
Abstract
Cerebral vasospasm after subarachnoid hemorrhage (SAH) is because of smooth muscle contraction, although the mechanism of this contraction remains unresolved. Membrane potential controls the contractile state of arterial myocytes by gating voltage-sensitive calcium channels and is in turn primarily controlled by K+ ion conductance through several classes of K+ channels. We characterized the role of inwardly rectifying K+ (KIR) channels in vasospasm. Vasospasm was created in dogs using the double-hemorrhage model of SAH. Electrophysiological, real-time quantitative reverse-transcriptase polymerase chain reaction, Western blotting, immunohistochemistry, and isometric tension techniques were used to characterize the expression and function of KIR channels in normal and vasospastic basilar artery 7 days after SAH. Subarachnoid hemorrhage resulted in severe vasospasm of the basilar artery (mean of 61% ± 5% reduction in diameter). Membrane potential of pressurized vasospastic basilar arteries was significantly depolarized compared with control arteries (+-46 ± 1.4 mV versus −29.8 ± 1.8 mV, respectively, P < 0.01). In whole-cell patch clamp of enzymatically isolated basilar artery myocytes, average KIR conductance was 1.6 ± 0.5 pS/pF in control cells and 9.2 ± 2.2 pS/pF in SAH cells (P = 0.007). Blocking KiR channels with BaCI2 (0.1 mmol/L) resulted in significantly greater membrane depolarization in vasospastic compared with normal myocytes. Expression of KIR 2.1 messenger ribonucleic acid (mRNA) was increased after SAH. Western blotting and immunohistochemistry also showed increased expression of KIR protein in vasospastic smooth muscle. Blockage of KIR channels in arteries under isometric tension produced a greater contraction in SAH than in control arteries. These results document increased expression of KIR 2.1 mRNA and protein during vasospasm after experimental SAH and suggest that this increase is a functionally significant adaptive response acting to reduce vasospasm.
Introduction
It has been reported that smooth muscle cells are depolarized during vasospasm after subarachnoid hemorrhage (SAH) (Harder et al, 1987; Zuccarello et al, 1996). This might be important because the membrane potential (Em) of smooth muscle cells, which is modulated primarily by potassium (K+) conductance, is an important determinant of vascular tone because of its role in regulating conductance of L-type calcium (Ca2+) channels (Nelson and Quayle, 1995). Changes in Em of only a few millivolts can cause large changes in vascular diameter (Faraci and Heistad, 1998). In pressurized arteries, the resting Em of smooth muscle cells is approximately 30 to 45 mV positive to the equilibrium potential for K+ (EK), which means that the opening or closing of K+ channels will contribute substantially to vasorelaxation or vasoconstriction, respectively (Standen and Quayle, 1998). Hyperpolarization of the smooth muscle cell membrane caused by opening of K+ channels will close voltage-gated Ca2+ channels, reduce intracellular Ca2+, and cause vascular relaxation. Cerebral vascular smooth muscle cells have four classes of K+ channels: voltage-dependent (Kv), large conductance Ca2+ -activated (BK), adenosine triphosphate sensitive (KATP), and inward rectifier (KIR) K+ channels (Nelson and Quayle, 1995).
We showed that vasospasm 7 days after SAH in dogs is associated with increased KIR 2.1 (the main smooth muscle KIR subtype; Zaritsky et al, 2000), messenger ribonucleic acid (mRNA) and protein in the vasospastic arteries (Aihara et al, 2004). In that previously published data, the endothelium was not removed from the arteries and thus the cellular localization of the increase in KIR remained unclear. Furthermore, no data were obtained to determine the functional significance of the alteration. These experiments determined the functional role and cellular localization of KIR channels in the basilar artery after SAH.
Materials and methods
Animal Model
Female mongrel dogs weighing 15 to 25 kg were randomly assigned to provide normal, control basilar artery (n = 13) or to undergo SAH created by injection of autologous blood, 0.5 mL/kg, into the cisterna magna on the day of baseline angiography (day 0) and 2 days later (day 2, n = 12). Methods for angiography and SAH have been described (Macdonald et al, 1995). Briefly, dogs were sedated by intravenous injection of sodium pentothal (15 mg/kg) and then intubated and ventilated on oxygen and 1% to 2% isoflurane. Baseline cerebral angiography was performed on day 0. Control dogs then were euthanized. Control dogs did not undergo injection of saline or some other control fluid into the cisterna magna because this does not cause vasospasm and there are no published data to suggest that such injections cause any change in cerebral arterial function 7 days later (Macdonald et al, 2004). In SAH dogs, 0.3 mL/kg of cerebrospinal fluid was removed from the cisterna magna and 0.5 mL/kg of fresh autologous arterial nonheparinized blood was injected. The injection was repeated on day 2. Subarachnoid hemorrhage animals underwent angiography on day 7 followed by euthanasia. Procedures on animals were approved by the Institutional Animal Care and Use Committee.
Tissue Harvest
Animals were perfused under general anesthesia with 8L ice-cold phosphate-buffered saline (PBS, pH 7.4) at an intraluminal pressure of 25 to 30 mm Hg. This volume was found to be necessary to remove completely intraluminal blood from the cerebral arteries. The brain was excised and placed in 4°C PBS. The basilar artery was removed and cleaned of surrounding arachnoid tissue. Segments for measurement of mRNA were denuded of endothelium by passing a catheter guidewire of an appropriate size through the arterial lumen followed by flushing with 4°C PBS and then immediate freezing in liquid N2. Removal of adjacent arachnoid and brain tissue and of endothelium was confirmed by light microscopy (lack of immunohistochemical staining for glial fibrillary acidic protein and the endothelial cell marker, CD31, data not shown). The absence of endothelium also was confirmed by lack of endothelium-dependent contraction of partially contracted arterial segments to the Ca2+ ionophore, A23187 (data not shown). Segments for electrophysiological studies were placed in dissection solution (in mmol/L: NaCl 130, KCl 5, MgCl2 1.3, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) 10, glucose 5; along with 100 U/mL penicillin and 0.1 mg/mL streptomycin) and kept at 4°C until enzymatically dissociated. Segments for tension studies were placed in Krebs–Henseleit buffer (in mmol/L: NaCl 123, KCl 5.9, CaCl2 2.5, MgCl2 1.2, NaHC03 25, H2SO4 1.2, NaH2PO4 1.2, and glucose 11), bubbled with 95% O2 and 5% CO2 at 4°C. All SAH segments used for experiments showed angiographic vasospasm.
Pressurized Artery Membrane Potential Recordings
We first needed to ensure that the cell isolation procedure used below for single-cell electrophysiology did not damage the smooth muscle cells and, specifically, preferentially damage the SAH cells. This was performed in several ways. First, to confirm that smooth muscle cells in vasospastic SAH arteries were depolarized as reported previously (Harder et al, 1987), intracellular microelectrode recordings were made of smooth muscle membrane potential in intact, pressurized dog basilar arteries from control dogs and dogs 7 days after SAH. The side branches of short segments of basilar arteries were carefully tied with threads of silk under an operating microscope. The endothelium was preserved. The vessels were mounted on glass pipettes and tied with sutures and pressurized to the pressure that would be expected in the basilar artery in vivo (80 mm Hg). Glass microelectrodes were pulled on a Sutter P-97 programmable puller (Sutter Instruments, Novato, CA, USA) and filled with KCl, 2 mol/L. Tip resistances were 20 to 50 MΩ and tip potentials < 5 mV. Intracellular penetration was obtained under a microscope by advancing the glass electrode into the surface of the vessel with the aid of a hydraulic micromanipulator (MW-3, Narishige International USA, East Meadow, NY, USA). Membrane potentials were recorded on an Axopatch-1D preamplifier (Axon Instruments Inc., Foster City, CA, USA). Signals were filtered with a 2 kHz low-pass filter, digitized at 12 bits using a DigiData 1200 interface controlled by pClamp 6.0.4. The current-clamp mode with zero input was used to monitor membrane voltage. The criteria for a successful impalement include a sharp decrease in voltage from baseline on entry of the microelectrode into a cell, stable membrane potential for at least 2 mins, no change in tip resistance, and return to zero potential on removal of the electrode (Knot and Nelson, 1998). These measurements were made at room temperature (22°C to 24°C) and body temperature (37°C). Bath and intraluminal solutions were Krebs–Henseleit solution containing (in mmol/L): NaCl 113.7, KCl 4.7, NaHCO3 25, MgSO 4 1.2, KH2PO4 1.2, glucose 10, andCaCl2 2.5 bubbled with a mixture of 95% O2 and 5% CO2 (Jiang et al, 2001).
Smooth Muscle Cell Isolation
The presence of functional KIR channels in large cerebral arteries from dogs and whether they were altered after SAH could be determined best by electrophysiological recordings from isolated smooth muscle cells. To that end, basilar artery smooth muscle cells were isolated by cutting the artery into small pieces and incubating the pieces for 30 mins in enzymes (500 U/mL collagenase type IV, 50 U/mL elastase, 100 U/mL DNase I, and 1 mg/mL trypsin inhibitor (Worthington, Lakewood, NJ, USA) (Subramanian et al, 1991). Smooth muscle cells were then separated by gentle trituration while suspended in a buffer solution (dissection solution + 0.2% bovine serum albumin), stored at 4°C, and used within 12 h of isolation. Positive staining for α-actin and contraction of isolated cells in response to agonists (KCl, 60 mmol/L or serotonin, 1 μmol/L) confirmed that this method yielded smooth muscle cells.
Electrophysiology
Smooth muscle cells were allowed to adhere to a glass coverslip mounted at the bottom of the recording chamber before being patched with borosilicate glass pipettes with resistances of 2.0 to 3.5 mΩ. Once sealed, the membrane was ruptured to achieve the whole-cell configuration. Cells achieving stable access resistances under 10 mΩ were used. Current responses to hyperpolarizing voltage ramps (120 to + 20 mV) were recorded using an EPC9 amplifier with Pulse software (HEKA Electronik, Lambrecht/Phalz, GmBH, Germany) or an Axopatch-1D amplifier with attached DigiData 1200 using pClamp 6.0.4 software (Axon Instruments Inc., Foster City, CA, USA). The internal (patch pipette) solution consisted of (in mmol/L): KCl 142.6, KOH 2.4, MgCl2 2.5, BAPTA (1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid) 10, HEPES 10, pH 7.2. BAPTA was included in the pipette solution to buffer intracellular Ca2+, a procedure we have shown completely blocks BK currents (Jahromi et al, 2005). The bath solution contained (in mmol/L): NaCl 100, KCl 50, MgCl2 1, CaCl2 2, HEPES 10, glucose 10, pH 7.4. BaCl2, 0.1 mmol/L, which blocks KIR at this concentration, was added to permit the derivation of subtraction currents representing KIR current. Ba2+, however, has been reported to block KATP channels (Quayle et al, 1997). A contribution of these channels to recorded currents was excluded by showing a lack of effect of glibenclamide, 100 μmol/L. Extracellular K+ ([K+]ex) was 50 mmol/L, giving an EK of −27 mV, which was observed in current traces in these experiments. Two measures were undertaken to exclude the possibility that currents were being generated by chloride. First, additional data obtained in control and SAH cells with [K+]ex of 150 mmol/L showed currents with a reversal potential of 0 mV, which is the predicted EK in symmetric K+. Second, preliminary experiments showed no effect of the chloride channel blocker, 4,4'-diisothiocyanatostilbene-2,2'-disulfonic acid, 100 μmol/L, on recorded currents (Nelson et al, 1997).
The nystatin-perforated patch technique was used to measure Em in the current-clamp mode (I = 0). The internal solution contained (in mmol/L): KCl 145, NaCl 5, MgCl2 1, HEPES 10; nystatin 200 μg/mL; pH 7.2 with KOH. Nystatin stock was made fresh every 2 h by dissolving nystatin powder into dimethyl sulfoxide (DMSO, lmg nystatin in 5 μL DMSO). Cells achieving stabilize access resistances between 8 and 15 mΩ were used for experiments. The bath solution contained (in mmol/L): NaCl 100, KCl 5, MgCl2 1, CaCl2 2, HEPES 10, glucose 10, pH 7.4. Cells with stable Em after switching into current-clamp mode were used for experiments. The Em was recorded as the cell was perfused with the KIR blocker Ba2+, 0.1 mmol/L. Comparison of Em values before and after the addition of Ba2+ permit the elucidation of the degree to which KIR channels contributed to the Em. These experiments were performed at room temperature (22°C to 24°C).
Molecular Biology
Our prior experiments showed KIR mRNA in whole basilar artery from dogs (Aihara et al, 2004). Endothelial cells may contain these channels so as to determine whether KIR mRNA was present in smooth muscle cells; total RNA was extracted from individual basilar arteries from which the endothelium had been removed as described above, using TRIzol (Gibco BRL, Gaithersburg, MD, USA). RNasefree deoxyribonuclease (DNAse, Promega, Madison, WI, USA) was used to eliminate contamination with genomic DNA. Ribonucleic acid reverse transcription (RT) was catalyzed by SUPER SCRIPT First-Strand Synthesis System (Invitrogen Life Technologies, Carlsbad, CA, USA). Ribonucleic acid (5 μg) was used for RT in a final volume of 10 μL. The reaction mixture consisted of 50 ng of random hexamers per 1 μg of total RNA. To increase the sensitivity of the polymerase chain reaction (PCR) from first-strand cDNA, we used RNase H to remove the RNA template from cDNA after first-strand synthesis.
Dog sequences of mRNAs were obtained by searching the NCBI database (http://www.ncbi.nlm.nih.gov/Entrez) for KIR 2.1 (kidney and atrial sources) and β-actin (Aihara et al, 2004). Primers, probes, and length of PCR products were chosen to satisfy the requirements as specified in Primer Express (version 1.5) software (Applied Biosystems, Foster City, CA, USA) for quantitative real-time PCR. 18S ribosomal RNA (18S rRNA; TaqMan Ribosomal RNA Control Reagent, Applied Biosystems) was used as an endogenous control. Quantitative real-time PCR was performed with real-time TaqMan technology using a Sequence Detection System model 7700 (Applied Biosystems) (Aihara et al, 2004).
Protein was extracted along with mRNA using TRIzol as described above. Extracted protein (150 to 250 μg per basilar artery) was quantified by spectrophotometry (at 562 nm) with Micro BCA Protein Assay Reagents (Pierce Biotechnology, Rockford, IL, USA). Equal amounts of protein from each arterial sample were separated on 12% sodium dodecyl sulfate gels (Bio-Rad Laboratories, Hercules, CA, USA) and transferred onto nitrocellulose membranes. The membranes were blocked at room temperature for 2 h in 5% milk in Tris-buffered saline with 0.1% Tween-20, followed by incubation with primary antibody (rabbit anti-human KIR 2.1 (1:400 dilution), Alomone Laboratories, Jerusalem, Israel) at 4°C overnight. After washing, the membranes were incubated for 1 h with goat anti-rabbit horseradish peroxidase-conjugated antibody (1:5,000 dilution, Promega). The enhanced chemiluminescence Plus Western blotting detection system (Amersham Bioscience, Piscataway, NJ, USA) was used to visualize the KIR 2.1-specific band on radiographic film. Total protein on each lane was shown by staining the transferred membrane with GelCode Blue Stain Reagent (Pierce). Images of bands on film and stained membranes were acquired and analyzed using FluorChem8800 (Alpha Innotech, San Leandro, CA, USA) with KIR band density normalized to the total amount of protein present in the lane.
Immunohistochemistry
Confirmation that KIR protein was present in basilar artery smooth muscle cells was obtained by confocal imaging of enzymatically dissociated smooth muscle cells. These cells were placed on poly-
Isometric Tension Recordings
The second method for determining function of KIR channels is from measurements of segments of basilar artery either in vitro or in vivo (Chrissobolis et al, 2000). For these studies, arterial branches and loose connective tissue were removed from segments of basilar artery from which the endothelium had been removed. The artery was cut into rings 3 mm long and suspended on stainless-steel hooks in 5 mL water-jacketed tissue baths filled with Krebs-Henseleit buffer at 37°C. One stainless-steel hook attached the ring to the bottom of an organ bath chamber and the other was attached to a Grass FT-03 isometric force transducer (Grass Telefactor, West Warwick, RI, USA). Signals were amplified using TBM-4M differential amplifiers (WPI, Sarasota, FL, USA) and digitized at 16-bits resolution (PCI-6034E 8-channel analog to digital PCI board, National Instruments, Austin, TX, USA). Data were sampled at 2 Hz and analyzed using custom programs (D Ryan, University of Chicago, Chicago, IL, USA) written in Igor 4.0 and NiDAQ Tools 1.2 (Wavemetrics, Lake Oswego, OR, USA) running on a personal computer. All contractions were measured relative to their preceding baseline and were expressed as a percentage of the maximal KC1-induced contraction in each ring. The bath solution was changed every 15 to 20 mins. Arterial rings were stretched until a stable tension of 1.5 g was achieved. Contractile responses to KCl (60 mmol/L) were recorded and rings with stable contractions > 0.5 g were used. The absence of endothelium was documented by lack of relaxation to adenosine triphosphate (1 μmol/L) of rings precontracted with prostaglandin F2α, 1 to 3 μmol/L. Rings were then stretched to increasing tensions (increments of 0.5 g up to 3.0 g) and responses to KCl (60 mmol/L) assessed again. Experiments were performed on arteries stretched to the point where the response to 60 mmol/L KCl was the greatest and were begun after the segments had equilibrated in a solution of normal [K+]ext (5.9 mmol/L) for 60 mins. Responses to KCl then were assessed in the presence and absence of BaCl2, 0.1 mmol/L.
Data Analysis
Vasospasm was assessed by comparing diameters of basilar arteries on angiograms from day 0 and day 7 at five predetermined points by two masked observers using an optical micrometer. Electrophysiology data analysis was performed using Igor Pro 4.07 (WaveMetrics Inc., Lake Oswego, OR, USA). Conductance (pS) was determined by fitting a line to the inward current between −100 and −40 mV and calculating the slope of the fit. Conductance data (pS) were normalized to cell capacitance (pF). Student's t-test or paired t-tests (SigmaStat, SPSS, Chicago, IL, USA) were used to determine significance, which was set at P < 0.05. All values are means ± standard error.
Results
Angiographic Vasospasm, Pressurized Artery Membrane Potential
Subarachnoid hemorrhage resulted in severe vasospasm of the basilar artery (mean of 61% + 5% reduction in diameter, P < 0.01, Student's t-test, Figure 1). Smooth muscle cell membrane potential was measured in arteries pressurized to the pressure they were estimated to be subjected to in vivo. Membrane potential was significantly depolarized in SAH arteries compared with control arteries at room temperature (–31.1 ± 1.5 mV (n = 97 cells from four dogs) versus −27.0 ± 1.2 mV (n = 72 cells from five dogs), respectively, P < 0.05, Student's t-test) and at 37°C (–46 ± 1.4 mV (n = 78 cells from four dogs) versus −29.8 ± 1.8 mV (n = 31 cells from five dogs), respectively, P < 0.01, Student's t-test, Figure 2).
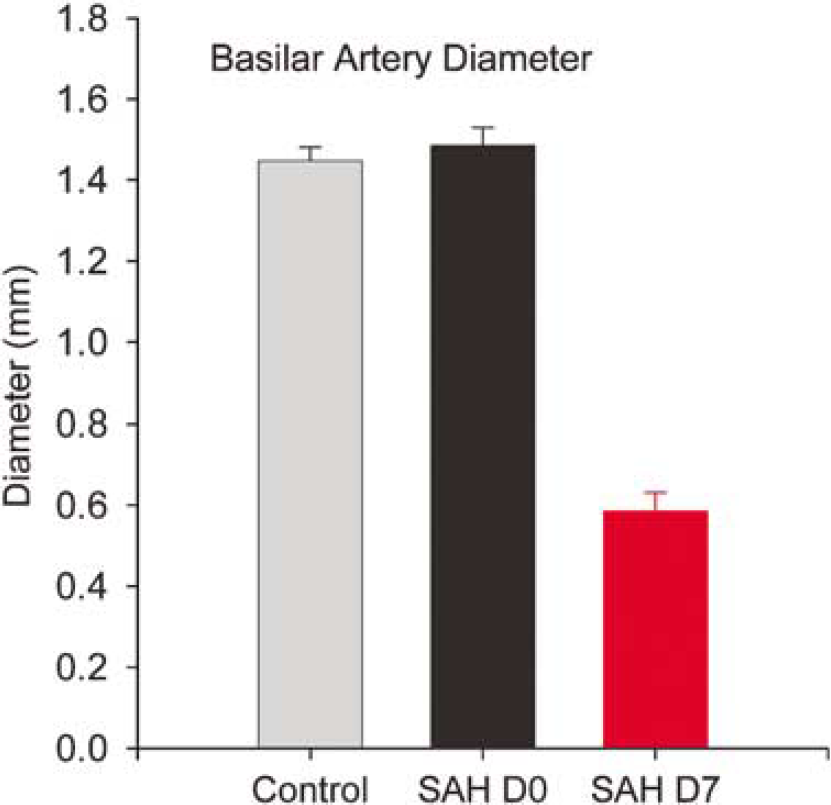
Measurements of basilar arterial diameter were taken from angiograms at day 0 and day 7. There was no significant difference in the diameter of the basilar artery of control and subarachnoid hemorrhage (SAH) dogs on day 0 (n = 13 control, n = 12 SAH). Subarachnoid hemorrhage caused severe vasospasm by day 7, reducing basilar artery diameter by 61% ± 5% (P < 0.01, paired t-test).
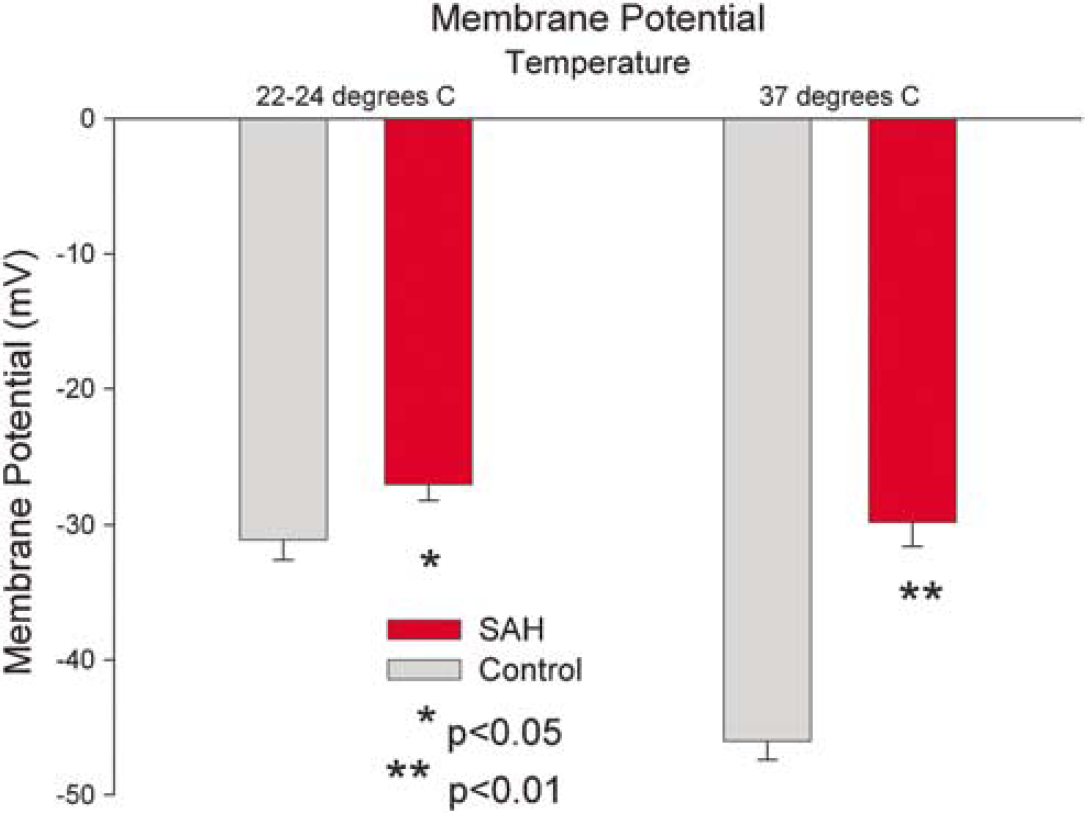
Membrane potential of smooth muscle cells of pressurized basilar artery from control and subarachnoid hemorrhage (SAH) dogs at 22 to 24°C and at 37°C. Membrane potential was significantly depolarized in SAH arteries compared with control arteries at room temperature (−31.1 ± 1.5 mV (n = 97 cells from four dogs) versus −27.0 ± 1.2 mV (n = 72 cells from five dogs), respectively, P < 0.05, Student's t-test) and at 37°C (−46 ± 1.4 mV (n = 78 cells from four dogs) versus −29.8 ± 1.8 mV (n = 31 cells from five dogs), respectively, P < 0.01, Student's t-test).
Whole-Cell Inwardly Rectifying K+ Currents are Increased After Subarachnoid Hemorrhage
Patch-clamp experiments were performed on isolated vascular smooth muscle cells originating from the basilar artery of six control and four SAH dogs. Average currents were obtained from four voltage ramps per cell. Average untreated and Ba2+ -treated currents for representative control and SAH cells show inward rectification characteristic of KIR in the SAH cells, which was not as evident in control cells (Figure 3). Currents obtained by subtracting currents obtained before and after application of Ba2+, 0.1 mmol/L, represent the KIR component of the total current and were larger in SAH than in control cells (Figure 3). In control cells, subtracted traces displayed little current at potentials negative to EK. In SAH cells, at potentials negative to EK, substantial Ba2+ -sensitive inward currents were observed. A significant increase in KIR conductance (pS/pF) was observed in SAH compared with control cells (Figure 4). Control cells (n = 30 cells from six dogs) showed an average conductance of 1.6 ± 0.5 pS/pF and SAH cells (n = 21 from four dogs) had an average conductance of 9.2 ± 2.2 pS/pF (P = 0.007, Student's t-test). These data are consistent with an increase in functional KIR current in vascular smooth muscle cells after SAH.
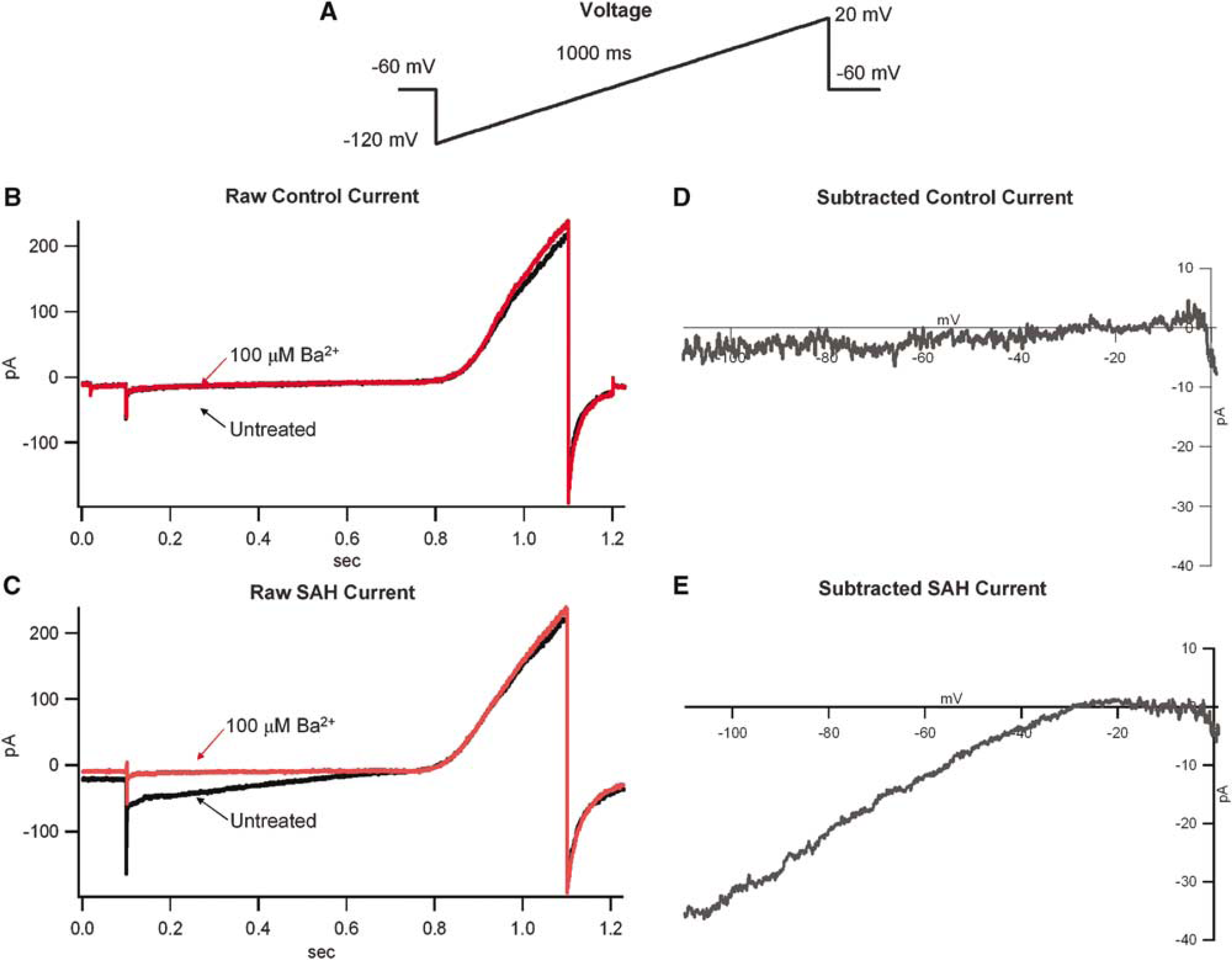
Inward-rectifying currents in isolated basilar artery myocytes. The external concentration of K+ was 50 mmol/L giving a Keq of −27 mV. (
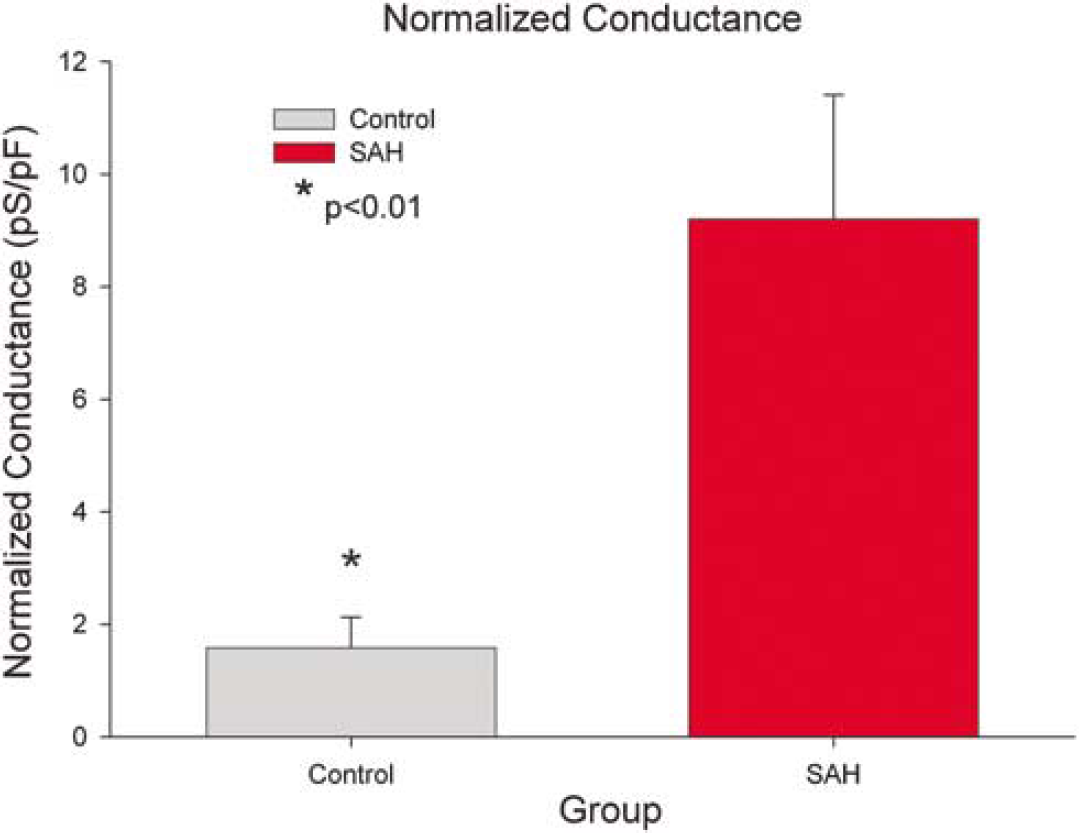
Comparison of inwardly rectifying K+ (KIR) conductance in control (n = 21 cells from six dogs) versus subarachnoid hemorrhage (SAH) cells (n = 30 cells from four dogs). A significant increase in inwardly rectifying current was observed in isolated basilar artery myocytes after SAH (from 1.6 ±0.5 pS/pF in control to 9.2 ± 2.2 pS/pF in SAH, P < 0.01).
The Contribution of Inwardly Rectifying K+ Channels to Resting Membrane Potential is Increased After Subarachnoid Hemorrhage
The contribution of KIR current to the baseline Em was assessed by determining the effect of blocking KIR channels in myocytes current clamped in the nystatin-perforated patch configuration. Baseline Em was recorded for 20 to 30 secs and then 0.1 mmol/L Ba2+ was added to the bath solution. Depolarization after application of Ba2+ was larger in myocytes from vasospastic arteries than it was for control myocytes (Figure 5). The depolarization in response to Ba2+ was 3.21 ± 0.46 mV for control cells (n = 20 cells from five dogs) and 5.07 ± 0.63 mV for SAH cells (n = 27 cells from five dogs, P = 0.03, Student's t-test). Another technique used to unmask KIR currents is to apply blockers for other K+ channels (4-aminopyridine (4-AP) to block Kv channels and paxilline to block BK channels) (Sanchez and McManus, 1996; Perillan et al, 1999). In our preparation, we observed a greater depolarization to 0.1 mmol/L Ba2+ after the application of 1 mmol/L 4-AP and 1 μmol/L paxilline in myocytes from vasospastic arteries compared with control cells (data not shown). These data indicate that the KIR channel plays a greater role in maintaining Em after SAH.
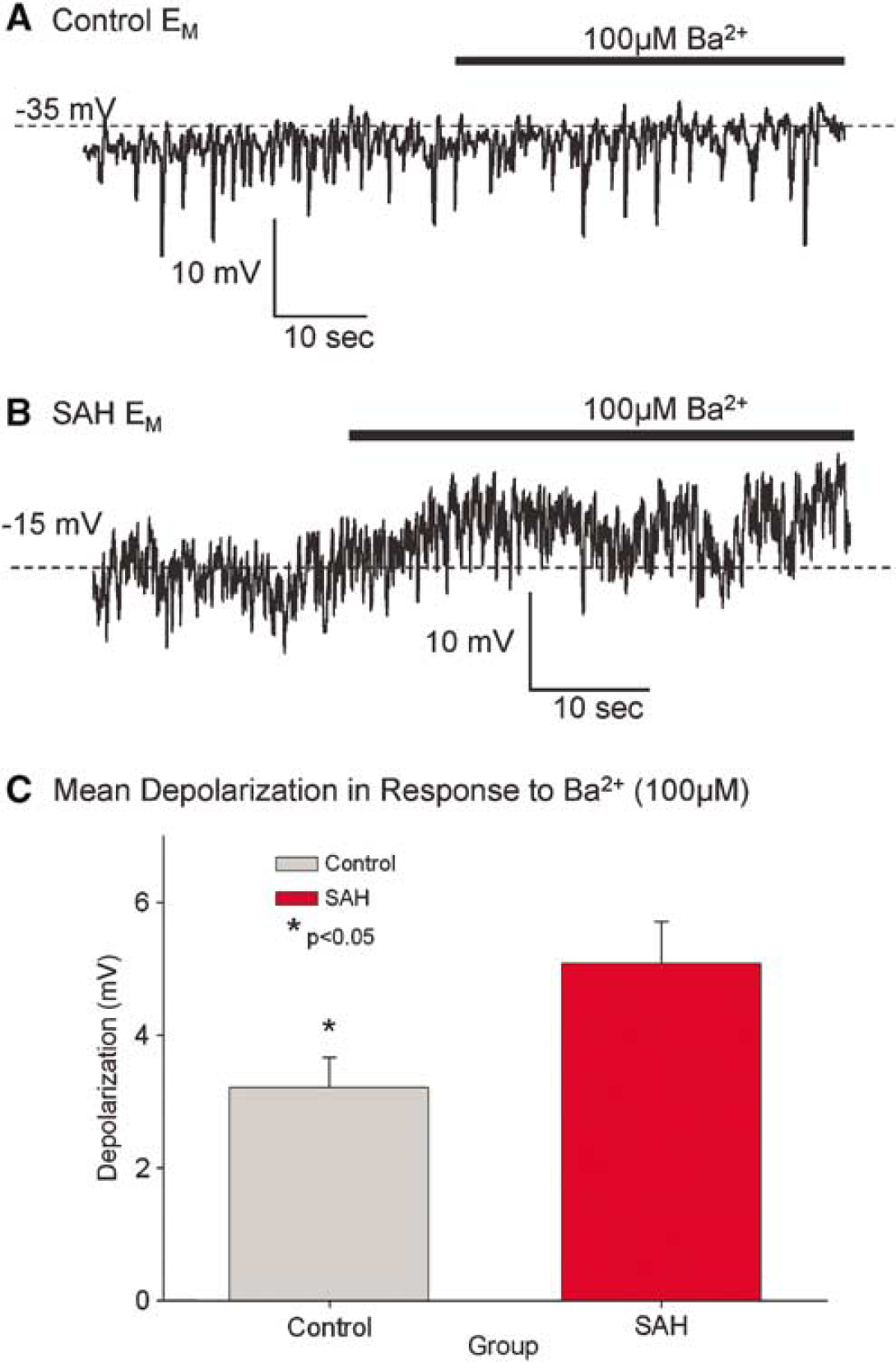
Recordings of membrane potential (Em) from isolated basilar artery myocytes. Cells were allowed to reach a stable Em and were then washed with 0.1 mmol/L Ba2+. (
Expression of Inwardly Rectifying K+ 2.1 Messenger Ribonucleic Acid and Protein is Increased After Subarachnoid Hemorrhage
The level of target gene (KIR 2.1 and β-actin) RNA was expressed as a ratio of the threshold cycle (CT) value for the target gene compared with that of 18S rRNA. These normalized ratios were then compared between the control and SAH groups. The normalization was performed on 18S rRNA because it was found that β-actin mRNA was significantly increased after SAH when compared with 18S rRNA (data not shown) (Ohkuma et al, 2001). The amount of KIR 2.1 RNA was significantly increased after SAH compared with control (300% increase relative to 18S rRNA, P < 0.001, Figure 6).
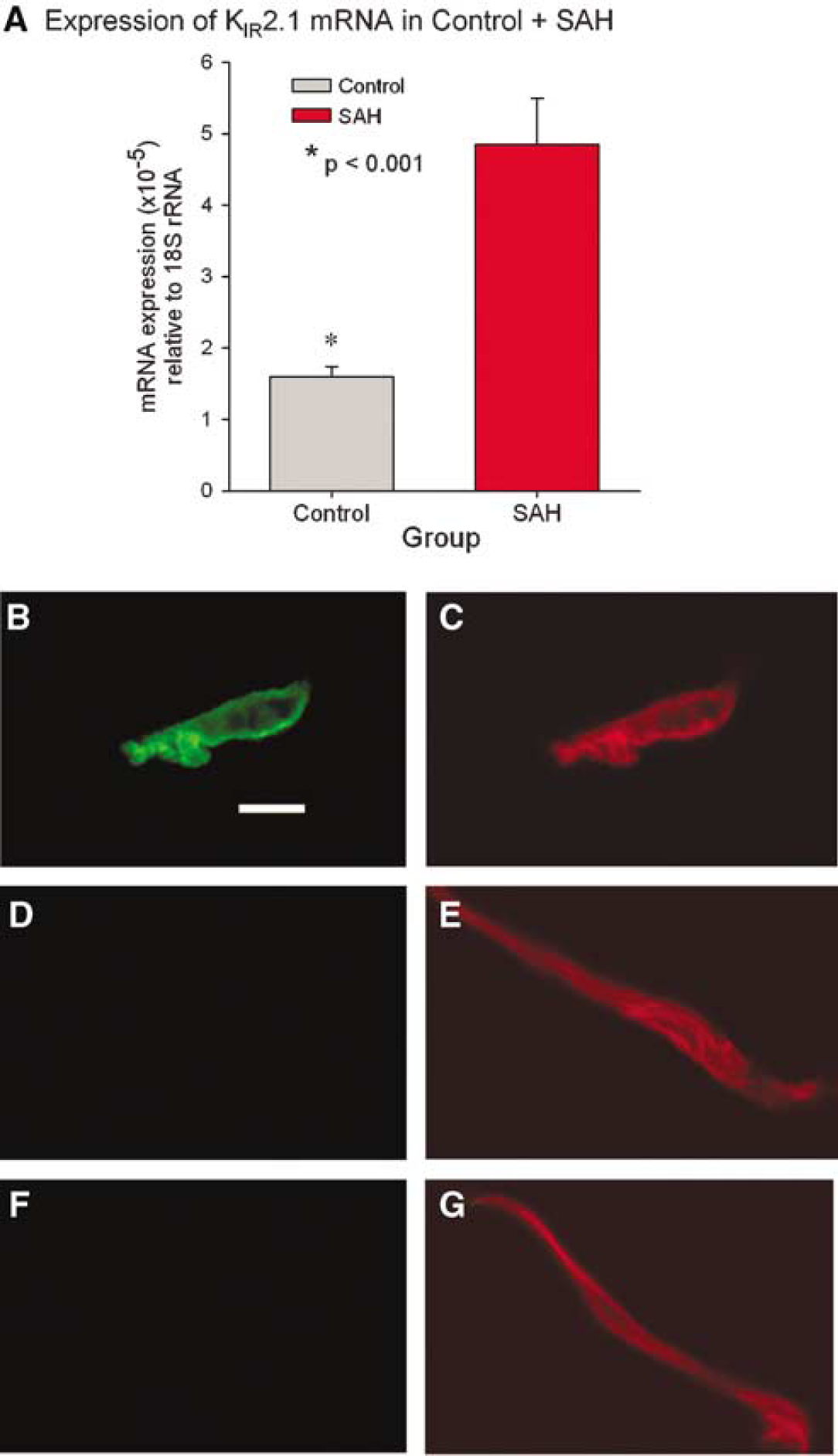
(
Immunohistochemistry showed an obvious increase in immunoreactivity for KIR with staining graded as marked in all SAH myocytes and absent/minimal or moderate in all control arteries (n = 10 per group, Figure 6). Western blotting of protein extracted from endothelium-denuded basilar arteries confirmed that there was a significant increase in KIR protein in basilar arteries after SAH (2365 ± 342 versus 1234 ± 253, n = 4 per group, P = 0.04, Student's t-test, Figure 7).
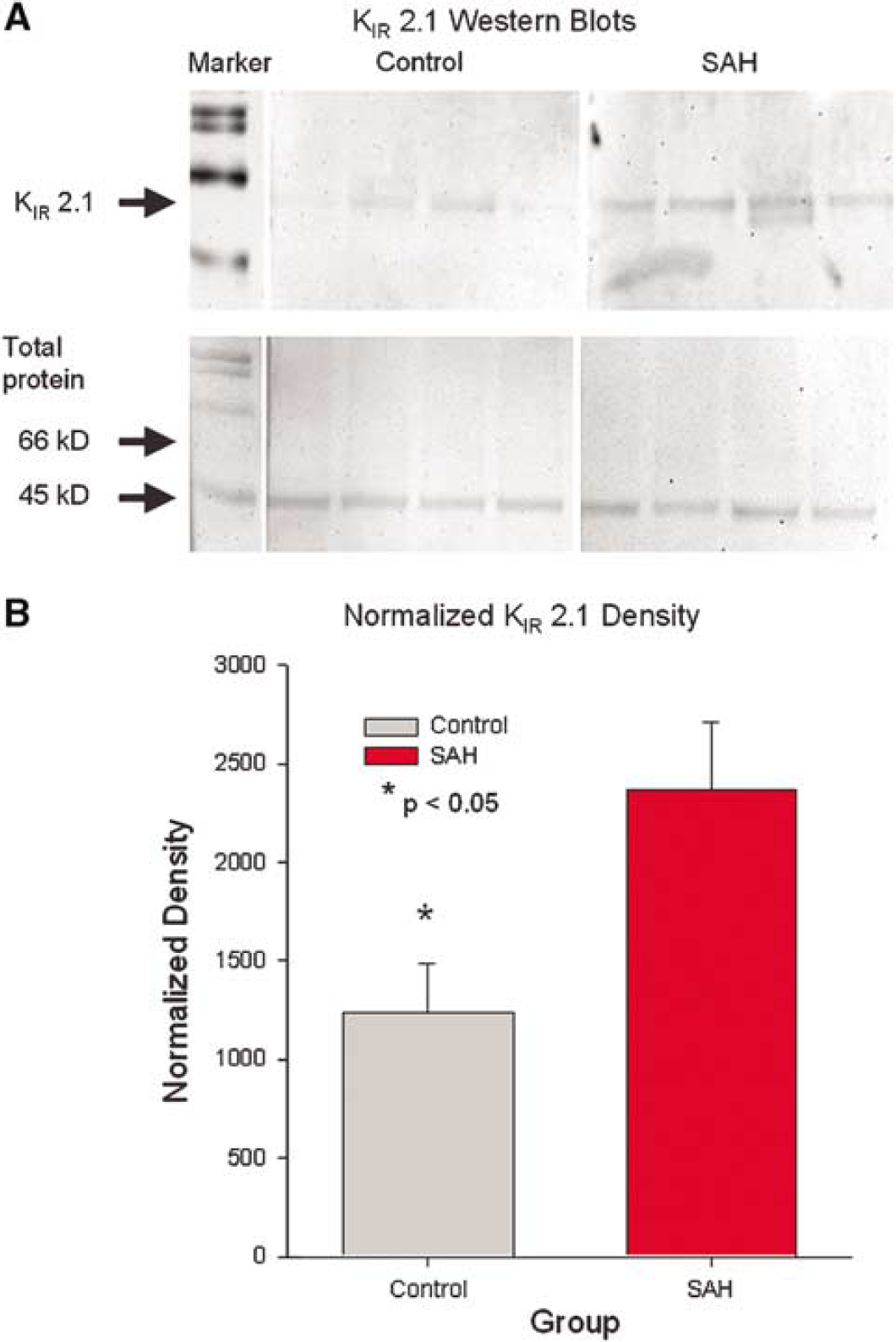
Western blot analysis of inwardly rectifying K+ (KIR) 2.1 in the dog basilar artery. (
Ba2+ Causes a Greater Contraction in Vasospastic Basilar Artery
Isometric tension recordings in vitro of rings of control and SAH basilar arteries in response to cumulative additions of KCl showed that there was significantly less contraction of SAH arteries compared with controls (3.65 ± 0.20 g for control and 2.19 ±0.17 g for SAH rings (n = 6 to 8 rings from five dogs per group, P < 0.001, Student's t-test). In addition, there was relaxation to KCl at low concentrations (5 to 10 mmol/L KCl) in the ring from the dog with SAH but not in the control dog, a response characteristically because of the activation of KIR channels (Figure 8). Concentration-contraction curves in response to cumulative additions of KCl showed that there was significantly less contraction to low concentrations of KCl (10 to 30 mmol/L) in arteries from dogs with SAH compared with those from control dogs (n = 6 to 8 rings from five dogs per group, P < 0.05, Student's t-test). Furthermore, there was greater contraction (as a % of the maximal contraction to 120 mmol/L KCl) of basilar artery rings to Ba2+, 1 mmol/L, after SAH than in control arteries (41% ± 11% versus 14% ± 3%, n = 23 rings from six and 24 rings from seven dogs, respectively, P = 0.02, Figure 8), while contractions to other agents such as paxilline (1 μmol/L, a blocker of BK channels) were significantly lower in SAH arteries compared with control (17% ± 5% versus 5% ± 4%, n = 9 rings from four dogs each, P = 0.04, Figure 8).
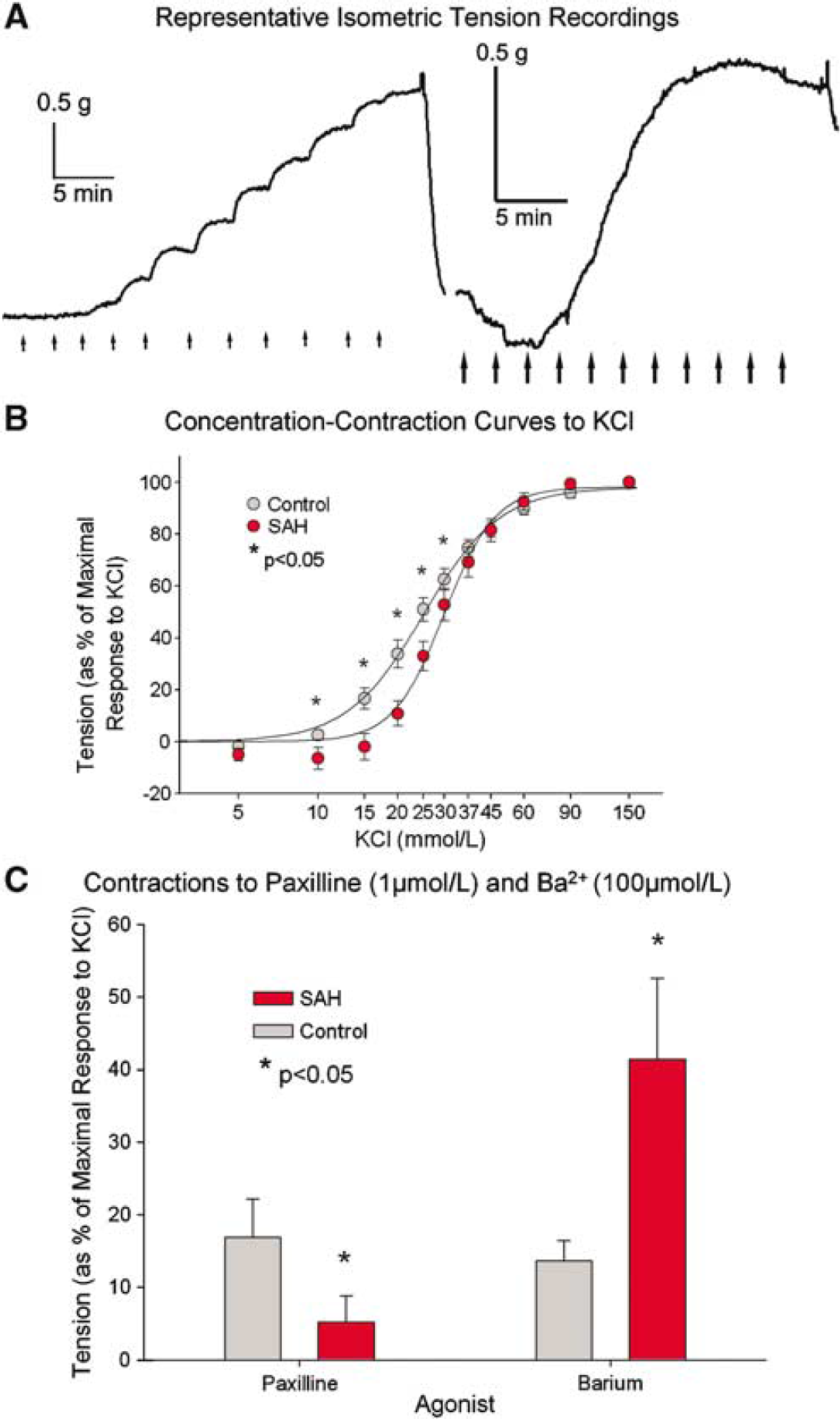
(
Discussion
Few studies have directly investigated alterations in K+ channels in vasospastic arteries. We reported that Kv 2 class channels were reduced functionally and molecularly in vasospastic dog basilar artery (Aihara et al, 2004; Jahromi et al, 2005), a finding consistent with indirect and less specific pharmacological evidence obtained in rats (Quan and Sobey, 2000). Pharmacological studies suggested an increased functional role for BK (Onoue and Katusic, 1998) or KATP (Sobey et al, 1996) channels in vasospastic dog or rat arteries, respectively. We found no electrophysiological evidence for altered BK channel function during vasospasm in dogs (Jahromi et al, 2005). Furthermore, in isometric tension recordings, SAH arteries contracted less to paxilline, a blocker of BK channels. This work provides the first evidence that the remaining K+ channel, KIR, is increased both functionally and at a molecular level during vasospasm in dogs.
KIR channels are named for their preference to conduct inward currents at potentials negative to EK. At physiological K+ concentrations (5 mmol/L), however, this characteristic would not have a significant impact on gK as the KEq (−84 mV) is strongly negative to observed Em (in the range of −30 to −40 mV)(Nelson and Quayle, 1995). This effect would be even more marked in vasospastic smooth muscle cells since they are depolarized by 10 to 17 mV relative to normal myocytes after SAH in rabbits, cats, and dogs (Harder et al, 1987; Zuccarello et al, 1996). KIR channels conduct small outward currents at potentials positive to KEq and it is these currents that are thought to contribute to gK and participate in the maintenance of Em (Quayle et al, 1997) This hyperpolarizing current would relax smooth muscle and is thought to be the basis for cerebral vasodilation in response to small increases in [K+]+ex, which occur during neuronal activation (Nelson and Quayle, 1995). KIR currents are believed to be more prominent in smaller resistance arteries than in the large conductance arteries of the circle of Willis (Edwards et al, 1988; Quayle et al, 1997; Zaritsky et al, 2000). The present work suggests that this channel functions in normal conductance arteries and is of greater importance during vasospasm.
Since vasospastic arteries are constricted, the question arises as to the importance of the increase in KIR channels. It could be postulated that vasospasm would be more severe if KIR current was not increased and that the increase is a compensatory mechanism opposing vasospasm. Time-course studies or experiments manipulating expression of KIR channels would be needed to determine this. KIR channels are found on endothelial cells and are upregulated by shear stress (Hoger et al, 2002). The present study, however, directly assessed smooth muscle cells so that any increase in KIR because of an increase in endothelial shear stress associated with vasospasm cannot be the explanation for the observed changes. The mechanism for the increase in KIR that was observed in this study is a matter of speculation. It is interesting that the synthetic smooth muscle phenotype may have increased KIR expression since it has been suggested that there might be some degree of phenotypic change in cerebral arterial smooth muscle after SAH (Ohkuma et al, 2003; Karkanis et al, 2003). It also should be noted that this study was conducted in female dogs. There is evidence that dilation of rat basilar artery in response to low concentrations of K+ is greater in female than male rats, although this was thought to be mediated indirectly by acetylcholine receptors (Chrissobolis and Sobey, 2004). Nevertheless, it would seem prudent to examine KIR in male dogs.