
Abstract
Transient cerebral ischemia following 1 to 2 hours of middle cerebral artery occlusion (MCAO) in the rat leads to infarction, which can be diminished by synaptic transmission modulators, implying aberrant cell signaling in the pathogenetic process. The authors report here changes in the levels of tyrosine phosphorylated proteins (PTyr) and calcium calmodulin kinase II (CaMKII) phosphorylation of Thr 286, in synaptosomal, particulate, and cytosolic fractions of different cortical areas following 1 or 2 hours of MCAO, or 2 hours of MCAO followed by 2 hours of reperfusion. At the end of 2-hour MCAO, PTyr, and in particular the pp180, indicative of NR2A/B subunit, increased in the synaptosomal fraction in less ischemic areas while it decreased in more severe ischemic regions. During reperfusion, phosphorylation increased at least 2-fold in all reperfused areas. During 2 hours of MCAO, the phosphorylation of CaMKII increased 8- to 10-fold in the synaptosomal fraction in all ischemic brain regions. During reperfusion, the phospho-CaMKII levels remained elevated by approximately 300% compared with the contralateral hemisphere (control). There was no increase in phospho-CaMKII in the cytosolic fraction at any time during or following ischemia in any of the brain regions examined. The authors conclude that both tyrosine kinase coupled pathways, as well as CaMKII-mediated cellular processes associated with synaptic activity, are strongly activated during and particularly following MCAO. These results support the hypothesis that aberrant cell signaling may contribute to ischemic cell death and dysfunction, and that selective modulators of cell signaling may be targets for pharmacological intervention against ischemic brain damage.
Ischemic brain damage due to middle cerebral artery occlusion (MCAO) can be modulated by glutamate receptors (Gill et al., 1992), calcium channel blockers (Minato et al., 1997; Yenari et al., 1996), adenosine analogs, and potassium channel activators (Takaba et al., 1997). Also, both during and following ischemia, intracellular calcium levels are elevated (Kristian et al., 1998), suggesting that intracellular signal transduction is severely disturbed, and may contribute to the final outcome of an ischemic insult and cause cell death (Wieloch et al., 1996).
CaMKII is a serine/threonine protein kinase ubiquitously found throughout the rat forebrain (Colbran, 1992; Hanson and Schulman, 1992). It is made up of 50- to 54-kd subunit (CaMKII-α) and 58- to 60-kd subunit (CaMKII-β). CaMKII is regulated by calmodulin and, among other factors, its activity is thought to be dependent on the autophosphorylation of CaMKII-α at Thr 286, which activates the enzyme, making it independent of Ca2+ (Bronstein et al., 1993; Miller et al., 1988). Glutamate receptor activation leading to an influx of Ca2+ into the neurons stimulates the translocation of CaMKII from the cytosol to the membrane of postsynaptic densities, where it becomes autophosphorylated at Thr 286 and subsequently independent of Ca +/calmodulin (Strack et al., 1997). Phosphorylated CaMKII on cell membranes may in turn phosphorylate proteins such as the GluR1 receptor (Barria et al., 1997), which enhances the neuronal firing rate (Shirke and Malinow, 1997). Similarly, it has been suggested that the translocation of CaMKII to membranes following cerebral ischemia may enhance neuronal firing and calcium influx (Hu and Wieloch, 1995).
Tyrosine kinases have been implicated in particular in the regulation of DNA transcription through the phosphorylation of transcription factors by mitogen-activated protein kinases and stress-activated protein kinases. However, tyrosine phosphorylation has also been implicated in signal transduction, and the NR2A/B N-methyl-
In vitro studies of protein kinase activities have been limited to measurements of enzymatic activity. With the advent of new antibodies directed against phosphorylated protein substrates for protein kinases, it has become feasible to study the steady-state phosphorylation of a protein and, therefore, the net effect of protein kinase/phosphatase activities in vivo. Consequently, in the present investigation we have studied changes in the levels of tyrosine-phosphorylated proteins, in particular 180-kd phosphoprotein, which might relate to the NR2A/B subunit, as well as the phosphorylation of CaMKII, a kinase of central importance for cell function. Since there is marked cross-talk among signaling pathways in the cell, we used PTyr and P-CaMKII as probes for monitoring the state of cell signaling in the ischemic brain and during reperfusion.
MATERIALS AND METHODS
Animal preparation and experimental groups
Three experimental groups were designed: 1 hour of MCAO (n = 3), 2 hours of MCAO (n = 5), and two hours of MCAO followed by two hours of reperfusion (n = 6). The procedures for transient MCAO were performed as described previously (Memezawa et al., 1992a; Zhao et al., 1994) and are summarized briefly below. All experimental procedures were approved by the Ethics Committee at Lund University under the supervision of the Swedish Department of Agriculture. Male Wistar rats (Möllegaards Breeding Center, Copenhagen), weighing 310 to 350 g, were fasted overnight but had free access to water. Anesthesia was induced by inhalation of 3% halothane in a N2O/O2 (70:30) mixture; thereafter, the animals were intubated. They were then ventilated on 1.0% to 1.5% halothane in the N2O/O2 mixture during surgery. The tail artery was cannulated for blood sampling and blood pressure monitoring. Blood pressure, Pa
Animals killed after 1 hour or 2 hours of MCAO, or 2-hour reperfusion following 2-hour MCAO were reanesthetized with halothane, tracheostomized, and ventilated. The brains were then frozen in situ (Ponten et al., 1973) with liquid nitrogen. Brain samples were dissected from coronal sections through anatomic regions of the neocortex and caudoputamen, as shown in Fig. 1. The samples were stored at −80°C until homogenization.
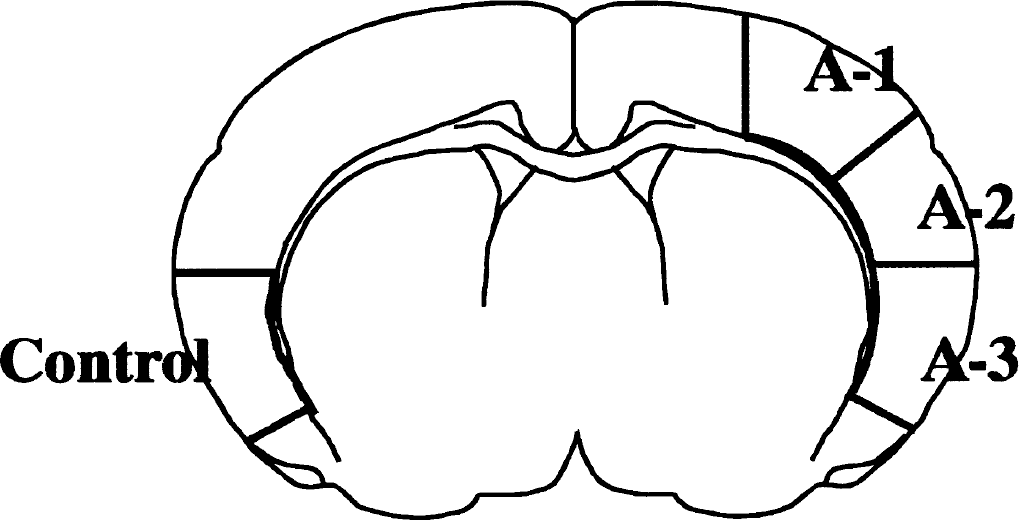
A coronal section of the cortex indicating the areas where tissues were sampled. Control samples were collected on the hemisphere contralateral to middle cerebral artery occlusion. The A-1 area is mildly ischemic, A-2 more severely ischemic, and A-3 is the core ischemic area.
Subcellular fractionation
Tissues were sonicated twice each for 10 seconds in 1:10 (wt/vol) homogenization buffer consisting of 50 mmol/L 3-(N-morpholino) propanesulfonic acid (MOPS; pH 7.4), 0.2 mmol/L dithiothreitol, 100 mmol/L KCl, 0.5 mmol/L magnesium chloride, 1 mmol/L sodium orthovanadate, 0.01 mmol/L EDTA, 1 mmol/L EGTA, 0.5 mmol/L ouabain, 50 g·mL−1 leupeptin, 10 g · mL−1 aprotinin, 5 g · mL−1 pepstatin, and 0.32 mol/L sucrose. The homogenates were centrifuged at 800 g for 10 minutes at 4°C, followed by centrifugation of the supernatant at 9,200 g for 15 minutes at 4°C in a Sorvall SE rotor. The resulting pellet, synaptosomal fraction (P2), was sonicated for 10 seconds in homogenization buffer containing 0.1% Triton X-100, and the supernatant was centrifuged at 165,000 g for 1 hour at 2°C in a TL100.2 rotor. The supernatant fraction (S3), consisting of the cytosol, and the resulting pellet, particulate fraction (P3), were reconstituted in homogenization buffer plus 1% Triton X-100. The protein concentration was determined (Lowry et al., 1951) before freezing the samples at −80°C for later analysis.
Electrophoresis and immunoblotting
Electrophoresis was carried out on 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) according to the method of Laemmli (1970). The samples (12.5 μg protein for synaptosomal and particulate fraction, 25 μg for cytosolic fraction) were mixed with a 5x SDS sample buffer of 0.3 mol/L Tris/HCl (pH 6.8), 25% mercaptoethanol, 12% SDS, 25 mmol/L EDTA, 20% glycerol, and 0.1% bromphenol blue, boiled for 4 minutes, and subjected to SDS-PAGE at a constant current of 40 mA (stacking gel) and 45 mA (separating gel). Following electrophoresis, proteins on the gel were electro-transferred onto polyvinylidene difluoride (PVDF) membrane (Bio-Rad Transblot; pore size, 0.2 μm) according to Towbin et al. (1979) with a constant current of 100 V for 90 minutes. After transfer, the PVDF membranes were washed in Tris-buffered saline (TBS), containing 0.1% tween 20 (TBST), and then preincubated with a blocking solution of 3% bovine serum albumin (BSA) in TBST for 1 hour at room temperature. Blots were then incubated with a solution containing primary antibodies against phosphotyrosine (anti-PTyr; Affinity, Nottingham, U.K.), and phospho-CaMKII (anti-phospho-CaMKII; Affinity) in TBST containing 3% BSA, overnight at 4°C. Finally, the PVDF membranes were incubated with a secondary antibody conjugated with horseradish peroxides in TBS containing 2% BSA for 1 hour at 4°C. Membranes were evaluated by ECL+Plus (Amersham, U.K.) Western blotting system. Auto-radiographs were quantified using a DIANA-II CCD camera system and TINA 2.0 program (Raytest Isotopenmessgeräte GmbH, Straubenhardt, Germany).
Statistical analysis
Data are presented as mean ± SD or mean values and 95% or 99% CI (lower confidence limit-upper confidence limit). Immunoblots were analyzed using the 95% or 99% CI (P < 0.05 or P < 0.01) in comparison to control values, and analysis of variance (ANOVA) followed by two-tailed Dunn-Bonferroni test (P < 0.05, P < 0.01). Physiological data were analyzed with ANOVA followed by Scheffé test (P < 0.05, P < 0.01).
RESULTS
Physiologic parameters
Physiologic parameters are listed in Table 1. Core temperature, blood pressure, arterial P
Physiologic parameters of rats subjected to middle cerebral artery occlusion
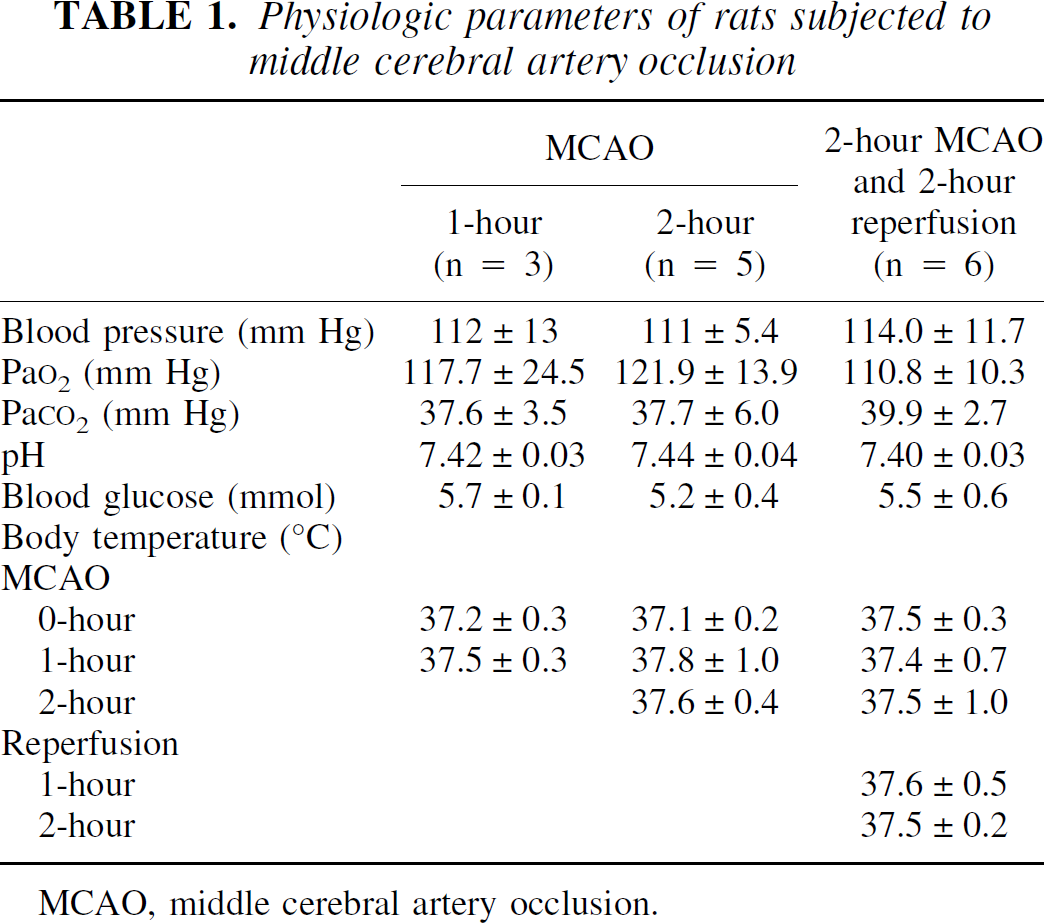
MCAO, middle cerebral artery occlusion.
Changes in phospho-CaMKII levels
Tissue was sampled from four brain areas—A-1 (weak ischemic penumbra), A-2 (strong ischemic penumbra), A-3 (ischemic core), and control (area contralateral to A-3)—as shown in Fig. 1. The tissue was homogenized into synaptosomal (P2), particulate (P3), and cytosolic (S3) fractions. Figure 2 shows immunoblots of these fractions with P-CaMKII antibody, at 1-hour MCAO, 2-hour MCAO, and 2-hour reperfusion following 2-hour MCAO. No P-CaMKII signal was seen in cytosolic fraction at any time point. Stronger signals were seen in the membrane fractions (synaptosomal and particulate fraction) from A-1, A-2, and A-3 areas at 1-hour and 2-hour MCAO, compared to the contralateral side. Also with 2-hour reperfusion following 2 hours MCAO, membrane fraction levels were higher, albeit lower than during the ischemic insult.
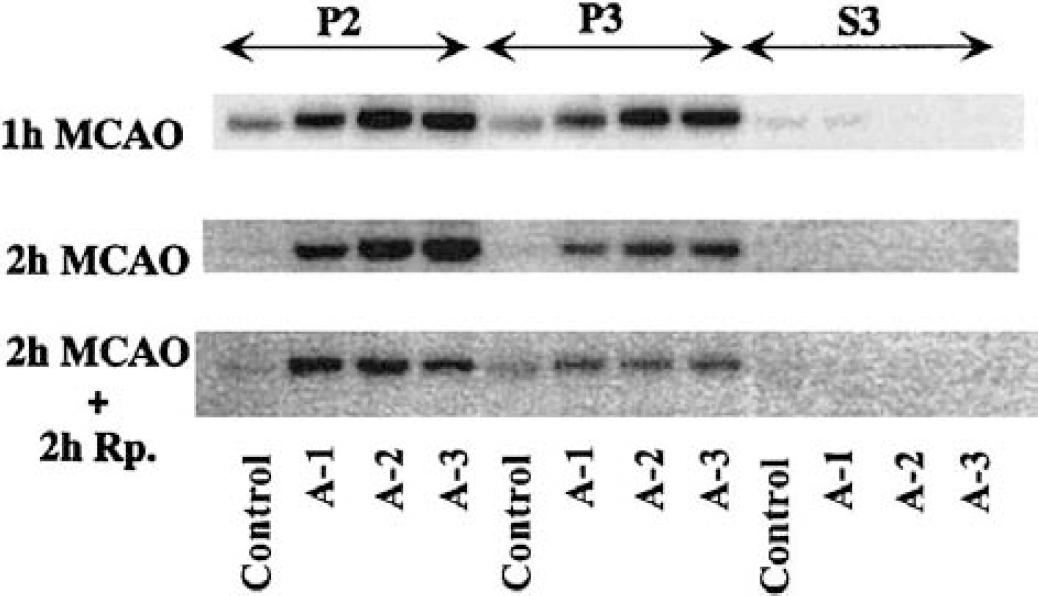
Phospho-CaMKII immunoblots from crude synaptosomal fraction (P2), particulate fraction (P3), and cytosolic fraction (S3) from different brain areas as indicated in Fig. 1. Tissues were sampled at 1 hour and 2 hours of middle cerebral artery occlusion (MCAO), and at 2 hours of reperfusion following 2 hours MCAO.
The protein levels in immunoblots were calculated and are presented as a percentage of that in the control area. Figure 3 shows the levels of P-CaMKII in synaptosomal and particulate fraction, at each time point. At 1-hour MCAO, in synaptosomal fraction, the levels of P-CaMKII significantly increased in all ischemic areas, A-1, A-2, and A-3 to 238% (99% CI: 197–279; P < 0.01), 370% (99% CI: 315–425; P < 0.01), and 343% (99% CI: 240–446; P < 0.01), respectively. There was significantly higher phosphorylation in A-2 compared with A-1 (Dunn-Bonferroni). Similarly, in particulate fraction, the levels of P-CaMKII increased significantly in all three areas.
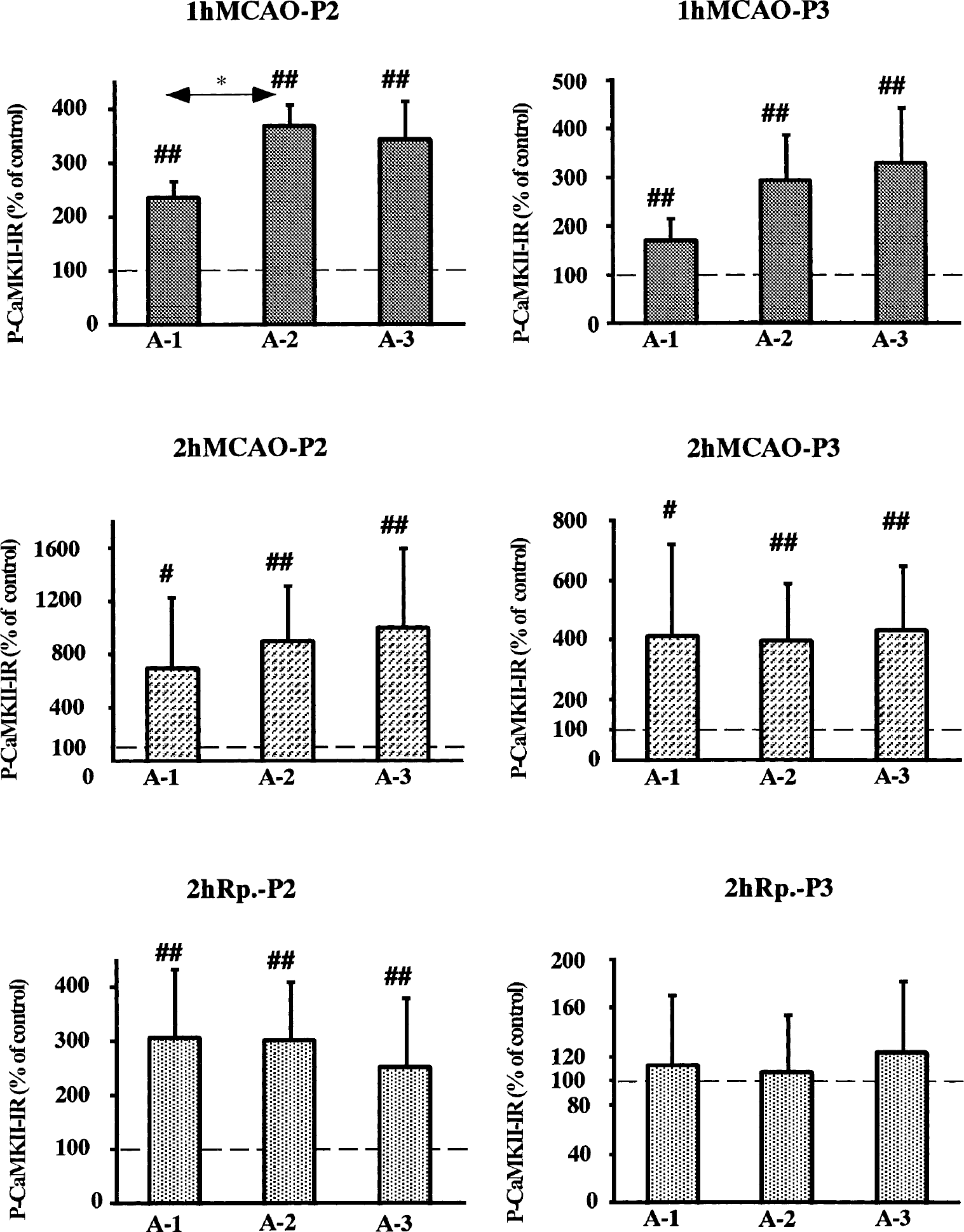
Changes in the levels of phospho-CaMKII (P-CaMKII) in crude synaptosomal fraction (P2) and particulate fraction (P3) from different brain regions as indicated in Fig. 1. The levels are presented as percent of the control side. The values represent means ± SD. # and ## denote statistical differences compared to the control side with 95% and 99% confidence intervals, respectively. *Denotes statistical difference at P < 0.05, Dunn-Bonferroni test. MCAO, middle cerebral artery occlusion.
At 2-hour MCAO, in synaptosomal fraction, the levels of P-CaMKII markedly increased in all ischemic areas, A-1, A-2, and A-3 to 698% (99% CI: 233–1,163; P < 0.01), 907% (99% CI: 443–1,371; P < 0.01) and 997% (99% CI: 318–1,676; P < 0.01), respectively. In particulate fraction, the levels increased fourfold compared with the control level in all ischemic areas.
In the synaptosomal fraction, there was significant threefold increase with 2 hours of reperfusion, in all ischemic areas compared with the control area. However, in particulate fraction, no significant changes were seen in any of the ischemic areas.
In summary, a marked increase in P-CaMKII levels in ischemic areas was observed in the synaptosomal and particulate fractions during 2-hour MCAO, and this elevation persisted during reperfusion in the synaptosomal fraction, but not in the particulate fraction.
Changes in the levels of tyrosine phosphorylated proteins
Figure 4 shows immunoblots of PTyr. A prominent band at 180 kd was revealed. In the synaptosomal fraction, PTyr levels in the A-2 and A-3 areas were lower than in both the control area and A-1 area at both 1-hour and 2-hour MCAO. In contrast, at 2 hours of reperfusion, PTyr levels in the ischemic areas were higher than that of the control area in the synaptosomal, but not particulate or cytosolic fractions.
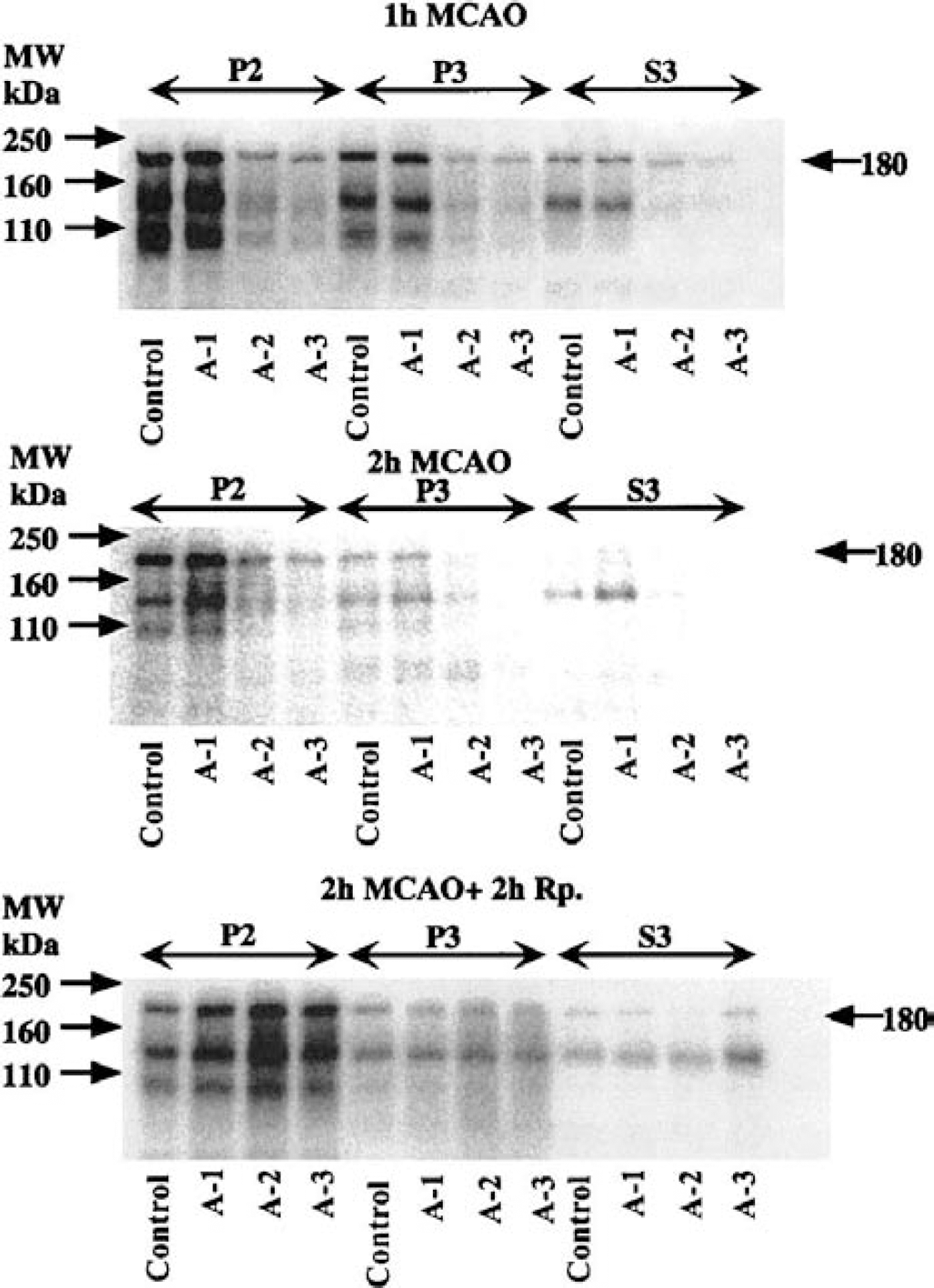
Immunoblots of tyrosine phosphorylated proteins in crude synaptosomal fraction (P2), particulate fraction (P3), and cytosolic fraction (S3) from different brain areas as indicated in Fig. 1. Tissues were sampled at 1 hour and 2 hours of middle cerebral artery occlusion (MCAO), and at 2 hours of reperfusion following 2-hour MCAO. A 180-kd protein containing the NMDA receptor NR2A/B subunit was revealed.
Since it has been shown that the 180-kd phosphoprotein (pp180) includes the NR2A/B subunit of the NMDA receptor (Takagi et al., 1997), we evaluated this band in greater detail. Figure 5 shows the levels of pp180 in synaptosomal fraction with 1-hour and 2-hour MCAO, and 2 hours of reperfusion. During MCAO, the level of PTyr significantly increased with 2-hour MCAO only in the A-1 area, to 180% (99% CI: 106–254; P < 0.01) of the control level. A similar tendency was seen in A-2 and A-3 areas.
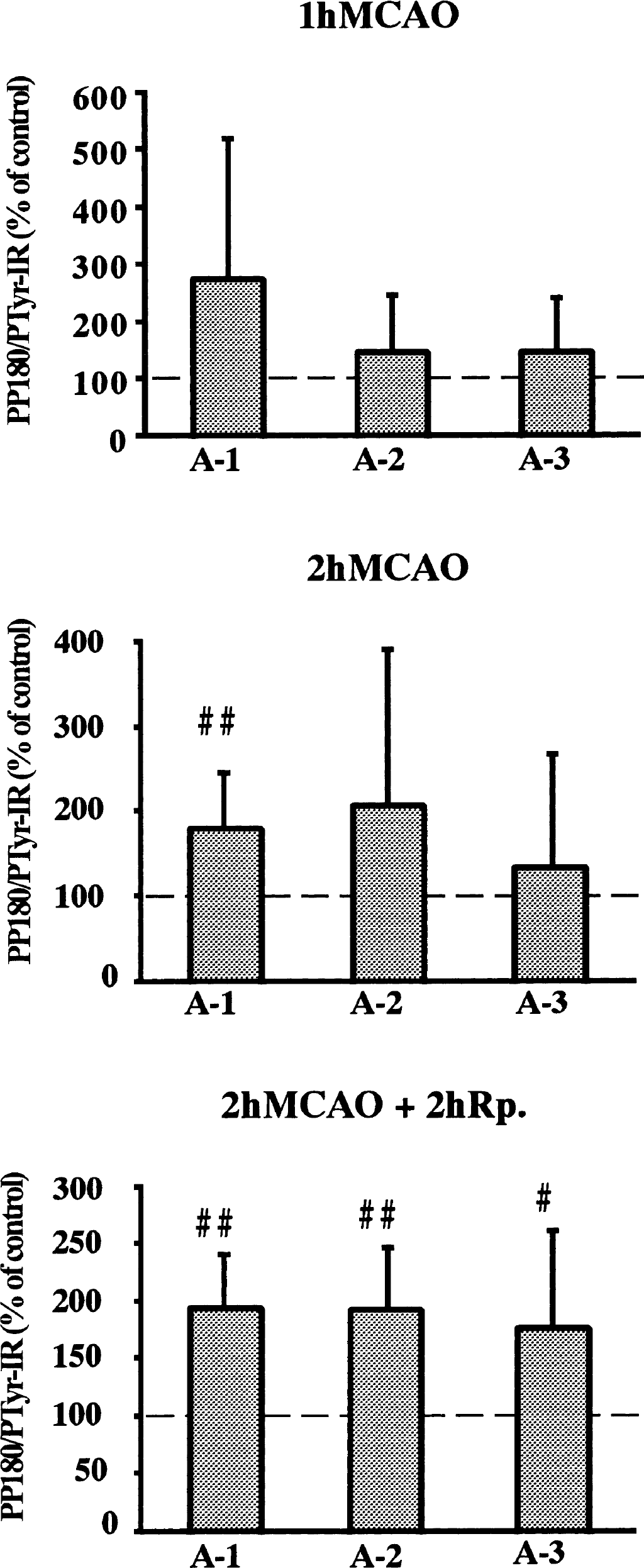
Changes in levels of tyrosine phosphorylated proteins in crude synaptosomal fraction (P2) from different brain regions as indicated in Fig. 1. The levels are presented as percent of the control side. The values represent means ± SD. # and ## denote statistical differences compared to the control side with 95% and 99% confidence intervals, respectively. MCAO, middle cerebral artery occlusion.
With 2 hours of reperfusion, the level of pp180 significantly increased in all ischemic areas to 194% (99% CI: 146–242; P < 0.01) in A-1, 192% (99% CI: 135–249; P < 0.01) in A-2, and 177% (95% CI: 110–244; P < 0.05) in A-3.
DISCUSSION
Our discussion below focuses on two findings in our investigation: (1) an increase in the phosphorylation of P-CaMKII during and following ischemia in synaptosomal fractions, and (2) the enhanced phosphorylation of PTyr (pp180) during reperfusion.
Protein phosphorylation and brain ischemia
Net protein phosphorylation is dependent on the balance between protein kinase and phosphatase activities. During ischemia, the limited supply of ATP is therefore a determining factor in the extent of protein phosphorylation. The model used in the present study has been characterized in terms of cerebral blood flow (CBF) and the levels of energy metabolites (Folbergrova et al., 1992; Memezawa et al., 1992b). During 60 and 180 minutes of ischemia, CBF decreases to approximately 40% of control levels in A-1, to 18% in A-2, and to 10% in A-3. The A-1 area is therefore a mild ischemic area called the penumbra (Astrup et al., 1981), while A-2 is subjected to severe ischemic insult and A-3 is the ischemic core. Graded oxygen and glucose supplies to brain tissues are also reflected in the levels of energy substrates. At 30 minutes of MCAO, the ATP levels decrease from 2.9 (μmol · g−1) to 1.9, that is, by approximately 35% in an area that is slightly larger than A-1, and to 0.8 (μmol · g−1), that is, approximately 70% in an area that includes both A-3 and the larger part of A-2. The A-1 neocortical area is therefore mildly ischemic and able to maintain significant energy production. Still, this area is included in the final infarct following 48 hours of reperfusion. On the other hand, the A-2 and A-3 areas are severely ischemic, and the low ATP levels may therefore be limiting for the kinase reactions.
Changes in tyrosine phosphorylation
The association between ATP levels and ischemia is clearly reflected in the phosphorylation of proteins during ischemia. In severely ischemic areas, tyrosine phosphorylation decreases, most probably owing to the lack of ATP. In contrast, in the mildly ischemic A-1 area phosphorylation increases, presumably owing to deregulation of receptor and neurotransmitter homeostasis, stimulating protein kinases (Anderson, 1997). Most notable was the increase in tyrosine phosphorylation during reperfusion, affecting all ischemic regions. This increase was seen only in the synaptosomal fraction, and was therefore a synaptic event, while proteins in the particulate fraction (plasma membrane, endoplasmic reticulum) or cytosolic proteins were essentially unaffected. Evidently, cell signaling is enhanced in synaptic areas, affecting membrane proteins such as receptors, ion channels, and cytoskeletal proteins. The increased tyrosine phosphorylation we observed is in agreement with recent findings using models of global cerebral ischemia (Shamloo and Wieloch, 1999; Takagi et al., 1997), where a persistent increase in tyrosine phosphorylation is seen in the vulnerable CA1 region. Of particular interest is the increased level of the 180-kd protein detected, earlier identified as the NR2A/B subunit. A persistent increase in this subunit may cause changes in ionic conductance, resulting in further deregulation of cellular homeostasis and cell death.
Changes in CaMKII phosphorylation
When calcium ions bind to calmodulin, the protein binds to CaMKII-α subunit and activates the enzyme (Soderling, 1993). This leads to autophosphorylation of Thr 286 on CaMKII-α (Miller et al., 1988). We report here that phosphorylated CaMKII-α is essentially absent in the cytosolic fraction, but present in the membrane fractions in the rat brain. This indicates that autophosphorylation occurs on membranes. The fact that the levels of phosphorylated CaMKII increased during ischemia in membrane fractions of all ischemic regions, and particularly in the more severe ischemic areas such as A-2, suggests that CaMKII pathways are notably activated. These results contrast with tyrosine phosphorylation, which ceased in the more ischemic regions. Our findings suggest that the Km for ATP may be low during autophosphorylation, or that phosphatases may not have access to the phosphorylation site counteracting a minor increase in phosphorylation. Similarly, as for PTyr, the increased levels of P-CaMKII persisted during reperfusion. In analogy with the elevated PTyr levels during reperfusion, continuous derangement of cellular signaling is also reflected in the increase in P-CaMKII. The increase in autophosphorylation, indicating enhanced CaMKII activity in the synaptosomal fraction, contradicts the observed marked decrease in CaMKII activity measured in brain homogenates under optimal assay conditions, i.e., in the presence of calcium chloride and calmodulin (Aronowski et al., 1992; Churn et al., 1990; Hu and Wieloch, 1995; Zalewska and Domanska-Janik, 1996). Since autophosphorylation increased during reperfusion, our data demonstrate that at least a fraction of CaMKII is activated.
Significance for ischemic brain damage
We propose that during ischemia, cellular signal transduction becomes activated due to enhanced receptor activation or to intracellular changes in second messenger homeostasis. This would lead to increased autophosphorylation of CaMKII-α at synapses and to increased tyrosine phosphorylation of synaptic membrane proteins. Enhanced protein phosphorylation in turn fuels a continuous cycle of further derangement that contributes to subsequent detrimental processes leading to cell death. For example, hyperphosphorylation of AMPA and NMDA receptors by CaMKII-α and NMDA receptor by an src-type kinase (Barria et al., 1997; Soderling, 1996) may occur during reperfusion, and result in increased sensitivity of the receptor to glutamate activation (Nellgard and Wieloch, 1992). Also, phosphorylated CaMKII may act as an amplifier of calcium signaling since it becomes partially Ca2+ independent (Colbran, 1992; Soderling, 1993).