
Abstract
Delayed cerebral vasospasm after subarachnoid hemorrhage (SAH) remains a significant cause of mortality and morbidity; however, the etiology is, as yet, unknown, despite intensive research efforts. Research in this laboratory indicates that bilirubin and oxidative stress may be responsible by leading to formation of bilirubin oxidation products (BOXes), so we investigated changes in bilirubin concentration and oxidative stress in vitro, and in cerebral spinal fluid (CSF) from SAH patients. Non-SAH CSF, a source of heme oxygenase I (HO-1), and blood were incubated, and in vitro bilirubin production measured. Cerebrospinal fluid from SAH patients was collected, categorized using stimulation of vascular smooth muscle metabolism in vitro, and information obtained regarding occurrence of vasospasm in the patients. Cerebral spinal fluid was analyzed for hemoglobin, total protein and bilirubin, BOXes, malonyldialdehyde and peroxidized lipids (indicators of an oxidizing environment), and HO-1 concentration. The formation of bilirubin in vitro requires that CSF is present, as well as whole, non-anti-coagulated blood. Bilirubin, BOXes, HO-1, and peroxidized lipid content were significantly higher in CSF from SAH patients with vasospasm, compared with nonvasospasm SAH CSF, and correlated with occurrence of vasospasm. We conclude that vasospasm may be more likely in patients with elevated BOXes. The conditions necessary for the formation of BOXes are indeed present in CSF from SAH patients with vasospasm, but not CSF from SAH patients without vasospasm.
Introduction
Cerebral vasospasm is a significant cause of mortality and morbidity in patients after subarachnoid hemorrhage (SAH). Approximately half of the patients who survive the initial SAH go on to develop vasospasm after 3 to 10 days (Weir et al, 1978). It is thought that the causative agent is located in the cerebrospinal fluid (CSF) of patients (Buckell, 1964). Despite intensive research efforts, the etiology of vasospasm has not yet been elucidated (Cadoux-Hudson et al, 1999; MacDonald, 1997; Pyne et al, 2000, 2001, 2003).
The delayed nature of vasospasm poses a problem: many of the putative causative agents investigated are not found in the CSF over a time course compatible with the onset of vasospasm (Buckell, 1964). However, the delay allows clinicians a valuable therapeutic window to prophylactically treat SAH patients.
Many CSF components after SAH have been placed under suspicion as the causative agents in this pathology, including hemoglobin (Wickman et al, 2003), bilirubin (Jia-Pei Miao and Jer-Fu Lee, 1989), endothelin (Zuccarello et al, 1995), peroxidized lipids (Takuwa et al, 1993), and bilirubin oxidation products (Clark et al, 2002; Lyons et al, 2004). Previous work from this laboratory has shown that the in vitro activity of CSF from SAH patients with vasospasm is not diminished by removal of protein from the CSF (Pyne et al, 2001). This excludes endothelin and hemoglobin and other proteins as causative agents. Bilirubin is produced over a time course comparable to vasospasm, but it does not cause contraction in smooth muscle in vitro or in vivo (Clark et al, 2002). Bilirubin oxidation products (BOXes) may have a similar time course to onset of vasospasm, and they are vasoactive both in vivo and in vitro (Clark et al, 2002). Evidence that oxidative stress may contribute to vasospasm (cerebral and coronary) has been published (Gaetani et al, 1998; Kahler et al, 2001; Matz et al, 1996; Polidori et al, 1997; Reeder et al, 2002); however, antioxidant therapy alone has not proven clinically effective, and is not standard of care (Asano et al, 1984; Asano et al, 1996; Germano et al, 1998; Luo et al, 1995). Thus, oxidative stress alone may not be the sole cause of vasospasm.
A perplexing question which is difficult to address regarding vasospasm after SAH, is why some SAH patients develop vasospasm, and some do not. In this study, we set out to investigate the conditions occurring in SAH patients' CSF, namely (i) presence of heme oxygenase, which is required for formation of bilirubin; (ii) formation of bilirubin over a comparable time course to vasospasm; (iii) presence of an oxidizing environment; and (iv) levels of BOXes. Our results suggest that increased BOXes, bilirubin, and oxidative stress are present in relatively high concentrations in the CSF of vasospasm patients.
MATERIALS AND METHODS
In Vitro Experiments
The in vitro incubation conditions are shown in Table 1. Control CSF was discarded after drainage for therapeutic purposes from ambulatory outpatients with hydrocephalus. Blood was obtained from healthy volunteers in red-capped BD Vacutainer Systems (Franklin Lakes, NJ, USA) tubes using standard sterile techniques.
Incubation conditions for in vitro bilirubin production
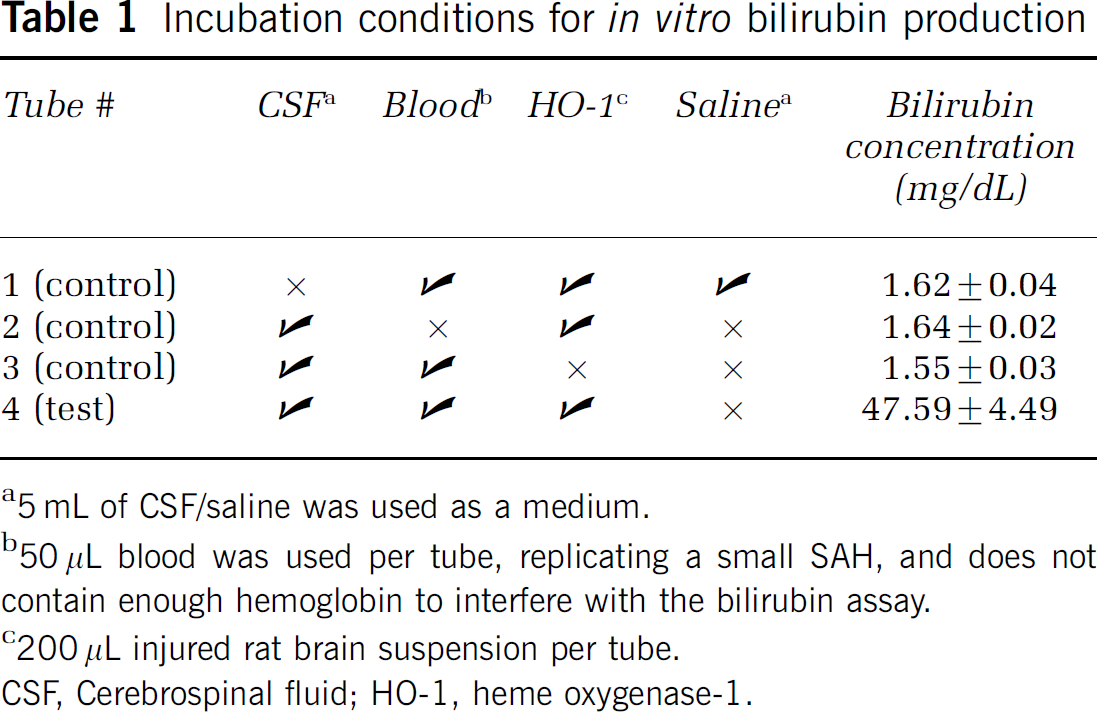
a5 mL of CSF/saline was used as a medium.
b50 μL blood was used per tube, replicating a small SAH, and does not contain enough hemoglobin to interfere with the bilirubin assay.
c200 μL injured rat brain suspension per tube.
CSF, Cerebrospinal fluid; HO-1, heme oxygenase-1.
Injured Rat Brain
The brain suspension was prepared as follows. Male rats were subjected to brain injury as previously described (Clark et al, 2002). Briefly, rats were anesthetized and 150 μL of autologous, whole blood was injected into the cisterna magna while animals were under general anesthesia. All surgical procedures were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the University of Cincinnati Animal Care and Use Committee. Autologous whole blood was collected from each rat from the femoral artery. The rats were allowed to recover for 24 h, and then killed. The rat brains were removed and homogenized, using a glass pestle, in a buffer containing (mmol/L) TRIS HCl, 20; sucrose, 0.25; EGTA, 10; EDTA, 1; β-mercaptoethanol, 10; phenyl-methyl-sulfonyl fluoride, 1, with 0.02% leupeptin, and 0.1% Triton X-100. One milliliter of buffer was used for each 100 mg of brain tissue. The suspension was gently centrifuged (1000g) to sediment large debris, and the supernatant used as a source of heme oxygenase-I (HO-1). This suspension was also used as a positive control for the HO-1 assay, which shows that there was a significant quantity of HO-1 protein present. Rat brain was used as a source of HO-1 (Matz et al, 1996).
Patient Cerebrospinal Fluid Selection
An IRB letter of exemption was obtained before the start of experiments. Cerebrospinal fluid obtained from SAH patients who received placement of temporary intraventricular or lumbar catheters as standard of care was collected. The CSF used in this study was selected according to the after criteria and has been described elsewhere (Cadoux-Hudson et al, 2001; Pyne et al, 2001):
(i) Control CSF: Non-SAH CSF containing no blood.
(ii) CSFc: From SAH patients whose CSF did not stimulate vascular smooth muscle as assessed by an increase in JO2 of >0.4 μmol/L O2/min g dry weight (dwt).
(iii) CSFv: From SAH patients whose CSF stimulated vascular smooth muscle as assessed by an increase in JO2 of >0.4 μmol/L O2/min g dwt.
The demographics of the patient population were obtained, and are described in the results section, in Table 2. The data were obtained adhering to HIPAA guidelines.
Table 2 contains a column headed vasospasm for completeness. This refers to the clinical symptomatic or asymptomatic presence or absence of vasospasm in the patients at the time the CSF was collected. This information was obtained in HIPAA compliant manner on a post hoc basis, and was not used as a criterion to classify CSF.
Bilirubin Determination
Patient demographics, biochemical and clinical parameters
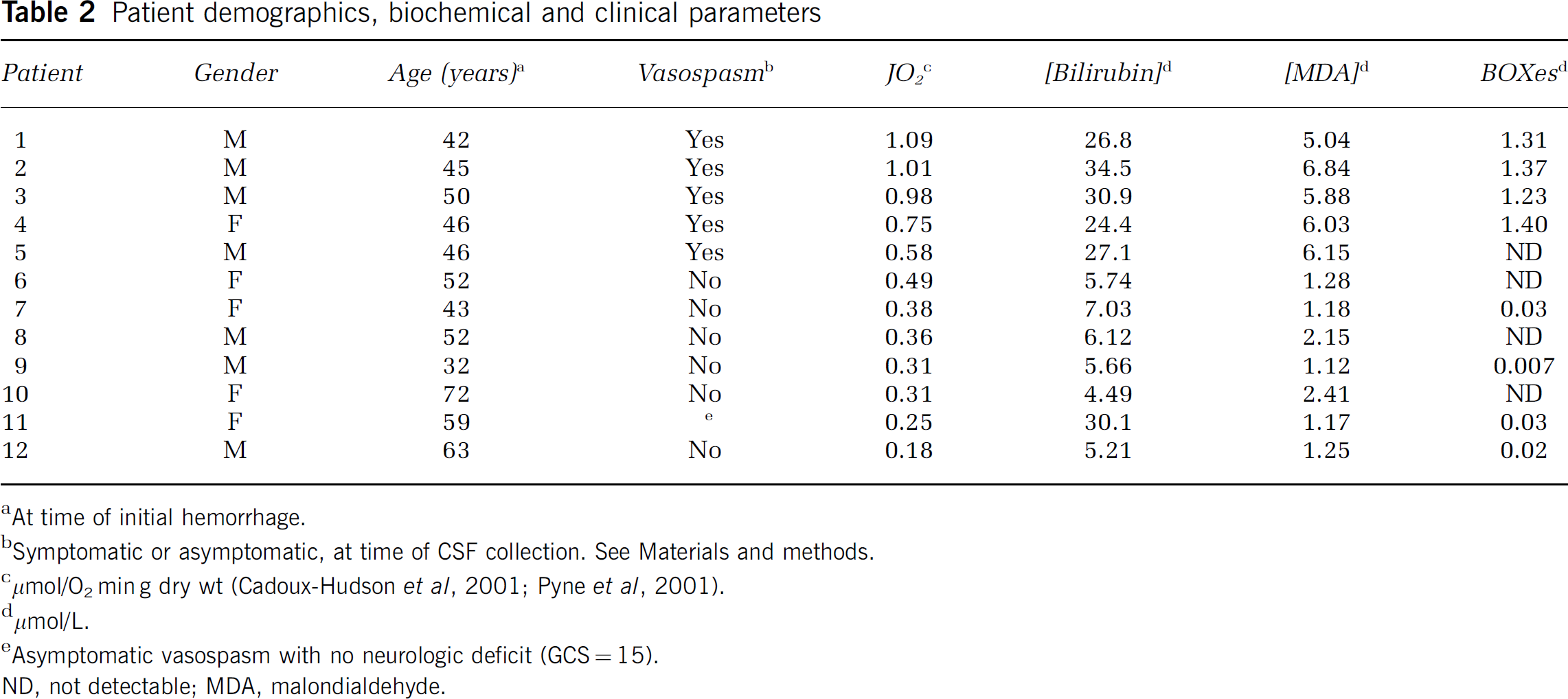
aAt time of initial hemorrhage.
bSymptomatic or asymptomatic, at time of CSF collection. See Materials and methods.
cμmol/O2 min g dry wt (Cadoux-Hudson et al, 2001; Pyne et al, 2001).
dμmol/L.
eAsymptomatic vasospasm with no neurologic deficit (GCS = 15).
ND, not detectable; MDA, malondialdehyde.
Total bilirubin was assayed using a method based on those developed by Michaelsson (1961) and Nosslin (1960) and adapted for use in a micro titer plate. To quantify bilirubin, the CSF (10 μL) was treated with a caffeine/benzoic acid reagent (25 g/L caffeine and 38 g/L sodium benzoate in 85 mmol/L sodium acetate solution) to release bound bilirubin. Diazo reagent (75 μmol sulfanilic acid and 6.6 μmol sodium nitrite dissolved in 6 mL of 0.1 mol/L HCl) was added to yield azobilirubin. This moiety was stabilized with a cysteine reagent (100 mg cysteine dissolved in 10.5 mL of dH2O) and alkalinized with alkaline tartrate reagent (100 mg cysteine dissolved in 10.5 mL of dH2O) to yield a blue coloration, which is less prone to hemoglobin interference. The resultant solution was analyzed spectrophotometrically at 600 nm, and the samples compared with a concomitantly run standard curve of Lin-trol bilirubin prediluted samples (Sigma, St Louis, MO, USA) to determine total bilirubin concentration.
Hemoglobin Determination
The sample is exposed to Drabkin's reagent (NaHCO3:K3Fe (CN)6:KCN, 100:20:5) to convert all the hemoglobin moieties to cyanmethemoglobin (Schoen and Solomon, 1962). A surfactant (Brij-35) was added to prevent protein or lipid-induced turbidity. The cyanmethemoglobin absorbs light at 540 nm, so the concentration can be determined by comparison with a standard curve constructed using Drabkin's reagent-treated lyophilized methemoglobin (Cat # 525–18; Sigma Diagnostics, St Louis, MO, USA).
Protein Determination
The BCA protein assay by Pierce (Rockford, IL, USA) was used to assess total protein concentrations. This assay is described in detail in Smith et al (1987).
Lipid Peroxidation Determination
Total peroxidized lipids were assayed using a commercially available kit from Calbiochem (San Diego, CA, USA). Peroxidized lipids are detected by their conversion of ferrous ions to ferric ions in acidic solution. The ferric ions then bind with the indicator dye, xylenol orange, and cause a color change, which can be monitored by measuring absorbance at 560 nm. The concentration of peroxidized lipids is then calculated using the molar extinction coefficient of the xylenol orange product (Nourooz-Zadeh et al, 1994).
Malondialdehyde Determination
Malondialdehyde (MDA) was assessed using a commercially available kit from Calbiochem (San Diego, CA, USA). Malondialdehyde reacts with N-methyl-2-phenylindole in acetonitrile and ferric ions to form a colored molecule that has a maximum absorbance at 586 nm (Esterbauer and Cheeseman, 1990).
Heme Oxygenase 1 Concentration Determination
Heme Oxygenase 1 was determined using a commercially available kit from StressGen Bioreagents (Victoria, BC, Canada). The assay is an ELISA containing human HO-1 antibodies. We specifically looked at HO-1 (as opposed to the constitutively active HO-2), as HO-1 is inducible by hemorrhagic means, and is therefore the most likely isoenzyme to be involved in conversion of heme to biliverdin after an SAH. We also wanted to confirm that the rat brain used as a source of HO-1 in the in vitro experiments did indeed contain HO-1 protein.
Bilirubin Oxidation Products Quantification Methods
Each sample was scanned spectrophotometrically between 280 and 500 nm, and a peak at 320 nm is clearly discernable. This is the wavelength at which BOXes absorb. The absorbance at 320 nm is compared with a standard curve of BOXes.
Statistical Analysis
Data were analyzed using ANOVA and P≤0.05 was considered to be statistically significant.
RESULTS
In Vitro Bilirubin Production
Figure 1 shows the production of bilirubin in vitro. It can be seen that the conditions necessary for bilirubin production not only include blood and HO-1, as one might expect, but require CSF under these conditions. The assessment of in vitro bilirubin production was also performed using lysed blood and anticoagulated blood, but no bilirubin production was observed (data not shown). The bilirubin concentration was significant at 24 h and continued to rise for the next 96 h.
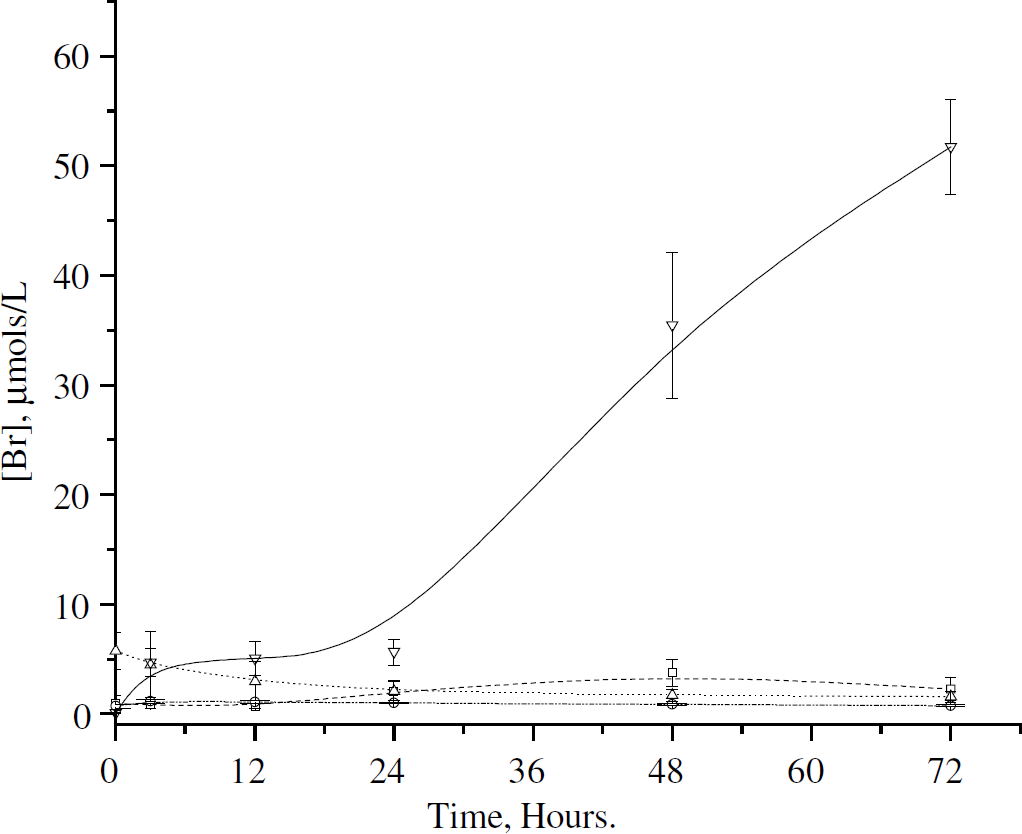
Production of bilirubin in vitro. Bilirubin production in in vitro mixtures of CSF/saline, heme oxygenase-I (HO-1), and blood. Please see Table 1 for tube contents and incubation conditions. □ = Tube 1, n = 3; = Tube 2, n = 3; ▵ = Tube 3, n = 3; ▿ = Tube 4, n = 3. The only incubation conditions resulting in bilirubin production included whole blood, HO-1,
Patient Population
Table 2 lists the patient demographics and biochemical parameters. The study patients comprised seven men and five women. The median age was 46 years, and the ages ranged from 32 to 72 years at the time of hemorrhage.
Bilirubin Concentration in Patients' Cerebrospinal Fluid
Figure 2A shows that the bilirubin concentration in the CSF samples increases over time; that is, the longer the SAH blood remains in the CSF, the more bilirubin is produced. Control (non-SAH) CSF contained levels of bilirubin that were at or below the level of sensitivity of the assay (0.01 μmol/L) (data not shown).
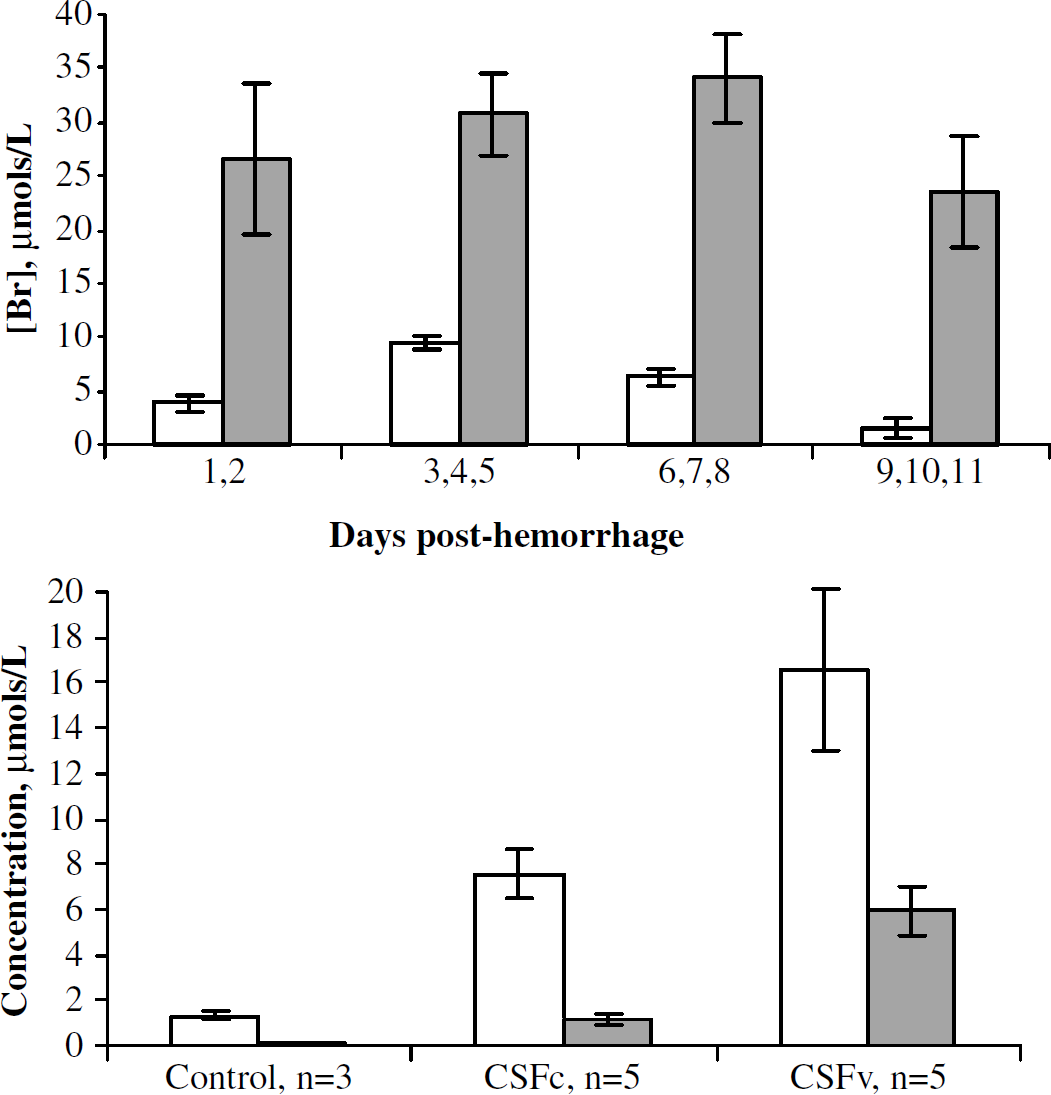
Bilirubin and oxidative environment in patient cerebrospinal fluid (CSF). (
The CSFc samples averaged 0.92±0.31 g/dL total protein and 0.39±0.06 g/dL hemoglobin, compared with 0.94±0.37 and 0.41±0.07 g/dL in the CSFv samples. Neither total protein concentration nor hemoglobin concentration was significantly different between CSFc (stimulated rate of O2 consumption to <0.4 μmol O2/min g) and CSFv groups (stimulated rate of O2 consumption to ≥0.4 μmol O2/min g). When bilirubin concentration is compared for patients with and without clinical vasospasm, there is significantly greater bilirubin in the vasospasm patients (5.6±0.93 versus 29.4±3.9 μmol/L).
Assessment of an Oxidizing Environment
Next, we assessed the level of oxidative stress in the CSF by measuring total oxidized lipids (LOOH), and MDA (a specific marker of lipid oxidation) (Figure 2). We found that total peroxidized lipids (LOOH) are significantly elevated in vasospasm patients compared with control (18.24±4.4 versus 7.5±0.53 μmol/L, respectively).
Heme Oxygenase Concentration
Figure 3 shows the HO-1 concentration in the CSF samples. It can be seen that HO-1 concentrations are significantly higher (P<0.05) in CSFv, compared with CSFc. Injured rat brain homogenate was used as a positive control for HO-1 production (Maines, 2000). There is a baseline level of HO-1 protein in the non-SAH CSF samples, but this is significantly (P<0.05) less than CSFc expression. It should be noted here that this antibody-dependent method for measurement determined concentration, not activity.
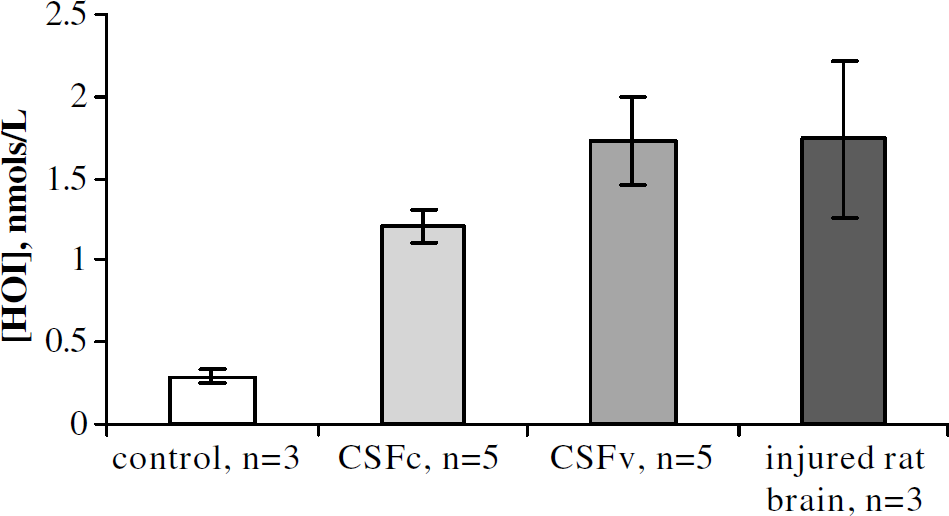
Heme oxygenase-I (HO-1) concentration in patient cerebrospinal fluid (CSF). Concentration of HO-1 is increased in CSF from vasospastic patients (▪, n = 6) compared with CSF from patients without vasospasm (▪, n = 6) and control CSF (□, n = 3). Rat brain, injured 24 h previous to homogenization, was used as a positive control (▪, n = 3).
Bilirubin Oxidation Products Results
We have found that the concentration of BOXes in CSFv of patients after SAH is significantly elevated compared with CSFc (1.33±0.07 versus 0.02±0.004 μmol/L, respectively). As seen in Table 2, the significantly higher BOXes concentration is concomitant with elevations in both bilirubin and oxidative stress.
DISCUSSION
Previously, we have reported that CSF from SAH patients stimulated vascular smooth muscle and that this method for assessing CSF correlated to clinical parameters (Cadoux-Hudson et al, 2001; Pyne et al, 2001). We have also suggested that bilirubin oxidation products, BOXes (Kranc et al, 2000) were found in the CSF of SAH patients and that these BOXes might contribute to vasospasm (Clark et al, 2002; Lyons et al, 2004). There are independent reports suggesting that oxidative stress (Cook, 1995; Macdonald and Weir, 1994; Watanabe et al, 1988; Weir, 1995) and bilirubin (Jia-Pei Miao and Jer-Fu Lee, 1989; Page et al, 1994) may correlate or contribute to vasospasm based on measurements in CSF (Page et al, 1994). But the thesis that oxidative stress leading to the oxidation of bilirubin (resulting in BOXes formation) as a contributing factor for vasospasm has been little studied. In this paper, we report that BOXes and the chemical conditions required to produce BOXes are present in the CSF of SAH patients with vasospasm and propose that these observations can, in part, account for the delay observed with SAH-induced vasospasm.
Our in vitro data show that, given the right conditions, bilirubin is indeed likely to be produced in the chemical environment present in the CSF after SAH. These data also complement the in vivo time course for bilirubin production we have recently reported (Morgan et al, 2004). These observations in the pig and rat encouraged us to further investigate the chemical conditions present in human CSF after SAH and to determine whether there was a difference in the chemical components in the CSF from patients with and without vasospasm after SAH. To address this question, we performed a retrospective analysis of the CSF of patients who had had SAH. Because our thesis is that BOXes cause or contribute to vasospasm, we quantified BOXes as well as the major chemical constituents required for the formation of BOXes. These constituents are: heme oxygenase, bilirubin, and oxidative stress (as determined by measuring MDA and LOOH). Having determined that the concentration of bilirubin was increased in CSFv, we wanted to determine whether HO-1 concentration was increased in the same CSF samples. Increased HO-1 activity may explain the increased bilirubin production, despite the same availability of substrate (as assessed by the similar concentrations of total protein and hemoglobin measured in the CSF).
We reported above that the increase in bilirubin concentration has a time component (Figures 1 and 2), and that there is a correlation between concentration of bilirubin and the occurrence of vasospasm as assessed in vitro (Cadoux-Hudson et al, 2001; Pyne et al, 2001) (Table 2). This is consistent with our previous work where we suggested that the hemorrhagic CSF from vasospasm patients correlated to clinical vasospasm when assessed in vitro (Cadoux-Hudson et al, 2001; Pyne et al, 2001). These observations also suggest that vasospasm is likely to be caused by vasoactivity present in the hemorrhagic CSF.
We found that there is a striking correlation between elevated BOXes, bilirubin, and oxidative stress. It is important to note that the total protein concentration was not significantly different between CSFc and CSFv. This, we believe, indicates that the volume of blood is not exclusively responsible for vasospasm, because the protein concentration would be greater with increased hemorrhage volumes. Thus, the incidence of vasospasm is a reflection of the chemical environment present in the CSF. Moreover, the increase in bilirubin is concomitant with increased HO-1. As we have recently shown in vivo using a pig model of SAH (Morgan et al, 2004), the production of bilirubin has a lag period that is consistent with the delay observed with vasospasm after SAH. Thus, we believe that the delay seen with vasospasm is because of the time it takes for HO-1 to be induced and for the bilirubin levels to be elevated. Next, when the bilirubin is elevated there must also be an increase in oxidative stress to lead to the formation of BOXes. Therefore, our thesis for BOXes contributing to vasospasm after SAH accounts for the lag period to vasospasm. It can be seen from Table 2 that patient 11 had high bilirubin, but low MDA, BOXes, and JO2 values. We were able to obtain limited information regarding this patient's clinical progress, and found that he had no neurologic deficit (GCS = 15 at the time of CSF collection). Again, we hypothesize that an oxidizing environment and high bilirubin concentrations are required for BOXes formation, and therefore, for vasospasm.
A recent paper reported that bilirubin was, in fact, higher in nonvasospastic-patient CSF than in CSF from patients with vasospasm (Suzuki et al, 2003). This may, at first, appear contrary to the data we report here. The authors do, however, list a number of reasons for their observations, which may explain this discrepancy, and, in fact, support our hypotheses. The authors do not report the initial hemoglobin concentrations in their patient samples, which are an important assessment of substrate availability, and they do not measure HO-1 levels or activity. Therefore, they postulate, that clearance of heme could be either by metabolism intracranially by HO-1, which would result in elevated bilirubin, or by increased clearance of CSF, which would not yield increased bilirubin levels. Thus, if the vasospastic patients had increased CSF clearance, this would explain why their bilirubin levels were lower. Although we believe that increased bilirubin is a prerequisite for BOXes production, and therefore an important marker for vasospasm after SAH (Morgan et al, 2004), we also postulate that an oxidizing environment is just as important.
We believe that these data suggest that the after conditions must occur for vasospasm to result. First, the hemorrhage must result in the induction of HO-1. Then HO-1 will increase the concentration of bilirubin. When bilirubin levels are elevated, it must be exposed to a potent oxidizing environment that causes the production of significant amounts of BOXes. The BOXes will cause or contribute to the vasospasm. One may ask, however, that if oxidative stress is requisite in this scenario, why have the antioxidant therapies not been more successful? We believe that the potent oxidizing environment that would be needed to produce BOXes may be occurring in, for example, macrophages. As well as contributing towards the oxidizing environment by releasing ROS into the extracellular space (Mori et al, 2001), the macrophages may have phagocytosed hemorrhagic debris, and degraded and oxidized the bilirubin. We posit, therefore, that the antioxidant therapies previously tested were not effective in preventing oxidation within the phagocytosed vesicles inside macrophages. Hence, the antioxidant therapy would not have been effective. In this scenario, BOXes that may have been formed inside the peroxidizing vesicles of the macrophages and other peroxide-utilizing, phagocytosing cells may exit the cells as a result of those cells dying (because of the toxicity of BOXes) or, because of the small, hydrophobic nature of BOXes, simply exiting through the cell membrane in the extracellular space. BOXes production inside cells such as macrophages is not mutually exclusive to BOXes production extracellularly. However, the probability of BOXes production within such an environment is consistent with the difficulty in treating vasospasm.
Conclusion
Here we present data that the in vitro correlates of vasospasm are associated with the chemical conditions that would be needed for production of BOXes. The conditions that are required to form BOXes are present in CSF from SAH patients with vasospasm, but not in the CSF from SAH patients without vasospasm. Increased bilirubin and increased oxidative stress are both associated with indicators of vasospasm. Thus, it remains likely that BOXes being produced in this chemical environment in the CSF might cause or contribute to vasospasm after SAH. We conclude that vasospasm may be more likely in patients with elevated BOXes.
Footnotes
Acknowledgements
The authors gratefully acknowledge Robert Hill for his assistance with this project.