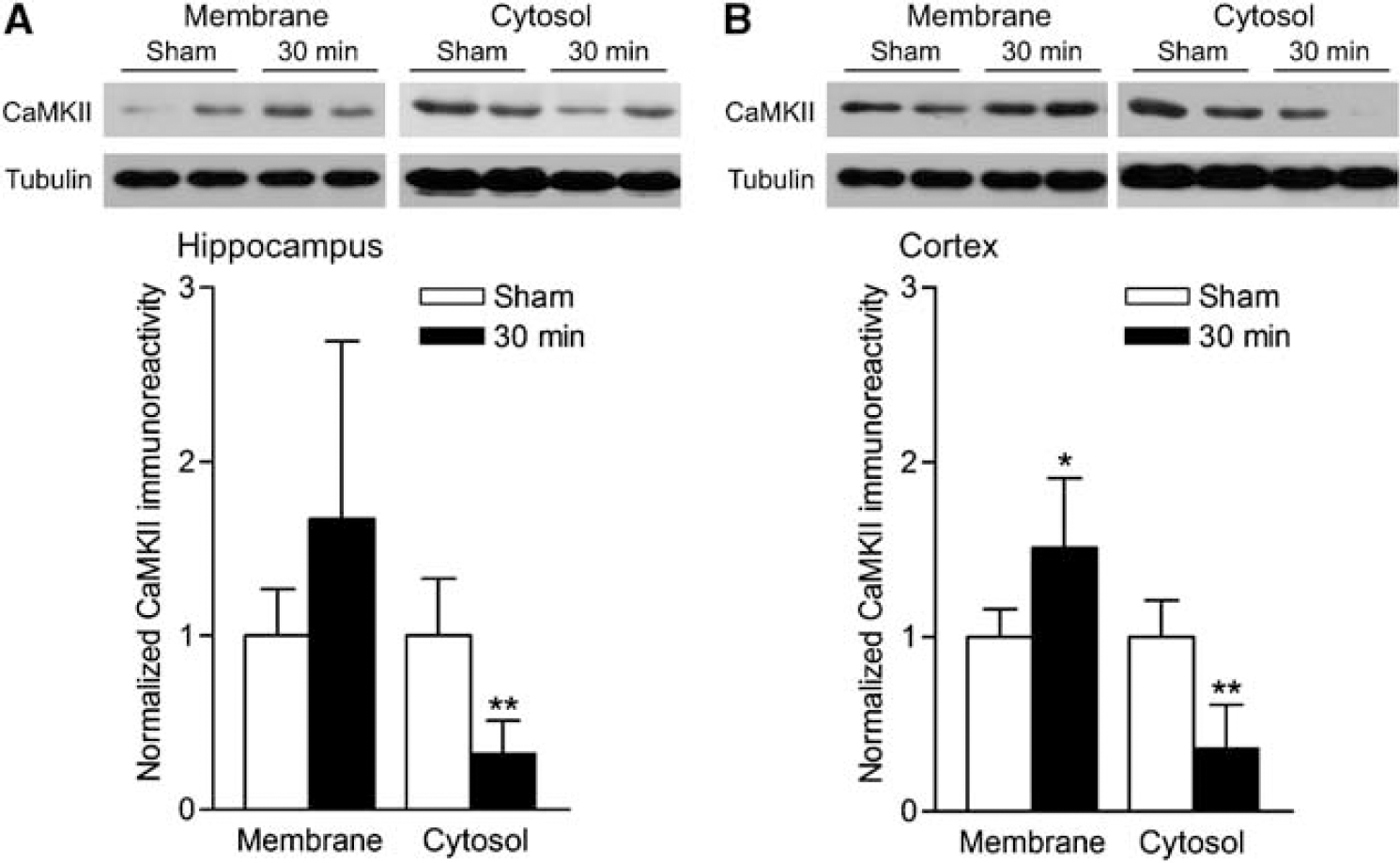
Redistribution of α-CaMKII localization 30 mins after TBI. (
Using confocal microscopy, we assessed the spatial localization of the changes in pCaMKII after TBI (Figure 3). In the hippocampus, pCaMKII increased in the somata and primary dendrites of neurons in all three major regions of the hippocampus, the dentate gyrus, CA3, and CA1 regions. This was most pronounced at 30 mins after TBI, although an increase was also seen 4 h after TBI. In the parietal cortex, pCaMKII increased in the primary dendrites and somata of neurons 30 mins after TBI. However, we did not observe any changes in total CaMKII immunoreactivity at any time point examined. These results indicate that the changes in pCaMKII after TBI are primarily in neurons, occur in all regions of the hippocampus, and throughout the parietal cortex.
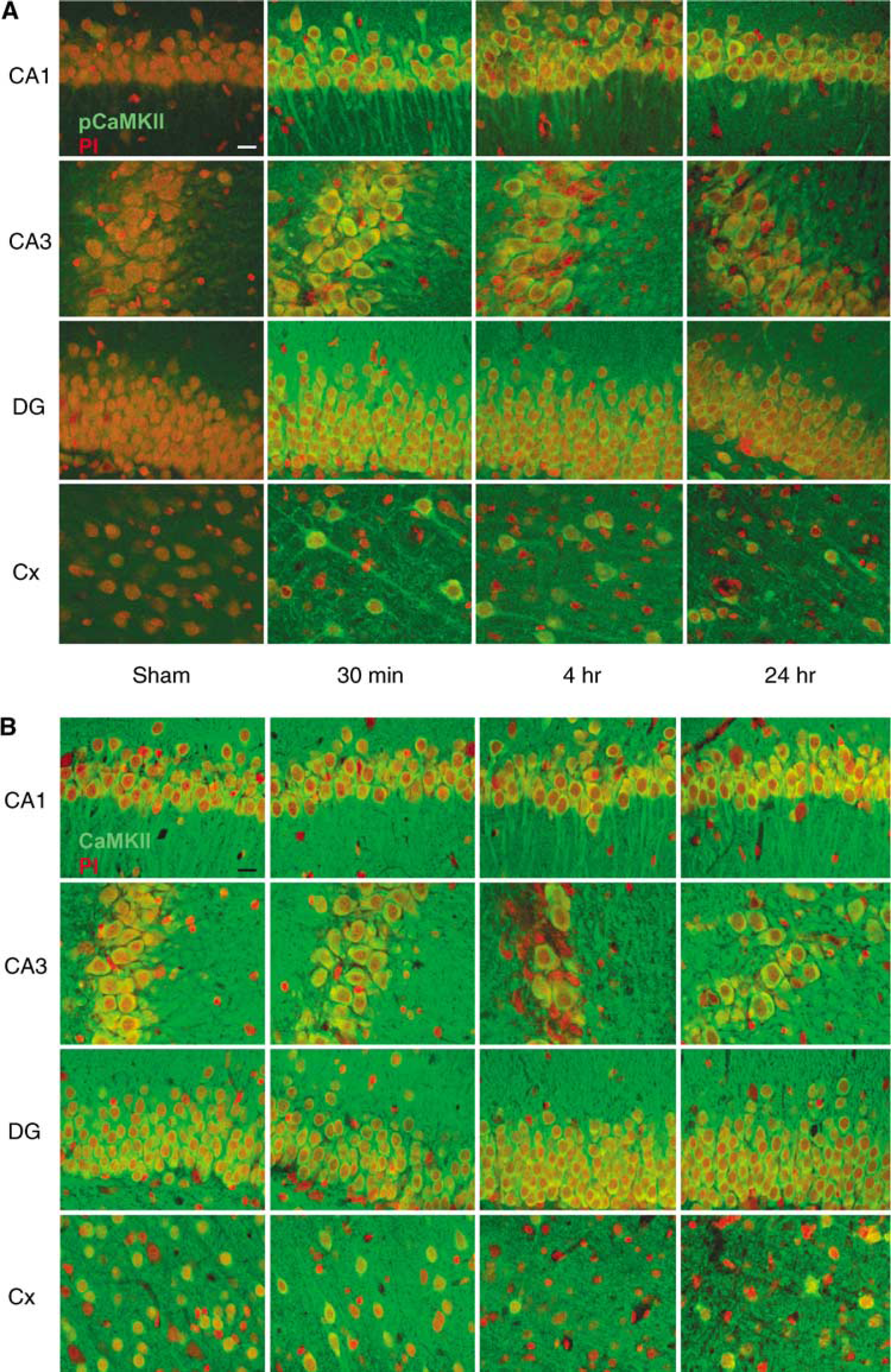
Localization of the changes in autophosphorylated, activated α-CaMKII after TBI. (
α-Calcium/calmodulin-dependent protein kinaseII regulates synaptic strength by phosphorylating the AMPA-type glutamate subunit 1 Ser831 (GluR1), which increases synaptic conductance (Derkach et al, 1999). This occurs during forms of synaptic plasticity, such as hippocampal long-term potentiation (LTP), and is also required for hippocampal-dependent learning and memory formation (Barria et al, 1997; Lee et al, 2000, 2003). During hippocampal LTP, α-CaMKII also phosphorylates the translation factor, cytoplasmic element binding protein (CPEB), which regulates the translation of dendritic mRNAs (Mendez et al, 2000; Atkins et al, 2004, 2005). To determine if the activation of α-CaMKII after TBI corresponded with phosphorylation of its downstream substrates, we probed hippocampal and cortical extracts with antibodies to phosphorylated GluR1 Ser831 (pGluR1), and antibodies to phosphorylated CPEB Thr171 (pCPEB). In total, cellular homogenates from the hippocampus, pCPEB increased at 1 to 4 h after TBI, returning to baseline levels by 24 h (Figure 4). In synaptosome subcellular fractions from the hippocampus, pGluR1 was also increased 1 h after TBI, and returned to basal levels by 4 h after TBI. Neither pCPEB nor pGluR1 significantly increased on the contralateral side of the hippocampus (data not shown). There were no changes in total GluR1 levels in synaptosome fractions or in nonfractionated total homogenates (not shown). In the parietal cortex, where the increases in activated α-CaMKII were lower and more variable, we observed only an increase in pGluR1 at 4 h after TBI, and no change in pCPEB levels. A small, significant increase in pCPEB and pGluR1 was observed in the contralateral parietal cortex at the 1 h time point (pCPEB 2.8 ± 1.1, n = 5, P < 0.001; pGluR1 1.8 ± 0.4, n = 5, P < 0.001). Thus, like the activation of CaMKII, substrates of CaMKII increased in phosphorylation rapidly after TBI (at 1 to 4 h), and began to return to baseline by 24h.
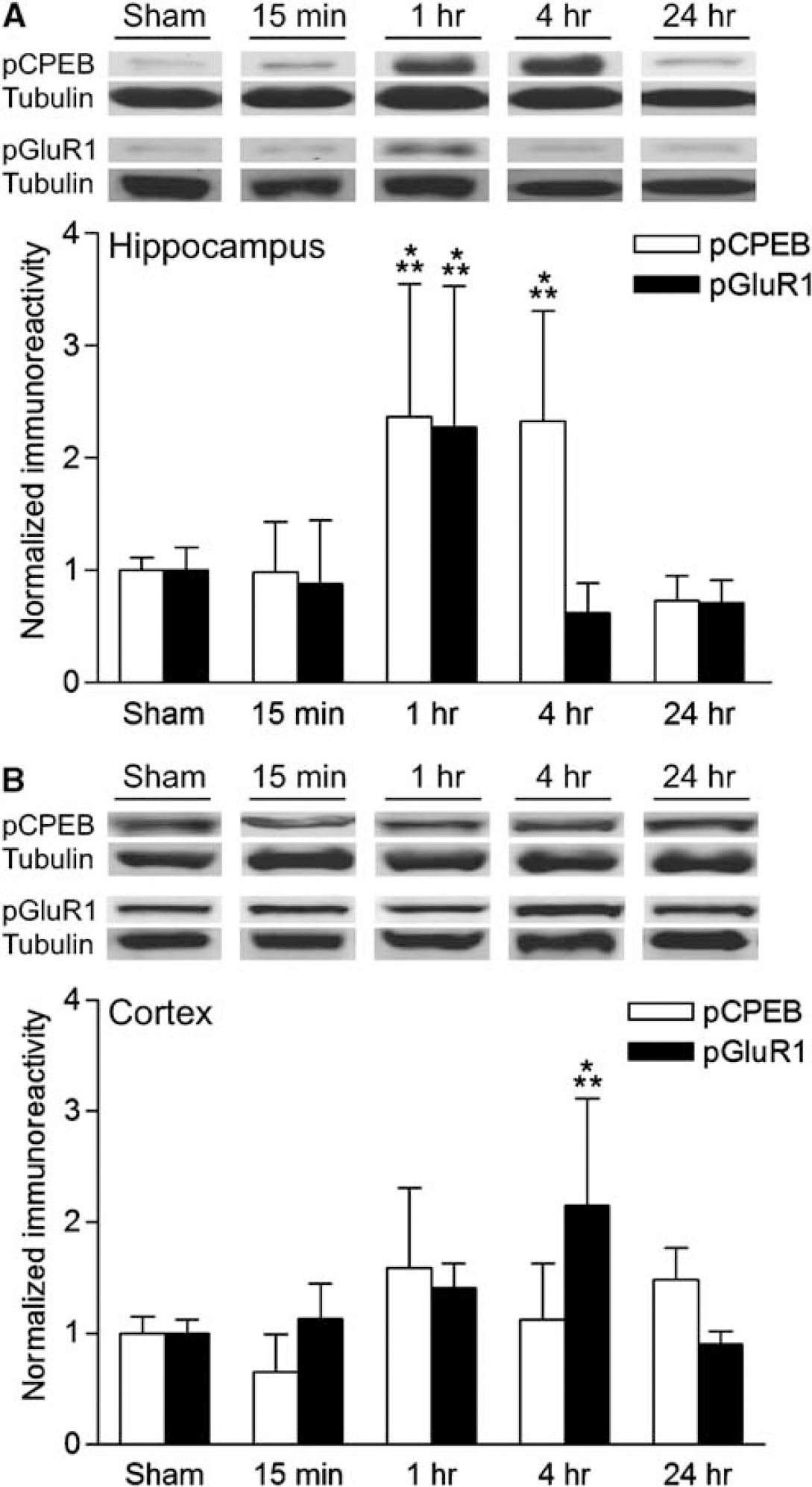
Regulation of α-CaMKII substrates after TBI. (
Calcium entry into neurons during memory formation and synaptic plasticity in the hippocampus triggers a multitude of biochemical signaling cascades (Elgersma et al, 2004; Mizuno and Giese, 2005). Besides activating α-CaMKII and increasing the phosphorylation of its downstream substrates, calcium entry also activates other members of the CaMK family (Kasahara et al, 2001; Schmitt et al, 2005). These include CaMKK, which phosphorylates and activates CaMKI and CaMKIV when bound to calcium/calmodulin (Soderling and Stull, 2001). CaMKI is a synaptically localized protein kinase that regulates the activation of MAPK during hippocampal LTP, and CaMKIV is a nuclear protein kinase that regulates transcription factors, such as CREB (Kang et al, 2001; Schmitt et al, 2005). To assess if other members of the CaMK family were regulated after TBI, we probed extracts from the cortex and hippocampus with antibodies to activated CaMKIV Thr196 (pCaMKIV) and antibodies to activated CaMKI Thr177 (pCaMKI). We observed a significant increase in both pCaMKIV and pCaMKI activation at 1 h after TBI in the hippocampus (Figure 5). On the contralateral side, there were no significant increases in pCaMKIV and pCaMKI in the hippocampus (data not shown). In the parietal cortex, only pCaMKI was significantly increased, and this occurred 1 h after TBI. A smaller increase in pCaMKI was also observed in the contralateral parietal cortex 1h after TBI (2.470.8, n = 5, P < 0.001). Thus, calcium entry during TBI triggers a range of calcium-dependent signaling cascades, activating several members of the CaMK family in the hippocampus.
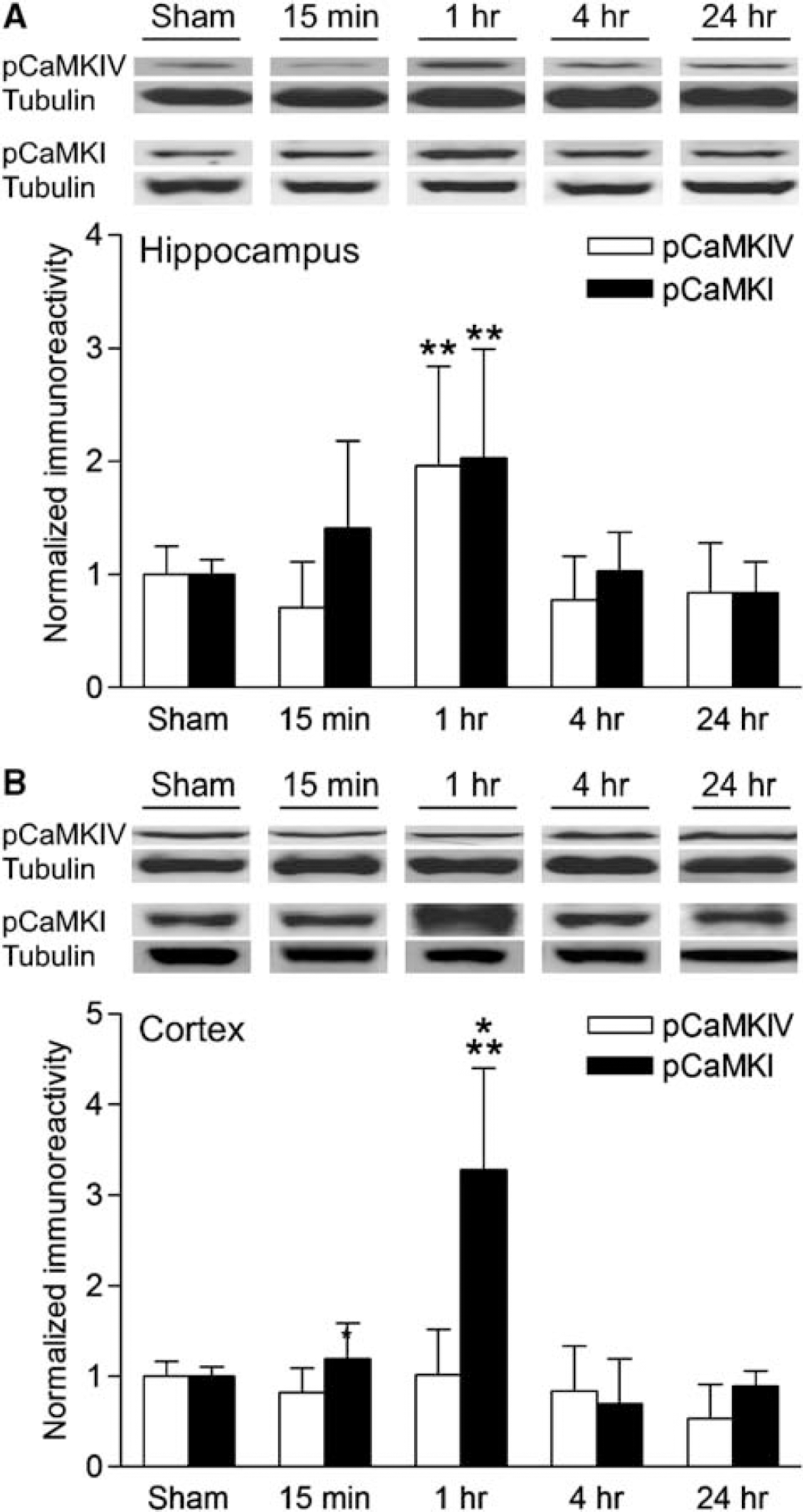
Activation of the CaM kinase family after TBI. (
Discussion
Traumatic brain injury causes significant behavioural impairments that are complex in both their nature and etiology (McAllister, 1992). Among the many behavioural problems seen in TBI patients, a typical and profound consequence is memory loss and in the ability to form new memories (Lovell and Franzen, 1994; Whyte et al, 1996; McAllister et al, 2004). This significantly interferes with patient recovery and quality of life after head injury (Ben-Yishay and Diller, 1993; Cicerone et al, 2000). Thus, to develop new therapeutic interventions, it is important to identify the molecular mechanisms underlying memory that are disrupted after TBI. We have now described a potential biochemical mechanism of why hippocampal-dependent memories are lost and impaired after TBI. α-Calcium/calmodulin-dependent protein kinase II, an important molecular decoder of hippocampal synaptic plasticity and memory formation, is significantly activated after TBI, as are two of its downstream substrates, GluR1 and CPEB. Confocal microscopy revealed that activation of α-CaMKII occurred throughout all subregions of the hippocampus and primarily in neurons after TBI. In addition, we have found that other members of the calcium/calmodulin-dependent protein kinase family, CaMKI and CaMKIV, are also activated in the hippocampus after TBI. These changes occurred on the ipsilateral, injured side of the brain, with little activation of CaMKs observed on the contralateral side. These results indicate that several of the calcium/calmodulin-dependent signaling pathways that are required for the formation of hippocampal-dependent memories are activated after TBI, and may possibly underlie in part, the molecular mechanisms of memory impairments after TBI.
During TBI, and in particular the FPI model, there is a large increase in intracellular calcium in the cortex and hippocampus that lasts for up to 4 days (Fineman et al, 1993; Nilsson et al, 1993; Nadler et al, 1995; Osteen et al, 2001). The activation of calcium/calmodulin-dependent protein kinases after moderate FPI peaked at 30 min, whereas total amounts of these protein kinases were unchanged after TBI. This suggests that protein phosphatases rather than degradation of the protein kinases play a major role in returning their phosphorylation levels back to baseline. Accordingly, activation of calcium-dependent protein phosphatases has been observed with moderate central FPI (Kurz et al, 2005). However, there was a substantial decrease in total CaMKII immunoreactivity in the cytosol, with a small increase of CaMKII in membrane subcellular fractions. α-Calcium/calmodulin-dependent protein kinase II is the major protein in the postsynaptic density (Kennedy et al, 1983; Goldenring et al, 1984; Kelly et al, 1984). Thus, an increase in CaMKII levels in the postsynaptic density may have been underestimated due to a large pool of CaMKII in the postsynaptic membrane. Furthermore, our immunohistochemistry confocal microscopy analysis did not reveal any significant loss of total CaMKII after TBI. Although this suggests that CaMKII redistributed to the membrane from the cytosol, we cannot rule out the possibility that there was also a degradation of cytosolic CaMKII after TBI.
The activation of CaMKII after TBI was rapid and correlated with phosphorylation of its downstream substrates, GluRl and CPEB. In the hippocampus, we observed a robust activation of CaMKII 30 mins after TBI, with a concurrent increase in phosphorylation of GluR1 and CPEB 1 h after TBI, which lasted for up to 4 h. In the parietal cortex, activation of CaMKII was more variable, as was the phosphorylation of GluR1 and CPEB. In some animals, pCaMKII levels were still at peak levels even at 24 h after injury. Thus, at even later time points after TBI, neurons may have difficulty activating the α-CaMKII pathway and its downstream substrates (Griesbach et al, 2004a). Deficits in hippocampal-dependent learning and LTP last for days to weeks after TBI and in particular FPI, suggesting a long-lasting impairment in the biochemical mechanisms that underlie synaptic plasticity and memory (Miyazaki et al, 1992; Bramlett et al, 1997a; Dixon et al, 1999; Sanders et al, 2000; Scheff et al, 2005). Whether the changes in CaMK-mediated cascades are involved in the long-term deficits in learning and LTP remains to be studied. Neuronal death after TBI contributes to the irreversible memory loss after TBI. However, overactivation of the CaMK signaling pathways after TBI may exhaust the cellular machinery for synaptic plasticity, thereby contributing to memory loss.
Classically, α-CaMKII is activated by calcium, autophosphorylates, and translocates to the postsynaptic density (Strack et al, 1997; Rodrigues et al, 2004; Bevilaqua et al, 2005). Once autophosphorylated and translocated to the PSD, α-CaMKII remains active in the absence of the initial calcium stimulus; this is considered to be a molecular mechanism utilized at synapses involved in forming memories (Fukunaga et al, 1995; Giese et al, 1998; Lisman et al, 2002). At the PSD, α-CaMKII binds multiple members of the PSD, such as the NMDA receptor subunit 2B and the scaffolding proteins densin-180 and a-actinin (Bayer et al, 2001; Robison et al, 2005). Similarly, we observed that TBI induced autophosphorylation of α-CaMKII and redistribution from cytosolic subcellular fractions to membrane subcellular fractions, suggesting translocation of the protein kinase to the membrane. If α-CaMKII translocated to synapses that were quiescent at the time of the TBI, this would imply a loss of synapse specificity in the activation of select synapses within a neuron. This is consistent with increases in the phosphorylation of the dendritic substrates of α-CaMKII, GluR1 Ser831 and CPEB. Clustering of α-CaMKII at synapses to phosphorylate GluR1 is thought to confer synapse specificity during synaptic plasticity (Frey and Morris, 1997; Hayashi et al, 2000; Merrill et al, 2005). The activation of α-CaMKII nonspecifically throughout the neurons, at both active and quiescent synapses, due to the large calcium influx globally throughout the neurons after TBI may disrupt the synapse specificity of synaptic plasticity. This may be one mechanism of how retrograde amnesia occurs at the time of injury in many TBI patients (Rees, 2003). Accordingly, there are significant impairments acutely and chronically in hippocampal-dependent memory formation after FPI (Lyeth et al, 1990; Bramlett et al, 1997a; Carbonell et al, 1998; Sanders et al, 1999; Griesbach et al, 2004b; Scheff et al, 2005).
After transient cerebral ischemia, α-CaMKII translocates to the PSD and is almost completely depleted in the cytoplasm of hippocampal CA1 neurons (Hu et al, 1995a, Hu et al, 1998). The changes in α-CaMKII after cerebral ischemia are much more robust than what occurs after TBI (Takagi et al, 2003; Fu et al, 2004; Tang et al, 2004; Yan et al, 2004). In comparison to TBI, the translocation of CaMKII to the PSD during cerebral ischemia could be more pathologic, sequestering α-CaMKII from the normal signaling pathways within the neuron (Hu et al, 1995a). Consistent with this, the CaM kinase inhibitor KN-93 attenuates neuronal death in a cell culture model of ischemia, oxygen, and glucose deprivation (Gao et al, 2005). In the parasagittal FPI model, there is selective neuronal death in the CA3 region of the hippocampus (Dietrich et al, 1994; Bramlett et al, 1997b). However in the present study, we observed an activation of α-CaMKII throughout surviving neurons in the hippocampus, not only in the CA3 region of the hippocampus. Thus, the activation of α-CaMKII after TBI in the dentate gyrus and CA1 region was not reflective of the selective neuronal death in the CA3 region of the hippocampus, but rather, a functional disruption in the normal signaling cascades of memory formation operative in the hippocampus after TBI.
Whether calcium/calmodulin-dependent protein kinase inhibitors would improve or worsen outcome after TBI is most likely dependent on the identity of the downstream substrates that these protein kinases phosphorylate after TBI, and their degree and duration of phosphorylation. For α-CaMKII, we observed an increase in the phosphorylation of the downstream substrates, GluR1 Ser831 and CPEB, in the cortex and hippocampus after TBI. These substrates may not in themselves suggest a pathologic activation of α-CaMKII after head injury. Both are physiologic CaMKII substrates during hippocampal synaptic plasticity, and were phosphorylated with comparable kinetics to that observed during LTP (Barria et al, 1997; Lee et al, 2000; Atkins et al, 2005). Furthermore, α-CaMKII was activated throughout all subregions of the hippocampus, even though the dentate granule cells and CA1 pyramidal neurons do not exhibit death after parasagittal FPI (Dietrich et al, 1994; Bramlett et al, 1997b). Accordingly, voluntary exercise during the first week after parasagittal FPI decreases CaMKII protein levels and worsens behavioural impairments on a hippocampal-dependent learning task (Griesbach et al, 2004aGriesbach et al, 2004 b). Thus, activation of α-CaMKII after TBI may be a functional change in synaptic plasticity within the neuron, and not a pathologic change.
In summary, we observed an activation of several calcium/calmodulin-dependent protein kinases after TBI; these included α-CaMKII, CaMKI, and CaMKIV. α-Calcium/calmodulin-dependent protein kinase II redistributed to membrane subcellular fractions, and this correlated with phosphorylation of its downstream substrates, GluR1 Ser831 and CPEB. Each of these protein kinase pathways has been implicated in the formation of hippocampal-dependent memories. Nonspecific activation of these protein kinases due to the widespread calcium influx into neurons that occurs after TBI may disrupt the machinery for CaMK-dependent memory formation after TBI. Thus, activation of the protein kinases that subserve memory formation during TBI may be one plausible mechanism underlying retrograde amnesia after head injury.
Footnotes
Acknowledgements
We thank TR Soderling and N Nozaki for the phospho-CaMKI and phospho-CaMKIV antibodies, A Oliva and D Liebl for critical reading of this manuscript, and the Dietrich and Hu labs for helpful discussions.