
Abstract
Many factors have been postulated to cause delayed subarachnoid hemorrhage (SAH)-induced vasospasm, including hemoglobin, nitric oxide, endothelin, and free radicals. We propose that free radicals (because of the high levels that are produced in the blood clots surrounding blood vessels after SAH) act on bilirubin, biliverdin, and possibly heme to produce BOXes (Bilirubin OXidized Products). Bilirubin oxidation products act on vascular smooth muscle cells to produce chronic vasoconstriction and vasospasm combined with a vasculopathy because of smooth muscle cell injury. This review summarizes recent evidence that BOXes play a role in SAH-induced vasospasm. The data supporting a role for BOXes includes (1) identification of molecules in cerebrospinal fluid (CSF) of patients with vasospasm after SAH that have structures consistent with BOXes; (2) BOXes are vasoactive in vitro and mimic the biochemical actions of CSF of patients with vasospasm; (3) BOXes are vasoactive in vivo, constricting rat cerebral vessels; and (4) there is a correlation between clinical occurrence of vasospasm and BOXes concentration in our preliminary study of patients with SAH. Since oxidation of bilirubin, biliverdin, and perhaps heme is proposed to produce BOXes that contribute to vasospasm, either blocking bilirubin formation, inactivating bilirubin or BOXes, or removing all of the blood clot before vasospasm are potential treatment targets.
Keywords
Introduction
The delayed vasospasm that occurs after subarachnoid hemorrhage (SAH) has been the subject of multiple studies, continuing controversies, and a continuing search for the substance or substances that cause this clinically frustrating disease (Dietrich and Dacey, 2000; Janjua and Mayer, 2003; Macdonald and Weir, 1991, 1994; Megyesi et al, 2000; Pluta, 2005; Roux et al, 1999). Once patients survive the initial SAH, and often have had the aneurysm adequately addressed by surgical and/or radiologi means, some patients develop vasospasm and have a stroke (sometimes referred to as a delayed neurologic ischemic deficit—DIND), by still unknown mechanisms (Dietrich and Dacey, 2000; Janjua and Mayer, 2003; Macdonald and Weir, 1991, 1994; Megyesi et al, 2000; Pluta, 2005; Roux et al, 1999). This review summarizes the evidence that oxidized forms of bilirubin cause or contribute to vasospasm associated with SAH. Oxidized forms of bilirubin (BOXes) fulfill all of the criteria for being a cause or contributor of vasospasm, and these will be addressed here. The purpose of this review is not to examine previously hypothesized mechanisms of vasospasm (Heros et al, 1983; Janjua and Mayer, 2003; Macdonald and Weir, 1991; Pluta, 2005), but rather to summarize recent evidence that oxidized species of bilirubin and biliverdin (and possibly heme) are found in cerebrospinal fluid (CSF) after SAH, and could be the cause of delayed vasospasm after SAH.
Two Classes of compounds that could cause Vasospasm
There are two general classes of compounds that could produce delayed vasospasm after SAH: (1) those that relate to something found in blood or metabolites of compounds found in blood (Figure 1); and (2) compounds that are regulated or induced as a consequence of blood around blood vessels. These classes of compounds could come from the blood, blood vessels, or brain, and be caused directly or indirectly by the blood from the hemorrhage. Examples of compounds in the first class would be hemoglobin (Hb)and BOXes, and examples in the second class of compounds would be endothelin or nitric oxide (Figure 2), which could be directly or indirectly induced by the BOXes. The rationale that blood itself or some metabolite of components of blood cause vasospasm is compelling since this helps explain why vasospasm tends to occur where large blood clots occur around vessels at the base of the brain (Harrod et al, 2005; Kistler et al, 1983).
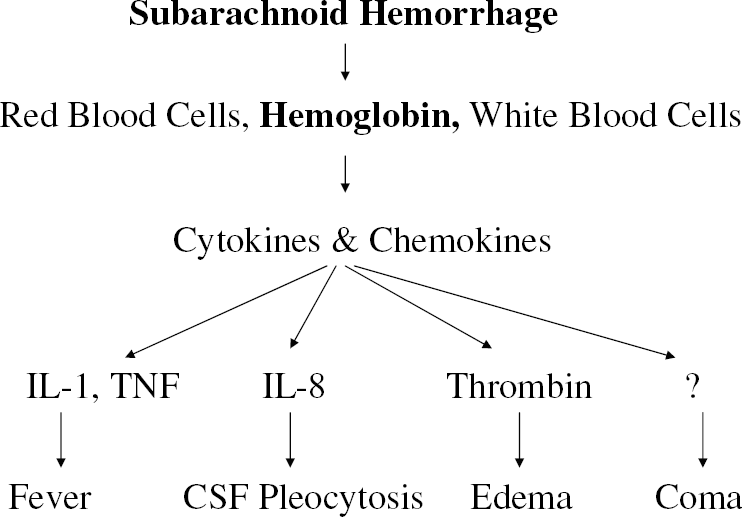
Systemic effects of Subarachnoid Hemorrhage (SAH). Subarachnoid hemorrhage results in a clot surrounding vessels at the base of the brain. The clot consists of red blood cells, white blood cells, and clotting-related proteins. The breakdown of red blood cells releases hemoglobin. An inflammatory response to the clot includes invasion of white blood cells and macrophages from the intact vessels into the clot. Those cells release cytokines and chemokines that include inter lenking-1 as well as other molecules that contribute to fever after SAH, influx of inflammatory cells into the CSF, edema, and possibly other systemic effects like hypertension and coma. The diagram proposes that IL-1 mediates fever, thrombin is one mediator of edema, and other factors mediate CSF pleocytosis and coma.
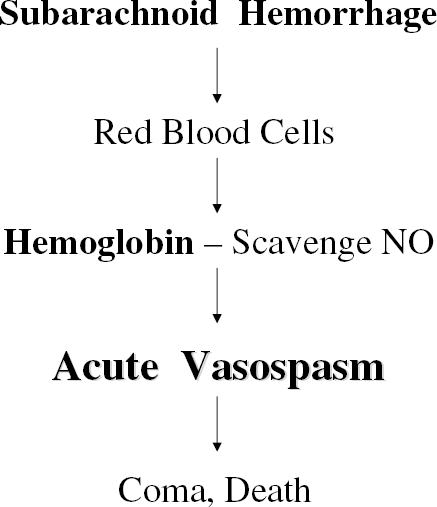
Hemoglobin scavenges nitric oxide (NO). Hemoglobin is released from red blood cells after SAH. The hemoglobin likely scavenges NO acutely to produce acute vasospasm that may contribute to coma and death. Since hemoglobin is also likely to be present in the blood clot for some time after SAH even at times when hemoglobin concentrations in CSF are low, it is also possible that hemoglobin itself and oxidation of hemoglobin (shown below) may contribute to vasospasm over time following SAH.
Hemoglobin has been a candidate for causing SAH-induced, delayed vasospasm for some time (Macdonald and Weir, 1991). It is present in high concentrations around blood vessels after SAH (Figures 1 and 2). Most importantly, it is a potent vasoactive compound because it scavenges nitric oxide, which will cause the vessels to constrict (Janjua and Mayer, 2003; Pluta, 2005). Although Hb may play a role in acute vasospasm after SAH, and perhaps in chronic vasospasm, the problem with implicating Hb has been the time course of delayed vasospasm. That is, delayed vasospasm is maximal at about a week after SAH at a time when CSF levels of Hb are low. Nevertheless, if Hb concentrations were very high in the clot immediately adjacent to blood vessels, and are substantially different than what is detected in the CSF, Hb could contribute to delayed vasospasm in brain after SAH (Janjua and Mayer, 2003; Macdonald and Weir, 1991).
Metabolites of Hb, including metabolites of heme or globin, would be candidates for producing delayed vasospasm since they would take time to form after SAH. Heme released from Hb is metabolized to bilirubin (Figure 3; Maines, 2000). It takes time for biliverdin and bilirubin to appear in the CSF after SAH. Indeed, the time course of bilirubin appearance in the CSF—appearing within a day, being maximum within 3 to 5 days, and disappearing after several weeks after SAH—roughly correlates with the delayed vasospasm after SAH (Pyne-Geithman et al, 2005). This phenomenological association with bilirubin had led to much interest in bilirubin as a player in SAH-induced vasospasm, but to our knowledge the consideration that a degradation product of bilirubin might contribute to vasospasm had not previously been thoroughly researched.
Heme Oxygenases and Bilirubin Production
There are two forms of heme oxygenase (HO) that are responsible for the production of bilirubin, heme oxygenase-2 (HO-2) and heme oxygenase-1 (HO-1) (Figures 3 and 4). Heme oxygenase-2 is constitutively expressed in many cells including neurons and vascular endothelial cells (Govindaraju et al, 2005; Maines, 2000). Heme oxygenase-1 is the inducible heat-shock protein that is induced by heme, iron, heat shock, free radicals, and other stimuli in a variety of cells, including macrophages, brain microglia, and vascular smooth muscle cells (Foresti et al, 1999; Govindaraju et al, 2005; Maines, 2000; Sharp et al, 1999; Wagner et al, 2003). Injection of blood (Matz et al, 1996a, b , c ), or injections of pure Hb into the subarachnoid space induces HO-1 in microglia throughout the brain (Turner et al, 1998). It was postulated that the heme from Hb was selectively transported into the microglia (endogenous brain macrophage), where the heme induced the HO-1 gene (Sharp et al, 1999; Turner et al, 1998, 1999). Heme oxygenase-1 in macrophages, microglia, and vascular smooth cells (Figure 4), and HO-2 in neurons and endothelium would metabolize the heme to biliverdin (Figure 4).
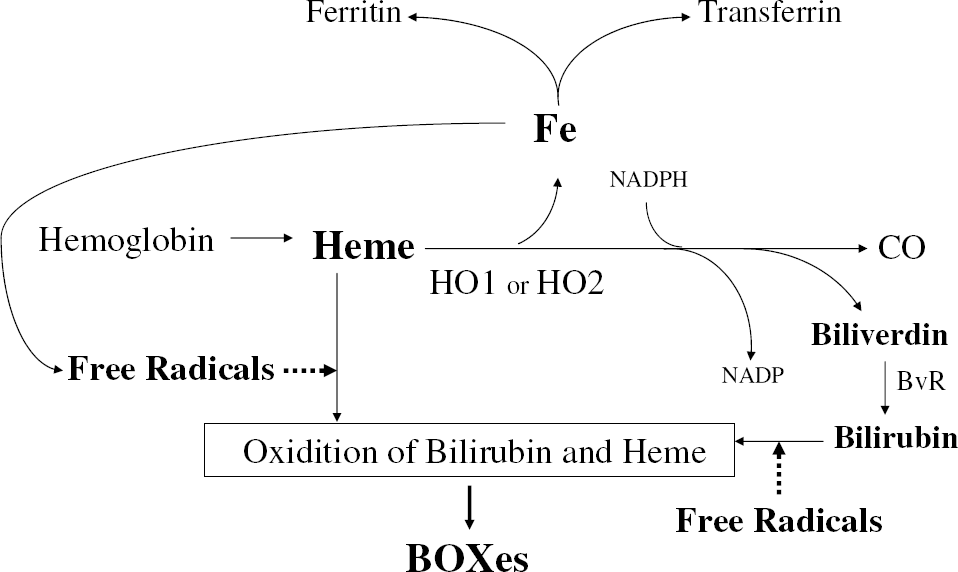
Heme metabolism. Heme from hemoglobin is metabolized to biliverdin by the heme oxygenase (HO) enzyme. Heme oxygenase enzyme activity is accounted for by two separate proteins: one is the constitutive heme oxygenase-2 (HO-2) found in neurons and vascular cells, and the other is the inducible heme oxygenase-1 (HO-1) protein that is induced in marcrophages and microglia and other cells. Biliverdin is metabolized to bilirubin via biliverdin reductase. Free radicals act on bilirubin, biliverdin, and possibly heme to produce BOXes. Iron released by metabolism of heme or breakdown of heme can be bound by ferritin intracellularly or transferrin extracellularly. Any free iron can interact with H2O2 to produce hydroxyl free radicals and oxidize bilirubin, biliverdin or heme.
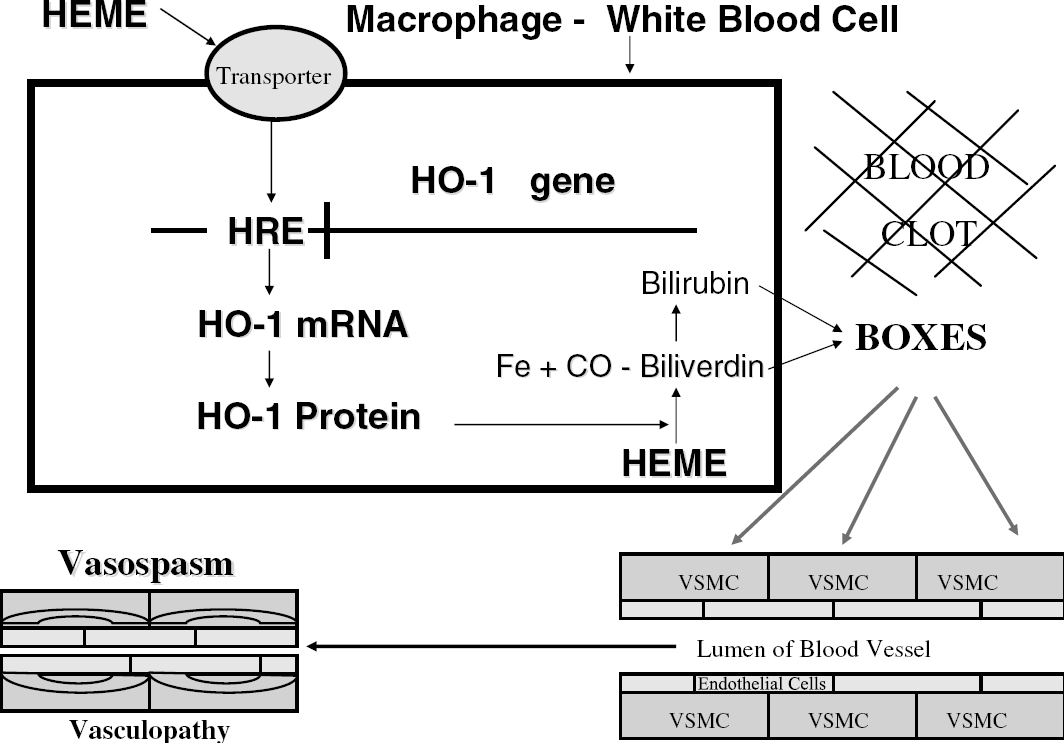
Formation of BOXes—most likely mechanism. Heme is transported into inflammatory cells including macrophages, inflammatory cells, and microglia via a membrane transporter. Heme, once inside the nucleus of the cell, acts on the heme response element (HRE) of the HO-1 gene to induce heme oxygenase-1 mRNA (HO-1 mRNA). Heme oxygenase-1 protein then metabolizes heme to biliverdin, carbon monoxide (CO), and iron. Biliverdin is metabolized to bilirubin by biliverdin reductase. Bilirubin and biliverdin are acted on by free radicals to produce BOXes. Bilirubin oxidation products diffuse into vascular smooth muscle cells (VSMC) to produce vasospasm and produce damage to the contractile elements of the VSMC cells (vasculopathy).
The localization of HO-1 and HO-2 dictates the source of bilirubin in brain, CSF, and around cerebral vessels. Heme oxygenase-2 is localized to neurons throughout brain and is constitutively expressed (Maines, 2000). Heme oxygenase-2 is also constitutively expressed in endothelial cells of blood vessels (Govindaraju et al, 2005). It seems unlikely that HO-2 in neurons is the source of bilirubin after SAH. Heme oxygenase-2 in endothelial cells could metabolize heme to bilirubin, but their physical location away from the clot would seem to make them a lesser source of heme-derived bilirubin.
Perhaps, the source of bilirubin from the clot is from macrophages and monocytes. Macrophages, white blood cells, and microglia selectively take up heme and induce HO-1 (Sharp et al, 1999), which would metabolize heme to biliverdin, which is then metabolized to bilirubin and even have the oxidative capabilities to form BOXes (Figures 4 and 5). Once bilirubin is formed in a macrophage, microglial cell, inflammatory cell, or other cells, it can exit cells fairly easily since it is stable and quite lipid soluble. The second and perhaps most direct source of bilirubin would be heme induction of HO-1 within smooth muscle cells. Heme from an adjacent clot could be transported into vascular smooth muscle cells, where it would induce HO-1 within those cells that would then metabolize the heme to biliverdin, which is then metabolized to bilirubin.
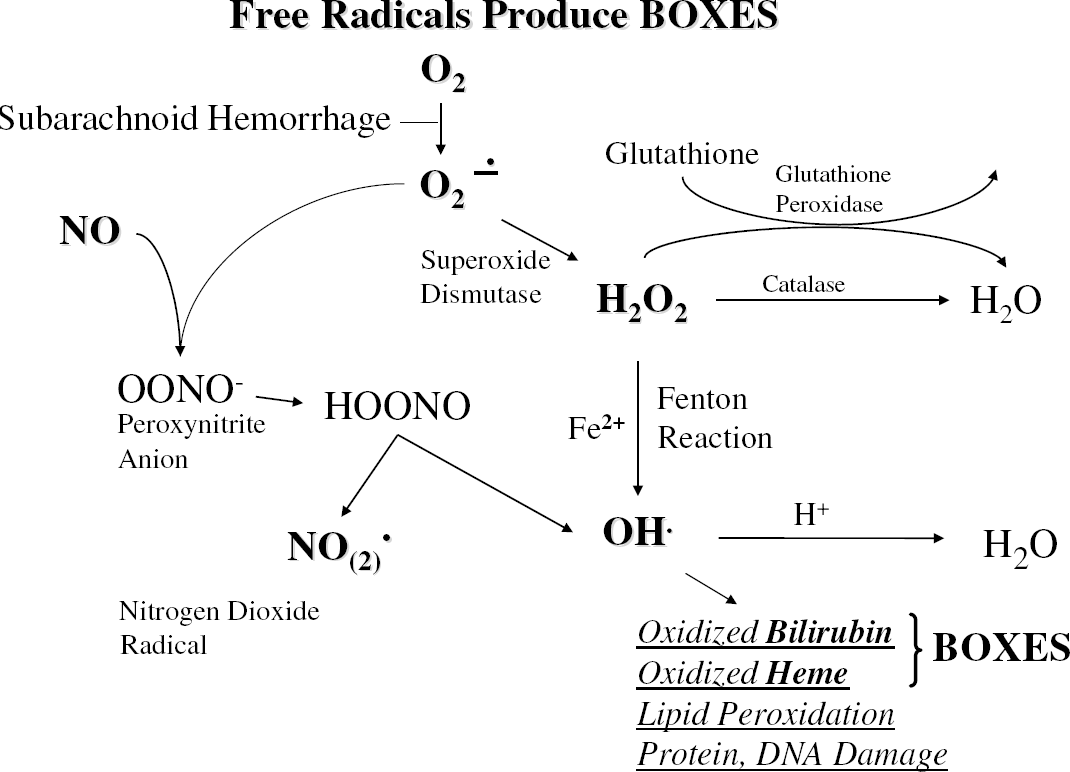
Free radicals that could produce BOXes. The pathways for the generation of superoxide, peroxynitrite, and hydroxyl ions are shown. Hydrogen peroxide in the presence of iron can produce hydroxyl ions that, in addition to the free radicals, act on bilirubin and biliverdin to produce BOXes.
The Discovery of BOXes
A search for vasoactive compounds in CSF of SAH patients was undertaken by Clark and co-workers, including Kranc, Cadoux-Hudson, and Pyne (Clark and Pyne-Geithman, 2005). This culminated in the identification of BOXes, compounds that are metabolites of bilirubin (Kranc et al, 2000). Bilirubin oxidation product was coined as a term for the compounds because they were formed by bilirubin oxidation. The BOX designation is also fitting since the BOXes were first identified in the Department of Biochemistry, OXford University in collaboration with the Department of Chemistry. Because there are isomers of the BOXes formed from the two ends of the bilirubin molecule (differing by the position of the methyl and vinyl groups; see also Figure 6), we have adopted the term BOXes as the plural form of BOX A and BOX B (Kranc et al, 2000). To date, we have observed no significant difference in the biologic activity of BOX A versus BOX B and consistently observe them to be present in equimolar concentrations.
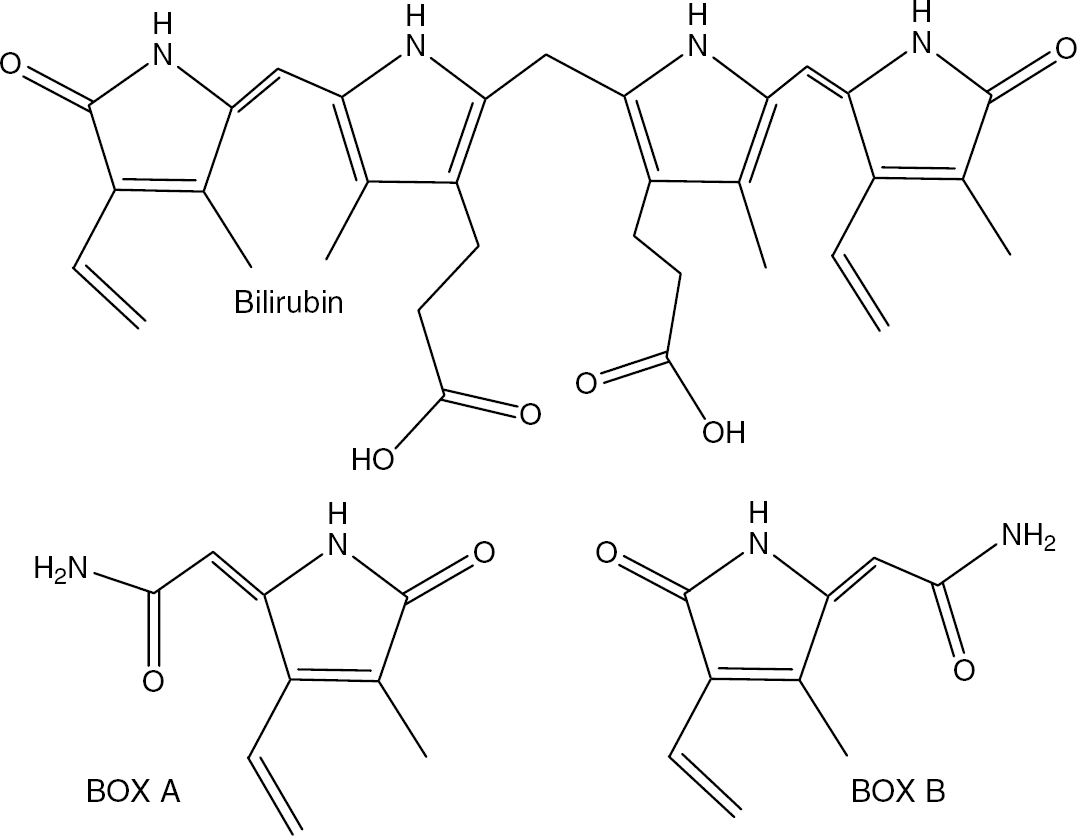
Structures of bilirubin, BOX A, and BOX B. Oxidative attack on the ring structure of bilirubin can produce BOX A and BOX B. Bilirubin oxidation product A and BOX B are derived by the cleavage of bilirubin at either end of the bilirubin molecule. The middle two pyrols are broken with the amide remaining with its nearest neighbor. The result is the formation of BOXes that are chemical isomers at the methyl and vinyl positions on the pyrole. It is because they are formed by the oxidation of bilirubin that they have been referred to as bilirubin oxidation products or BOXes.
Identification, Characterization, and Structure of BOXes
The process that led to the identification of BOXes included several bioassays and several different approaches. One bioassay included porcine carotid artery rings that displayed contraction/tension generation (vasospasm) in vitro when CSF from SAH patients with vasospasm was applied compared with CSF from SAH patients without vasospasm (Clark et al, 2001). The porcine carotid artery rings were also used to assess oxygen consumption when CSF from patients was applied. Oxygen consumption in vascular smooth muscle is intimately linked to contractile work (Krisanda and Paul, 1988; Lynch and Paul, 1987; Paul, 1983; Paul et al, 1984). Therefore, we utilize the rate of oxygen consumption as a surrogate marker for contractile stimulation (Clark et al, 1993, 2000a, b; Lynch and Paul, 1985; Paul, 1983). Cerebrospinal fluid from patients with SAH who had vasospasm produced marked increases of oxygen consumption by the carotid artery rings compared with CSF from SAH patients without vasospasm (Pyne et al, 2001b). The same CSF fractions from SAH patients with vasospasm that produced increases of oxygen consumption also produced increased tension generation of the carotid artery rings. These two bioassays helped confirm which CSF samples contained the active compound associated with vasospasm (Kranc et al, 2000; Pyne et al, 2000, 2001a, b).
These studies required sufficient CSF from SAH patients, which was possible because of the dedicated efforts of the Oxford Neurosurgeon Thomas Cadoux-Hudson (Cadoux-Hudson et al, 2001; Kranc et al, 2000; Pyne et al, 2000, 2001b). By serendipity, the samples were collected so that they were protected from light (wrapped in foil) and air (frozen). Protection from light was critical for the success of these studies.
The nature of the molecule(s) that contributed to vasospasm was clarified by extraction of the active molecules. The finding that the vasoactive compounds were extracted in chloroform suggested lipid solubility and tended to rule out proteins or peptides as the active compound. The finding that the extracted compounds were sensitive to light suggested that bilirubin or derivatives of bilirubin could be the vasoactive compounds. In addition, the finding that the biologic activity of the vasoactive compounds was markedly reduced by light helped explain many difficulties in previous studies where attempts at purification of causative molecules using in vitro assays appeared to fail or were not very fruitful.
These biochemical features of the extraction of the vasoactive compounds from CSF of SAH patients led us to consider whether oxidation of bilirubin or biliverdin might produce vasoactive compounds. Therefore, we examined the effects of hydrogen peroxide (H2O2) on purified bilirubin. Hydrogen peroxide was chosen mainly because it is easy to work with, and it is formed in vivo via reactions shown in Figure 5. Hydrogen peroxide, in the presence of iron, forms hydroxyl ions (Fenton reaction) that would be expected to oxidize bilirubin, biliverdin, and heme—all of which would be found in high concentration in clots surrounding cerebral blood vessels after SAH.
The initial studies showed that treatment of pure bilirubin with H2O2 (BOX) produced compounds that increased oxygen consumption and increased contractile tension of porcine carotid artery rings to a similar degree or even greater than CSF from SAH patients with vasospasm (Kranc et al, 2000). In addition, the oxidized bilirubin compounds were extractable with chloroform and they were light sensitive—just as the vasoactive compounds in CSF were (Kranc et al, 2000).
BOXes and Contraction of Vessels In Vitro
We have unpublished preliminary data that when isolated vascular rings in the absence of endothelial cells and absence of myogenic tone or exogenous stimulation have no contractile response to BOXes alone (Pyne et al, 2004). However, BOXes potentiated the contraction to KCl depolarization as well as to agonist-induced contractions to histamine and phenylepherine. Thus, the paradox appears to be that rather than initiating the contractile events, BOXes in vitro shift the system to greater degrees of contraction and subsequent lumenal narrowing. This increased lumenal narrowing is obfuscated by the apparent slowing of relaxation observed by isolated vascular rings exposed to BOXes (Pyne et al, 2004). The putative result of potentiation of contraction along with inhibition of relaxation could indeed lead to the constriction to occlusion observed during vasospasm.
Characterizing the BOXes and Detecting in Cerebrospinal Fluid
The next steps were to characterize the compounds that are produced by the oxidation of bilirubin. Therefore, bilirubin treated with H2O2 was extracted with chloroform and applied to a reverse-phase HPLC column. Three main peaks were detected and isolated. Mass spectrometry identified one peak as MVM, which is 4-methyl-3-vinylmaleimide—a previously isolated photodegradation product of biliverdin. The other two molecules were novel, and were named BOX A and BOX B (Kranc et al, 2000), as are shown in Figure 6. Bilirubin oxidation product A and BOX B were vasoactive in vitro, and lost all biologic activity when exposed to sunlight (Kranc et al, 2000).
Importantly, CSF from SAH patients with vasospasm was extracted with chloroform and applied to the HPLC. Three peaks that migrated at the approximate positions as MVM, BOX A and BOX B were identified. In addition, when BOX A was added to the CSF-SAH (doping experiments), the retention time of the representative peak in the CSF-SAH comigrated with BOX A (Kranc et al, 2000).
These data provided the first definitive evidence that BOX A was produced by oxidation of bilirubin or biliverdin, and that BOX A was present in the CSF of SAH patients.
Chemistry and Oxidative Stress after Subarachnoid Hemorrhage
We have shown that treatment of either bilirubin or biliverdin with H2O2 produced MVM, BOX A, and BOX B (Figures 6 and 7; Kranc et al, 2000). However, based on structural considerations alone, it is also possible that direct oxidation (or peroxidation) of heme within Hb could also produce BOXes or at least BOX-like, vasoactive molecules (shown schematically in Figure 7). Mechanistically, it is extremely important whether BOXes come exclusively from bilirubin and biliverdin (Figure 4) or whether they could also come from heme (Figures 3 and 7). This is because bilirubin and biliverdin are products of HO metabolism, and HO is found mainly intracellularly (Figure 7). If BOXes can also be formed directly form oxidation of heme, then free radicals in the extracellular milieu acting directly on heme in the clot could produce BOXes extracellularly (Figure 7). If, however, BOXes are derived mainly from bilirubin and biliverdin, then inhibiting HO enzyme activity should slow the production of bilirubin and possibly decrease the concentrations of BOXes that are produced (Figure 7).
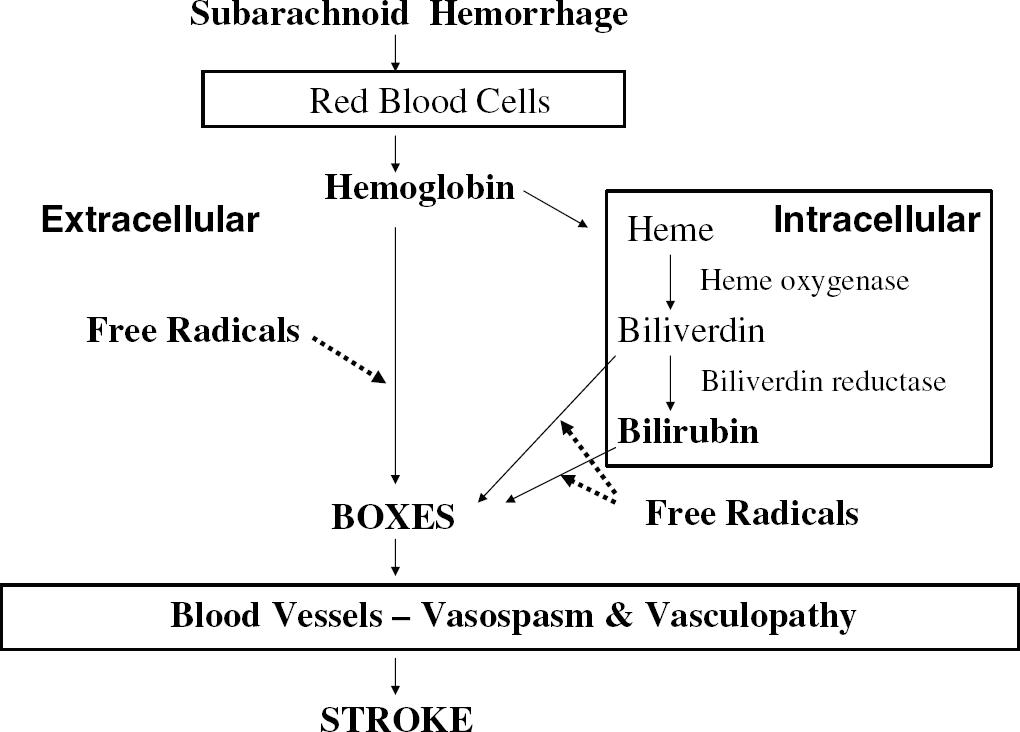
Production of BOXes via intracellular versus extracellular routes. The likely route for the formation of BOXes is via oxidation of bilirubin. Heme is transporter intracellularly where heme oxygenase metabolizes heme to biliverdin, and biliverdin reductase metabolizes biliverdin to bilirubin. Free radicals act on bilirubin and biliverdin to produce BOXes. Alternatively, heme from hemoglobin could be acted upon by free radicals extracellularly to produce BOXes. The relative roles for these two routes for production of BOXes are unknown. The intracellular route of production of biliverdin and bilirubin and oxidation of biliverdin and bilirubin to BOXes is the most likely route. The intracellular route for the production of BOXes is important since inhibiting heme oxygenase or biliverdin reductase could decrease bilirubin production and production of BOXes. Once BOXes are formed, they diffuse into vascular smooth muscle cells to produce chronic vasospasm and a vasculopathy that can result in stroke after subarachnoid hemorrhage.
There is growing evidence that oxidative stress contributes to, or is associated with, vasospasm after SAH (Macdonald et al, 2004; Macdonald and Weir, 1994; Morgan et al, 2004; Pyne-Geithman et al, 2005). Features of SAH that contribute to free radicals and high oxidative stress include the presence of large amounts of clot and Hb that contains large amounts of iron. The Fenton reaction using Iron can contribute to the production of highly reactive hydroxyl species (Figure 5). Inflammatory cells that respond to and help clear the blood and clot are a tremendous source of superoxide and peroxynitrite anions (Figure 5). Nicotinamide adenine dinucleotide phosphate (reduced form) oxidase activity often occurs at endosomes and vesicles, and could be involved, in the production of BOXes. Based on chemical modeling of heme, biliverdin, and bilirubin, the BOXes could be produced via nucleophilic attack with H2O2, superoxide, nitric oxide, or peroxynitrite radicals (Figures 5 and 6). Thus, the basic hypothesis is that a very strong oxidative environment—found in the clot around cerebral vessels—combined with oxidation of bilirubin and biliverdin found in high concentrations in the clot—acts on the cerebral vessels to produce vasospasm.
Additional evidence of increased oxidative stress after SAH includes increased lipid peroxides and peroxidized lipids (Okamoto et al, 1997) and increased malonyldialdhyde (Macdonald et al, 1992) and reactive oxygen species. However, these reports are relatively nonspecific concerning the mechanism by which oxidative stress might occur, since the oxidative stress could cause or contribute to vasospasm or simply be the result of vasospasm. Further, recent clinical trials with antioxidants such as tirilazad (Haley et al, 1995; Kassell et al, 1996; Lanzino et al, 1999) have not been extremely effective in treating the vasospasm patient. These negative reports should not, however, be interpreted as indicating that oxidative stress is not involved in the etiology of SAH-induced cerebral vasospasm. The antioxidants may not have reached the therapeutic target in the clots and vessels, or the antioxidants used may not have targeted the appropriate free radicals. The relative membrane-specific antioxidant effect of tirilazad makes it unlikely to prevent BOXes formation, if the BOXes are formed in the vesicles of macrophages, for example, Hall (1996).
We have recently confirmed evidence of oxidative stress using CSF of SAH patients. Oxidative stress increased, as assessed by lipid peroxides and malonyldialdehyde (MDA) measurements, in the SAH patients with vasospasm compared with SAH patients without vasospasm (Pyne-Geithman et al, 2005). Notably, the temporal profile of the reactive oxygen species in the SAH patients with vasospasm was similar to the time course of the incidence of vasospasm (Pyne-Geithman et al, 2005). Similarly, we also reported that the production of bilirubin in the CSF after a porcine model of SAH produced bilirubin with a similarly delayed time course (Morgan et al, 2004). Again these data does not answer whether the measured increases of oxidative stress are a cause or an effect of SAH-induced vasospasm. The data are simply consistent with the hypothesis that a strong oxidizing environment exists that could contribute to the oxidation of bilirubin to produce BOXes in the clot around vessels after SAH.
Finally, the role of bilirubin in vasospasm is likely to be complex. We propose that oxidation of bilirubin produces molecules (BOXes) that cause or contribute to vasospasm. However, bilirubin itself has been shown to be an antioxidant (Dohi et al, 2003; Stocker et al, 1987, 1990) and could therefore protect from vasospasm by decreasing oxidative stress. If bilirubin protects against oxidative stress and protects against vasospasm, and if bilirubin oxidation produces molecules that contribute to vasospasm, then whether vasospasm occurs may be of some balance between these two factors—BOX concentrations versus bilirubin concentrations. Therefore, some consideration of relative BOXes concentrations to bilirubin may correlate better with vasospasm rather than just BOXes concentrations (Pyne-Geithman et al, 2005).
BOXes concentration in Patients—Preliminary Data
With the discovery of BOXes, the Clark group has begun to assess levels of BOXes in the CSF of patients (Pyne-Geithman et al, 2005). Examining CSF using a spectrophotometeric technique and scanning at a 320 nm made it is possible to detect the BOXes in CSF of these patients. This was accomplished using CSF that was extracted to remove proteins and the quantification performed using a standard curve and the BOXes extinction coefficient. In SAH patients with vasospasm, the BOXes concentrations started at the lower limits for our current ability to detect them and increased to significant levels (1.2–1.4 μ/l) at about 6 to 8 days after SAH. Although much work remains to be performed correlating the occurrence of vasospasm and BOXes concentrations detectable in the CSF, an important finding was that the BOXes concentration was only elevated in those patients where the bilirubin and oxidative stress were increased (Pyne-Geithman et al, 2005). Thus, it appears that a combination of oxidative stress and high bilirubin are required to produce BOXes, which in turn produce SAH-induced vasospasm. This multifactorial requirement (for increased oxidative stress and increased bilirubin) being necessary for BOXes production and the induction of vasospasm is consistent with the published data, suggesting that multiple factors may contribute to SAH-induced cerebral vasospasm (Dietrich and Dacey, 2000; Janjua and Mayer, 2003; Kaye et al, 1984; Lanterna et al, 2005; Macdonald et al, 1994, 2004; Macdonald and Weir, 1991, 1994; Pluta, 2005; Sobey and Faraci, 1998; Zimmermann and Seifert, 1998).
Although BOXes and oxidative stress may be necessary or at least very important for producing vasospasm, they may not be singularly sufficient for producing vasospasm. Bilirubin oxidation products, oxidative stress, and perhaps one other additional factor—Hb, endothelin, or another—may all be necessary to produce SAH-induced vasospasm— and no one factor may be sufficient to produce clinical vasospasm after SAH. More work will be required to provide the final answers to these questions.
BOXes and Vasospasm In Vitro and In Vivo
With the identification of BOXes and their correlation with SAH-induced vasospasm in patients, other experiments determined whether BOXes produced vasospasm using in vitro and in vivo models (Clark et al, 2002; Lyons et al, 2004). These experiments generally confirm the idea that the BOXes are biologically active and can produce vasospasm. As briefly described above, application of BOXes to porcine carotid artery rings in vitro caused increased tension in the vascular rings (Pyne et al, 2000, 2003). This correlate of vasoconstriction was associated with increased oxygen consumption and ATP consumption (Cadoux-Hudson et al, 2001; Lyons et al, 2004; Pyne et al, 2000, 2001a, b). The time course and metabolic responses of the carotid artery rings after application of the BOXes was similar to that after application of CSF from SAH patients with vasospasm (Kranc et al, 2000).
An important in vivo confirmation of these results was performed by applying BOXes directly to surface vessels of cortex using a cranial window in rats (Clark et al, 2002). The BOXes produced a dose-dependent vasospasm that occurred within 30 mins and was prolonged, lasting at least 24 h. The vasospasm was associated with increased stress gene expression in subcortical gray matter. We concluded that ‘BOXes produce a severe and prolonged vasospasm of dural and cerebral vessels. This finding supports the possibility that the oxidized products of bilirubin found in the CSF of patients with vasospasm after SAH directly contribute to and/or are the primary cause of SAH-induced cerebral vasospasm.’ Using this in vivo model, however, direct application of BOXes did not produce a stroke or cortical infarction seen in humans with SAH-induced vasospasm (Clark et al, 2002). Thus, BOXes are not sufficient to produce vasospasm-induced stroke in vivo, at least using the parameters that were used in those studies (Clark et al, 2002). Another factor, or factors must be present and active. Alternatively, the prolonged exposure of the brain and blood vessels to BOXes may be required for the development of cortical infarction since our in vivo experiments have only used a single application of BOXes.
BOXes may not be the sole cause of vasospasm. Our data strongly suggest that BOXes are associated with SAH-induced vasospasm, but their causative role still needs to be established. For example, it is possible that BOXes act to inhibit relaxation, but that endothelin stimulates constriction. Such distinctions for the events associated with vasospasm, constriction, maintenance of constriction, and DIND, have yet to be determined.
Boxes Injure Smooth Muscle Cells in Culture
Not only does SAH produce vasospasm but also the vessel walls may be histologically, metabolically and functionally abnormal, suggesting that factors associated with SAH produces a vasculopathy in combination with the vasospasm (Alksne and Branson, 1980; Boullin and Mohan, 1977; Boullin et al, 1976, 1981; Findlay et al, 1991; Kim et al, 2000; Mayberg, 1998; Ogihara et al, 1999; Sobey and Faraci, 1998; Stoodley et al, 2000). Therefore, we studied the effects of BOXes on vascular smooth muscle cells in culture (Lyons et al, 2004). Vascular smooth muscle cells from porcine carotid arteries were grown in culture, exposed to increasing doses of BOXes, and analyzed. Oxidative phosphorylation was assessed in the cells using the water-soluble tetrazolium salt (WST) assay for succinate dehydrogenase. Succinate dehydrogenase is a key regulatory step in the Krebs cycle and is part of the electron transport chain via Complex II. Confluent vascular smooth muscle cells exposed to BOXes for 48 h showed a dose-dependent increase in succinate dehydrogenase activity. The increase in oxidative metabolism is reminiscent of the increased oxygen consumption that we observed in porcine carotid artery rings exposed to BOXes or to CSF from SAH patients or to BOXes (Cadoux-Hudson et al, 2001; Pyne et al, 2001b). The increased metabolism could mean either that there is increased ATP flux or mitochondrial dysfunction/uncoupling. Because BOXes do not appear to stimulate directly mitochondria, we proposed that BOXes increase ATPase activity including the myosin ATPase, which is the most active ATPase in the muscle, to produce metabolic stress on the vessels (Lyons et al, 2004). We also showed that (LDH) was released from smooth muscle cells exposed to BOXes, likely indicating increased metabolic/anaerobic stress on the cells, and cell injury and release of LDH through leaky cellular membranes (Lyons et al, 2004).
In addition, the BOXes produced morphologic reorganization of the contractile proteins as seen with the alpha smooth muscle actin staining (Lyons et al, 2004). Vascular smooth muscle is a contractile tissue that is normally fusiform with parallel filaments making up its cytoarchitecture. The thin filaments are largely composed of actin and the alpha smooth muscle actin is considered a reliable marker of vascular smooth muscle cells. Normally, the alpha smooth muscle actin is aligned in parallel in vivo and to a certain extent in vitro as well. When the vascular smooth muscle cells were exposed to BOXes, there was a profound reorganization of the alpha smooth muscle actin (Lyons et al, 2004). This abnormal cytoarchitecture would make the cells less contractile and would be detrimental to vascular function (Lyons et al, 2004). Future studies are required to determine if BOXes produce similar structural changes in vivo. The data suggest that BOXes stress cells, alter actin architecture, increase LDH release and stimulate respiration by activating ATPases such as myosin ATPase (Butler et al, 1996; Takagi et al, 1989). The occurrence of these events in vivo would contribute to vasospasm/lumenal narrowing, especially if the contractile protein myosin ATPase was stimulated (Butler et al, 1996; Frykholm et al, 2004; Petzold et al, 2003). Bilirubin oxidation products induced changes in contractile proteins in smooth muscle cells might explain that the vessels with chronic vasospasm after SAH are resistant to various types of vasodilatory treatments, so much so that angioplasty is now often used to physically dilate these narrowed vessels (Bejjani et al, 1998; Coyne et al, 1994; Janjua and Mayer, 2003; Zubkov et al, 1999).
Summary of Evidence that BOXes Cause or Contribute to Vasospasm
We propose that BOXes are a causative agent of SAH-induced cerebral vasospasm. The BOXes can explain why SAH-induced vasospasm is delayed by 3 to 7 days after SAH. After SAH and clot formation, it takes time for heme to be metabolized to bilirubin, with the peak bilirubin concentrations occurring at 3 to 4 days after SAH (Morgan et al, 2004; Page et al, 1994; Pyne-Geithman et al, 2005; Shuttleworth et al, 1977; Vermeulen and van Gijn, 1990; Vermeulen et al, 1983)}. Once the bilirubin were formed, it would take additional time for oxidation of the bilirubin to occur, helping to explain the delay of vasospasm from 4 to 11 days After SAH. Many studies have suggested that oxidative stress might cause or at least contribute to vasospasm. If BOXes do cause vasospasm, then free radical/oxidative stress would certainly contribute to the formation of BOXes.
We have published four critical observations that support a role for BOXes as a cause of SAH-induced cerebral vasospasm. First, we discovered oxidized forms of bilirubin and determined the structure of these new molecules in the CSF of SAH patients (Kranc et al, 2000). Second, we showed that BOXes formed by the peroxidation of bilirubin in vitro stimulated vessel contraction and metabolism, and that this was similar to the metabolic and vasoactive properties of CSF from patients with SAH-induced vasospasm (Lyons et al, 2004). Third, we showed that the BOXes were vasoactive in vivo using a rat cranial window model for assessing vessel diameters (Clark et al, 2002). Fourth, we have presented preliminary data showing that SAH patients with vasospasm generally had high CSF concentrations of bilirubin, oxidative stress and BOXes compared with SAH patients without vasospasm that generally did not have BOXes detectable in CSF (Pyne-Geithman et al, 2005). These data support a role for BOXes in causing vasospasm.
Future Therapy
Assuming that BOXes are at least one of several factors that cause vasospasm after SAH, several therapeutic approaches could be considered and important questions concerning these approaches addressed along the way. The importance of BOXes production needs to be established by examining the time course of BOXes production and clearance in the CSF to the clinical condition after SAH. Then, bilirubin production could be slowed or reduced using current HO inhibitors (metal protoporphyrins) that could decrease precursor bilirubin levels and decrease BOX concentrations. This assumes, as mentioned above, that direct oxidation of heme is not a major source of BOXes (Figure 7). If BOXes are being produced, after SAH, the localization of this production will be important to devise appropriate strategies. If BOXes are produced in the lysosomes of macrophages, then an extracellular antioxidant therapy might not be appropriate, but an antioxidant therapy that works in cells might be beneficial. Further, preventing macrophage activation might be beneficial. Alternatively, new attempts at devising specific antioxidants that prevent oxidation of bilirubin, and that might be effective at diffusing through clot and into inflammatory cells including white blood cells and macrophages, might be sought and tested. Bilirubin oxidation products specific antibodies could be considered to decrease their activity, although delivery to areas surrounding blood vessels might be problematic. Equally, the ability to manufacture specific BOXes antibodies is problematic because of their size and structural similarity to bilirubin. The sensitivity of the BOXes to sunlight could potentially be exploited by testing whether more penetrating types of radiation might also inactivate BOXes. Therapies designed to remove blood around vessels could be more aggressively pursued since removing blood should decrease heme, bilirubin and BOXes, and ultimately prevent vasospasm.
Footnotes
Acknowledgements
We acknowledge the substantial contributions made by numerous researchers who have built a substantial body of knowledge in this important field. To list all the substantial contributors would be prohibitive; however, we offer our special thanks to the contributions of Pickard, MacDonald, Wier, Zhang, Foresti, Dohi, Suzuki, Miao, and Lee. We also wish to acknowledge our collaborators who have contributed to this work: Chris Schofield, Thomas Cadoux-Hudson, Gail Pyne-Geithman, Mario Zuccarello, and the Cincinnati Northern Kentucky Stroke Team.