
Abstract
In cytoplasm, nuclear factor-k(NF-kB) is associated with the inhibitory protein, IkBα. On activation by H2O2, IkBα is phosphorylated and degraded, exposing the nuclear localization signals on the NF-kB heterodimer. Cyclooxygenase-2 (COX-2), which mediates prostaglandin synthesis during inflammation, is induced by oxidative stress mediated by NF-kB. We investigated whether the NF-kB signaling pathway affected cell death and COX-2 expression after hypoxia-induced oxidative stress in wild-type (WT) and copper/zinc-superoxide dismutase transgenic (SOD1 Tg) astrocytes. In WT astrocytes, phospho-IkBα was highly expressed after oxygen-glucose deprivation (OGD) and 2 h of reperfusion, concomitant with the decrease in kBa. The NF-kB p50 level increased similarly in WT and SOD1 Tg astrocytes (1.2-/1.4-fold) after OGD. Electrophoretic mobility shift assay showed higher DNA-binding activity of NF-kB p50 in WT than in SOD1 Tg astrocytes 6 h after 4 h of OGD. The COX-2 level was induced by 2.7- and 1.3-fold after OGD in WT and SOD1 Tg astrocytes, and an antioxidant protected both groups against OGD injury. Superoxide dismutase transgenic cells were 23% more protective against OGD injury than WTs when assessed by lactate dehydrogenase release. However, transfection of NF-kB small interfering RNAs in SOD1 Tg astrocytes aggravated cell death and increased COX-2 expression. These results suggest that the NF-kB signaling pathway induced COX-2 expression and promoted cell death in WTs after OGD injury; however, NF-kB activation protected cells and decreased COX-2 expression in SOD1 Tg astrocytes. This biphasic role of NF-kB might be coordinately regulated by reactive oxygen species levels in astrocytes, thereby functioning as a regulator of cell death/survival.
Introduction
Reactive oxygen species (ROS), including H2O2, are generated in increased amounts in pathologic biologic processes and can play a role in signal transduction. Reactive oxygen species have been increasingly recognized as critical components in disease- and stress-induced cellular injuries such as ischemia/reperfusion, ultraviolet irradiation, and inflammation. These ROS can cause direct cellular damage and can also act as intracellular messengers to modulate signal transduction pathways. One such redox-regulated transcription factor is nuclear factor-kB (NF-kB) (Schreck et al, 1991), a ubiquitous transcription factor that plays a major role in the regulation of stress-related genes and is activated during environmental hypoxia in the dorsocaudal brainstem of adult rats (Simakajornboon et al, 2001). Induction of NF-kB has been detected in traumatic brain injury (Yang et al, 1995; Bethea et al, 1998), spinal cord injury (Bethea et al, 1998), global forebrain ischemia (Clemens et al, 1997), and focal ischemia (Salminen et al, 1995; Schneider et al, 1999). In the resting state, NF-kB is sequestered in the cytoplasm through its close association with specific inhibitory proteins called inhibitors of NF-kB(IkB), belonging to a gene family consisting of IkBα, IkBβ, IkBε, IkBγ, Bcl-3, P100, and P105 (Karin and Ben-Neriah, 2000). Signals from various stimuli induce activation of upstream kinases, mainly the IkB kinase (IKK) complex that contains two catalytic kinase subunits, IKKα and IKKã, and a structural and regulatory component named IKKγ. Activated IKK phosphorylates IkBα and IkB/?, leading to their ubiquitination, degradation, and subsequent NF-kβ activation and nuclear translocation (Chen et al, 2003).
Nuclear factor-kB plays a central role in the cellular response of numerous genes to a variety of stress signals. On activation by agents such as tumor necrosis factor, IkBα is phosphorylated at serine residues 32 and 36 (Zandi et al, 1997), ubiquitinated at lysine residues 21 and 22, and degraded through the proteosomal pathway, thus exposing the nuclear localization signals on the p50–p65 heterodimer. Then p65 undergoes phosphorylation, leading to activation of NF-kB-mediated transcription.
Cyclooxygenase (COX) (prostaglandin G/H synthase) is the first committed step in the production of prostaglandin and thromboxanes. Cyclooxygenase-1 is constitutively expressed in many tissues, including platelets, gastrointestinal mucosa, and kidney (DeWitt and Smith, 1988). The inducible form, COX-2, is primarily expressed in leukocytes and brain (Kujubu et al, 1991). Neuronal excitation and increased intracellular calcium, two stimuli that induce expression of COX-2, are also important in the pathophysiology of neuronal death in ischemia and a variety of neurodegenerative diseases (Rothman and Olney, 1986). In non-neuronal cells, COX-2 activity mediates inflammatory injury (Seibert et al, 1995); however, COX-2 overexpression may prevent apoptosis in intestinal epithelium (Tsujii and DuBois, 1995).
Previous reports indicate that NF-kB can function upstream of COX-2 to control transcription of this gene through IKK pathway activation (Plummer et al, 1999). Two putative NF-kB motifs from the COX-2 promoter were found to bind p50/p65 NF-kB heterodimers in an interleukin-1/β-dependent manner (Crofford et al, 1997), whereas hypoxia induced COX-2 transcription via p65 binding to 3′ NF-kB consensus element in the enzyme upstream promoter region in vascular endothelial cells (Schmedtje et al, 1997). The role of NF-kB in COX-2 expression and cell survival after ischemic-like injury in astrocytes is unclear. In this study, we investigated the effect of oxygen-glucose deprivation (OGD) and reperfusion injury on IkBα phosphorylation, NF-kB activation, COX-2 expression, and cell survival in wild-type (WT) and copper/zinc-superoxide dismutase transgenic (SOD1 Tg) mice. We then showed that transfection of NF-kB small interfering RNAs (siRNA) significantly attenuated expression of COX-2 and cell death in WT astrocytes, whereas it aggravated cell death and increased COX-2 expression in SOD1 Tg astrocytes. Results from these studies indicate that the transcription factor NF-kB 107 has dual actions in WT or SOD1 Tg astrocytes, depending on the circumstances under which it is activated.
Materials and methods
Primary Cultures of Murine Cortical Cells
All procedures were performed according to a protocol approved by the Stanford University Animal Care and Use Committee and in accordance with the National Institutes of Health guide for the care and use of laboratory animals. Astrocyte cultures were prepared from the cerebral cortices of postnatal (days 1 to 3) SOD1 Tg mice and their littermates as described previously (Yu et al, 1989; Dugan et al, 1995). Briefly, cerebral cortices freed of meninges were incubated in 0.25% trypsin (Invitrogen, Carlsbad, CA, USA), diluted in Eagle's minimal essential medium (Invitrogen) for 30 mins at 37°C, and then mechanically dissociated. The dissociated neocortical cells were plated on Falcon Primaria 24-well plates (Becton Dickinson, Lincoln, IL, USA) at a density of two hemispheres per multiwell, in Eagle's minimal essential medium supplemented with 10% equine serum (HyClone, Logan, UT, USA), 10% fetal bovine serum (HyClone), 21 mmol/L (final concentration) glucose, and 10 ng/mL epidermal growth factor.
Transfection of Small Interfering RNA
Superoxide dismutase transgenic and WT astrocytes were plated on 24-well plates and grown for 6 days. At the time of transfection, the cells were 50% to 60% confluent. SiPORT Lipid reagent (Ambion, Austin, TX, USA) and NF-kB p50 siRNA (Ambion) or nonfunctional negative control siRNA (Ambion) were dissolved separately in Optimem I (Invitrogen). After 15 mins of equilibration at room temperature, each RNA solution was combined with the respective volume of the SiPORT lipid solution, mixed gently, and allowed to form siRNA liposome for a further 20 mins at room temperature. The transfection mixture was added to the antibiotic-free cell culture medium to a final concentration of 50 nmol/L siRNA and 2 μL/mL SiPORT lipid. After incubating for 4 h under normal cell culture conditions, Eagle's minimal essential medium supplemented with 10% equine serum and 10% fetal bovine serum were added to reach the final volume of the well.
Oxygen–Glucose Deprivation
Cultures were transferred to an anaerobic chamber (Coy Laboratory Products Inc., Grass Lake, MI, USA) in an atmosphere of 5% H2 and 90% N2. The culture medium was replaced three times with deoxygenated, glucose-free balanced salt solution (BSS0), pH 7.4, containing Phenol Red (10 mg/L) and the following (in mmol/L): NaCl 116, CaCl2 1.8, MgSO4 0.8, KCl 5.4, NaH2PO4 1, NaHCO3 14.7, N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid 10. Balanced salt solution 5.5 contained 5.5 mmol/L glucose. Cultures were placed in a humidified 37dGC incubator within the anaerobic chamber. Oxygen tension was monitored with an oxygen electrode meter and was kept under 0.02%. Oxygen–glucose deprivation was ended by adding glucose to the culture medium to a final concentration of 5.5 mmol/L and returning the cultures to the normoxic incubator for the indicated time period before sampling and determining the lactate dehydrogenase (LDH) activity.
Lactate Dehydrogenase Assay
Lactate dehydrogenase was measured as described previously (Koh and Choi, 1987). The medium was sampled at each time point after reperfusion. Astrocytes were then frozen/thawed to provide the maximum LDH release values (full-kill numbers). The percent death (% of LDH release) was calculated by dividing the experimental time point by the full-kill values × 100.
Ribonucleic acid Extraction and Reverse Transcriptase-Polymerase Chain Reaction
Total cellular RNA was extracted from cells using the RNeasy Mini kit (Qiagen, Valencia, CA, USA), according to the manufacturer's instructions. Complementary DNA was reverse transcribed from 2 mg of total cellular RNA using oligo (dT) primers and Moloney murine leukemia virus reverse transcriptase (RT). In all, 1 μg of complementary DNA was amplified for 30 cycles using mouse NF-kB p50 gene-specific primer sets (SuperArray, Bethesda, MD, USA). The polymerase chain reaction (PCR) included a 2-min denaturation at 94°C followed by 30 cycles, each consisting of denaturation at 94°C for 15 secs, annealing at 54°C for 30 secs, and extension at 72°C for 1 min, with a final extension phase of 2 mins. Polymerase chain reaction was performed on a programmable thermal controller instrument-thermal cycle model. Meanwhile, the same amount of complementary DNA was amplified for 30 cycles using specific glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primers (SuperArray). The product was visualized after electrophoresis on a 1% agarose gel and staining with ethidium bromide.
Nuclear Factor-kB Activation Assays
Nuclear extracts for electrophoretic mobility shift assay (EMSA) were prepared from the WT and SOD1 Tg astrocytes using ProteoExtract (Calbiochem, Darmstadt, Germany). Nuclear factor-kB binding consensus sequence (5′-AGT TGA GGG GAC TTT CCC AGG C-3′; Santa Cruz Biotechnology, Santa Cruz, CA, USA) was radiolabeled with [32P]-γ adenosine triphosphate by T4 polynucleotide kinase (Invitrogen) to produce double-stranded DNA probes. For binding reaction, 10 mg of nuclear extract was incubated with poly (dI-dU) and the 32P-labeled doublestrand oligonucleotide (100-fold molar excess) competitor was preincubated with nuclear extract for 20 mins at room temperature. A mutant NF-kB oligonucleotide used for the competition assay was as follows: 5′-AGT TGA GGC GAC TTT CCC AGG C-3′ (the mutant sequences are underlined; Santa Cruz Biotechnology). The hybridization signal on the X-ray film was scanned with a GS-700 imaging densitometer (Bio-Rad, Hercules, CA, USA) and the results were quantified using Multi-Analyst software (Bio-Rad).
Western Blot Analysis
Cells were washed twice with cold phosphate-buffered saline and collected by centrifugation at 1000g. Proteins were extracted using ProteoExtract Subcellular Proteosome Extraction Kit (Calbiochem). Equal amounts of whole cell extracts were denatured at 100°C for 5 mins in Laemmli sample buffer/5% 2-mercaptoethanol, electrophoresed on 4% to 12% Tris-Glycine gels (Invitrogen), and electrotransferred onto a membrane (Invitrogen). The membrane was incubated overnight at 4°C with rabbit polyclonal antibodies raised against NF-kB p50 (1:400; AbCam, Cambridge, MA, USA), NF-kB p65 (1:200; Cell Signaling Technology, Beverly, MA, USA), COX-2 (1:400; Cell Signaling Technology), IkBα (1:400; Cell Signaling Technology), and phospho-IkBα (1:500; Cell Signaling Technology).
Western blots were performed with horseradish peroxidase-conjugated anti-immunoglobulin G (Amersham International, Buckinghamshire, UK) and were detected by SuperSignal West Pico Chemiluminescent Substrate, according to the manufacturer's instructions (Pierce, Rockford, IL, USA). Equivalent sample loading was confirmed by stripping membranes with Restore Western Blot Stripping Buffer (Pierce) and reprobing with an anti-β-actin antibody. The film was scanned with a GS-700 imaging densitometer (Bio-Rad) and the result was quantified using Multi-Analyst software (Bio-Rad).
Viability Testing
Viability was tested after 4 h of OGD in the WT and SOD1 Tg astrocytes treated with N-tert-butyl-α-phenylnitrone (PBN). In each case, the cell cultures were stained with 2 μmol/L of calcein AM and 4 μmol/L of ethidium homodimer (EthD-1) using a Live/Dead kit (Molecular Probes, Eugene, OR, USA). Calcein and EthD-1 fluorescence was observed through a fluorescence microscope (excitation 470 to 490 nm, dichroic mirror 505 nm, emission 470 to 490 and 590 nm). As calcein AM is converted to green fluorescence by intracellular esterase, green staining indicated metabolically active cells. Ethidium homodimer is an indicator of membrane damage and reveals dead cells. The fluorescence image was obtained by × 200 magnification.
Immunostaining of Cyclooxygenase-2
The effect of OGD on the expression level of COX-2 was examined by immunocytochemical methods. Briefly, primary mouse astrocytes were plated on poly-D-lysinecoated glass slides. After the injury, the cells were permeabilized with 0.1% Triton X-100. After washing with phosphate-buffered saline, the slides were blocked with 5% normal horse serum for 1 h and then incubated with rabbit polyclonal COX-2 antibodies at a 1:250 dilution. After overnight incubation at 4°C, the slides were washed and incubated with goat anti-rabbit immunoglobulin G Texas Red (Vector Laboratories, Burlingame, CA, USA) at a 1:1000 dilution. Stained slides were mounted with mounting medium and analyzed under a fluorescent microscope (Zeiss, Oberkochen, Germany).
Measurement of Cyclooxygenase-2 Activity
The cytosolic fraction of the primary astrocytes was used to measure the peroxidase activity of COX-2. The peroxidase activity was assayed colorimetrically by monitoring the appearance of oxidized N,N,N,N -tetramethyl-p-phenylenediamine at 590 nm (Kulmacz and Lands, 1983), according to the procedure described by the manufacturer (Cayman Chemical, Ann Arbor, MI, USA). A COX-1-specific inhibitor, SC-560, was used to distinguish COX-2 activity from COX-1 activity.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism version 3.0 (GraphPad Software, San Diego, CA, USA). Analysis of variance (ANOVA), followed by a Student–Neuman–Keuls test, was used for multiple comparisons. The difference for each comparison was considered statistically significant at the P < 0.05 level.
Results
Oxygen–Glucose Deprivation Induces Degradation of Ikbα
Phosphorylation of IkBα at Ser32 and Ser36 and their subsequent degradation are suggested as key steps in the process of NF-kB activation (Karin, 1999). In a first approach to explore the upstream signaling mechanism by which NF-kB might be activated by OGD injury, we examined IkBα and phospho-IkBα levels by Western blot analysis in the WT and SOD1 Tg astrocytes. As shown in Figure 1A, the IkBα level decreased after OGD and slowly recovered to the base level during reperfusion. Accordingly, there was a rapid increase in phospho-IkBα levels after OGD and until 4 h of reperfusion. The SOD1 Tg astrocytes showed a decrease in IkBα after OGD, but there was no consistent increase in phospho-IkBα after OGD and at similar time points of reperfusion (Figures 1B, 1D, and 1F). Thus, OGD seemed to have a more significant effect on phosphorylation and degradation of IkBα in the WT astrocytes than in the SOD1 Tg astrocytes.
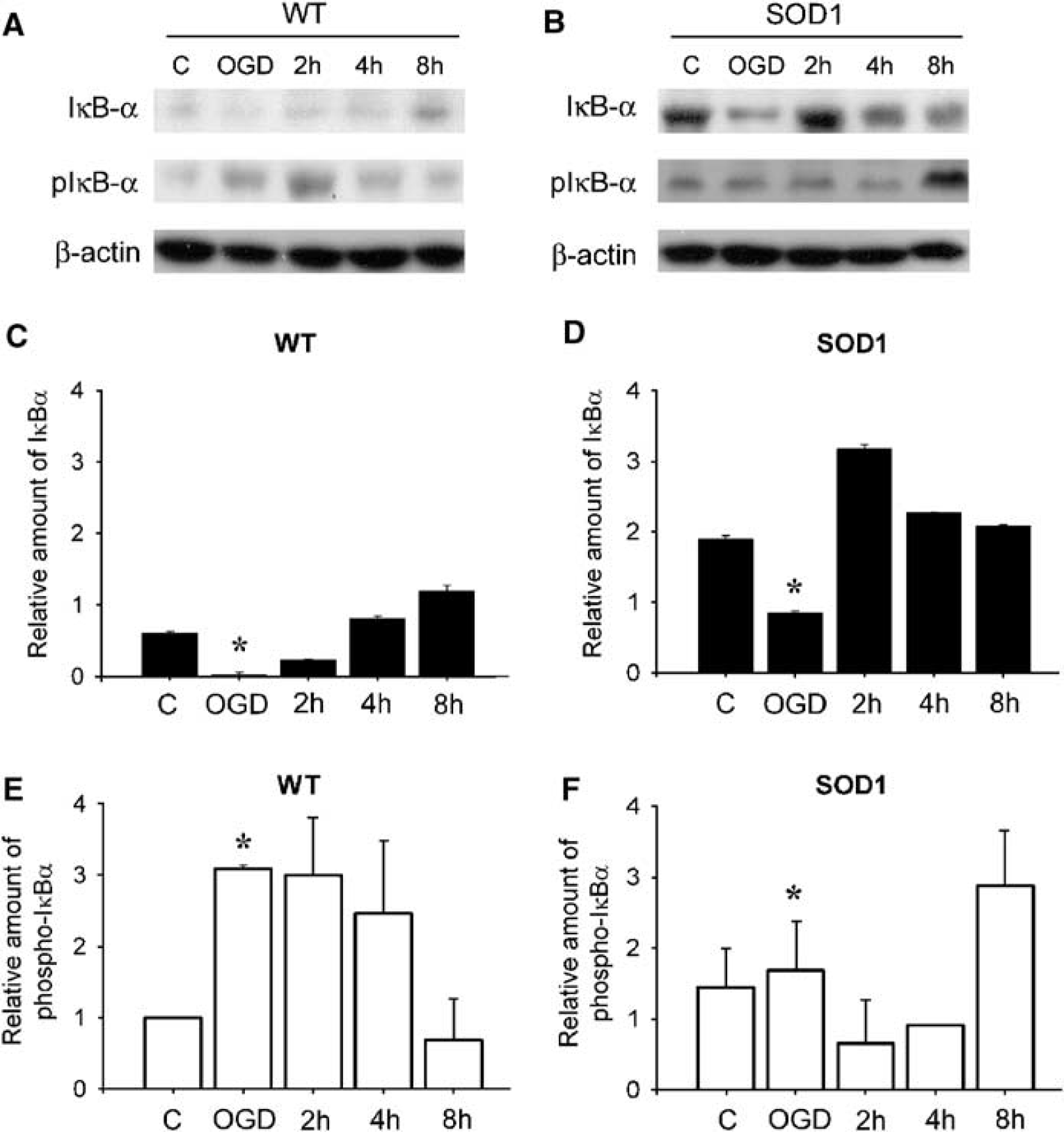
The protein level of IkBα and phospho-IkBα in WT and SOD1 Tg astrocytes.
Activation of Nuclear Factor-kB p50 in Primary Astrocytes After Oxygen-Glucose Deprivation Injury
Nuclear factor-kB activation requires the degradation of IkBα. To determine whether IkBα degradation can induce NF-kB activation, we first examined by EMSA the DNA-binding activity of NF-kB from nuclear extracts prepared from the WT and SOD1 Tg astrocytes after OGD injury. As shown in Figure 2, DNA-binding activity greatly increased 6 h after 4 h of OGD in the WT astrocytes. The SOD1 Tg astrocytes showed a gradual increase in DNA-binding activity 2 and 6h after 4 h of OGD. The relative NF-kB-binding activity was higher in the WT nuclear extracts than in the SOD1 Tg astrocytes at 6 h of reperfusion (Figure 2, lanes 4 and 8). To show that the band visualized by EMSA after OGD and reperfusion was indeed NF-kB, we incubated nuclear extracts with antibodies against the p65 subunit of NF-kB. This shifted the band to a higher molecular mass (Figure 2, lane 9), thus suggesting that this complex consisted of the p65 subunit. Adding an excessive amount of unlabeled consensus probe caused complete disappearance of the band, suggesting this band was specific for NF-kB DNA binding (Figure 2, lane 10).
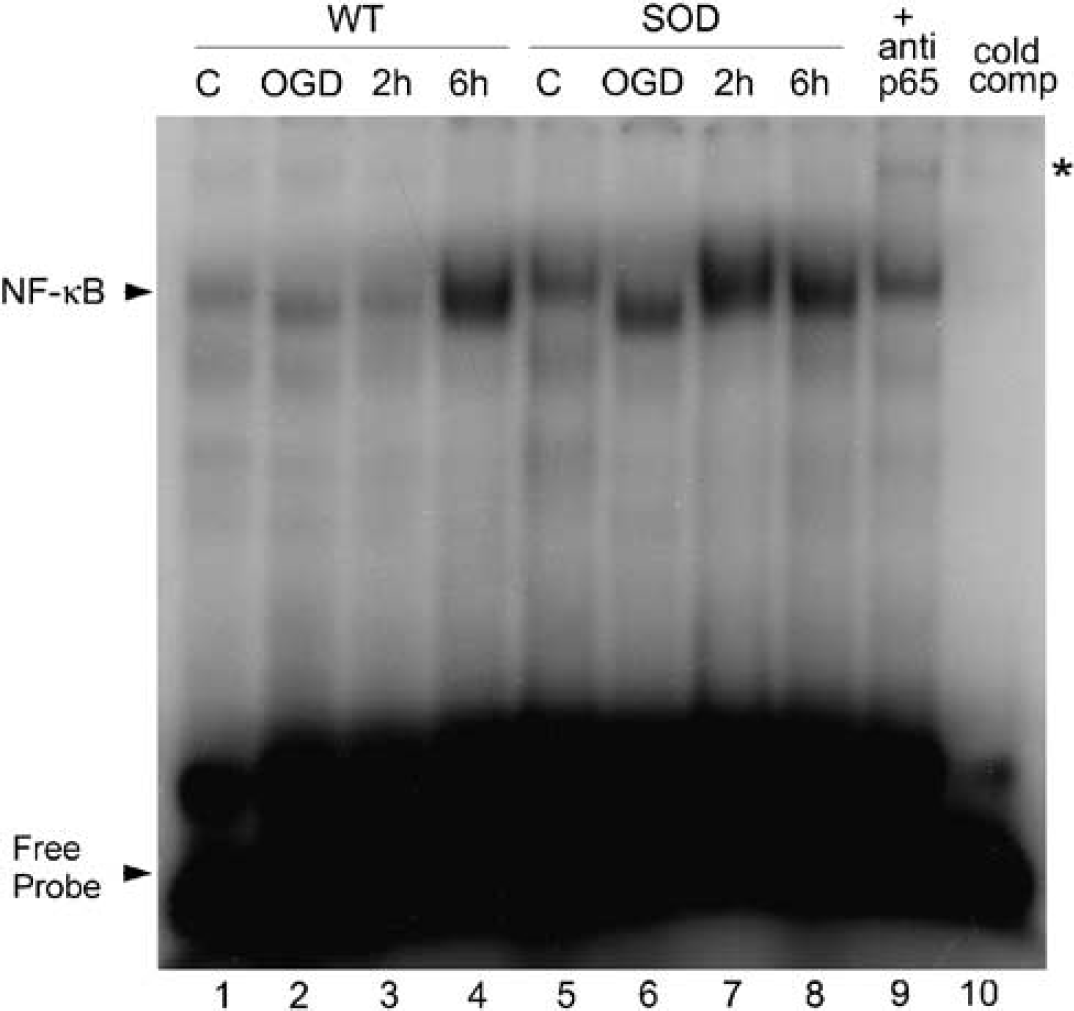
Changes in NF-kB activation after OGD injury in WT and SOD1 Tg astrocytes. WT and SOD1 Tg astrocytes were subjected to 4 h of OGD and reperfused for the indicated times. Nuclear extracts were analyzed for NF-kB activation by EMSA as described under Materials and methods. Lane 1, WT control; lane 2, WT after 4 h of OGD; lanes 3 and 4, WT after OGD, followed by 2 and 6 h of reperfusion; lane 5, SOD control; lane 6, SOD after 4 h of OGD; lanes 7 and 8, SOD after OGD followed by 2 and 6 h of reperfusion. Lane 9, the star marks the band of supershifted p65/DNA complex after incubating with anti-p65 antibody. Lane 10, 50-fold concentrations of unlabeled NF-kB oligonucleotides (50 × competitor) were used for the competition experiment.
The cellular level of NF-kB was examined by Western blot analysis using an anti-NF-kB p50 antibody (Figure 3B). The WT and SOD1 Tg astrocyte cultures were subjected to 4 h of OGD and the cells were harvested at various time points of reperfusion. Expression of the NF-kB p50 protein was increased by 1.2- and 1.4-fold in the WT and SOD1 Tg astrocytes after 4 h of OGD injury compared with the WT control astrocytes. To determine the definite role of NF-kB in OGD-injured WT and SOD1 Tg astrocytes, we specifically suppressed the expression of NF-kB by using an RNAi strategy. Reverse transcriptase-polymerase chain reaction results showed that NF-kB siRNA reduced the transcription of NF-kB mRNA. Nuclear factor-kB siRNA 3, which was more efficient, was used throughout our experiments (Figure 3A, left panel, lane 5). Levels of the housekeeping gene GAPDH were not affected by negative control siRNA and NF-kB siRNA did not affect GAPDH mRNA or β-actin protein levels (Figure 3A, right panel, and 3B). Transfection of NF-kB siRNA decreased the cellular level of NF-kB p50 to 40% 70.09% and 34% 70.3% in the control WT and SOD1 Tg astrocytes, respectively, after OGD injury. In addition, induction of NF-kB p50 was also significantly blocked by siRNA in both the WT and SOD1 Tg astrocytes at the end of OGD injury (Figure 3B and 3C). These results confirm that OGD injury induced the cellular level of NF-kB and activated NF-kB transcription activity in the WT and SOD1 Tg astrocytes. In addition, phosphorylation and degradation of IkBα had a more essential role in activating NF-kB in the WT astrocytes after OGD injury.
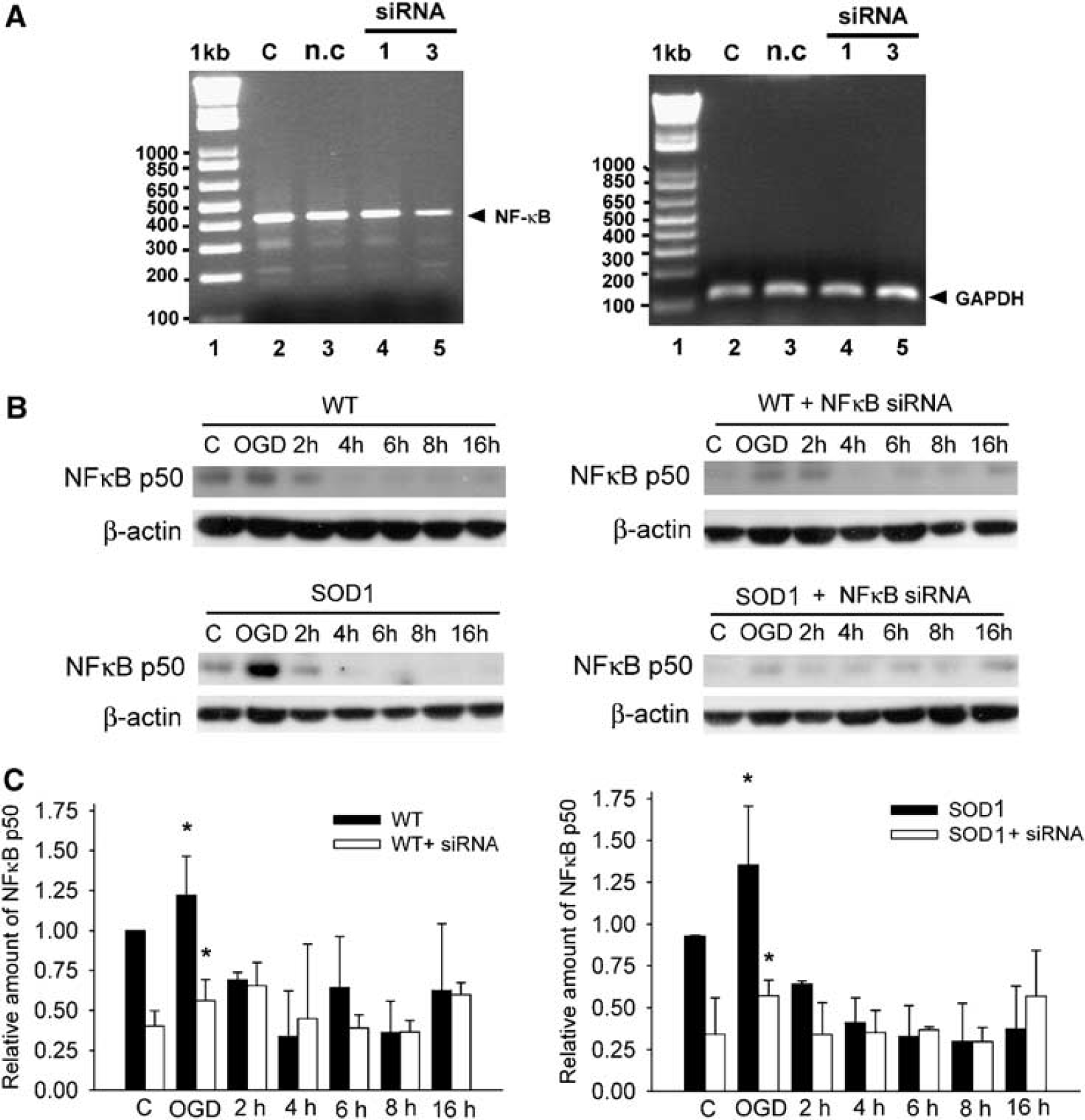
Reverse transcriptase-polymerase chain reaction analysis of NF-kB p50 mRNA and Western blot analysis of WT and SODI Tg astrocytes transfected with NF-kB p50 siRNA.
Expression of Nuclear Factor-kB p65 in Primary Astrocytes After Oxygen–Glucose Deprivation Injury
We analyzed the effect of OGD injury on induction of NF-kB p65 by Western blotting (Figure 4). Nuclear factor-kB p65 was rapidly increased after OGD and 2 h of reperfusion in the WT astrocytes. In the SOD1 Tg astrocytes, there was a rapid induction of p65 at the end of 4 h of OGD injury, but it declined rapidly to the basal level after reperfusion (Figure 4B). Transfection of NF-kB siRNA reduced the rapid induction of p65 after OGD injury, but did not completely block the expression of p65 in the SOD1 Tg astrocytes. Transfection of NF-kB p50 siRNA did not have a great effect on the expression of p65 in the WT astrocytes.
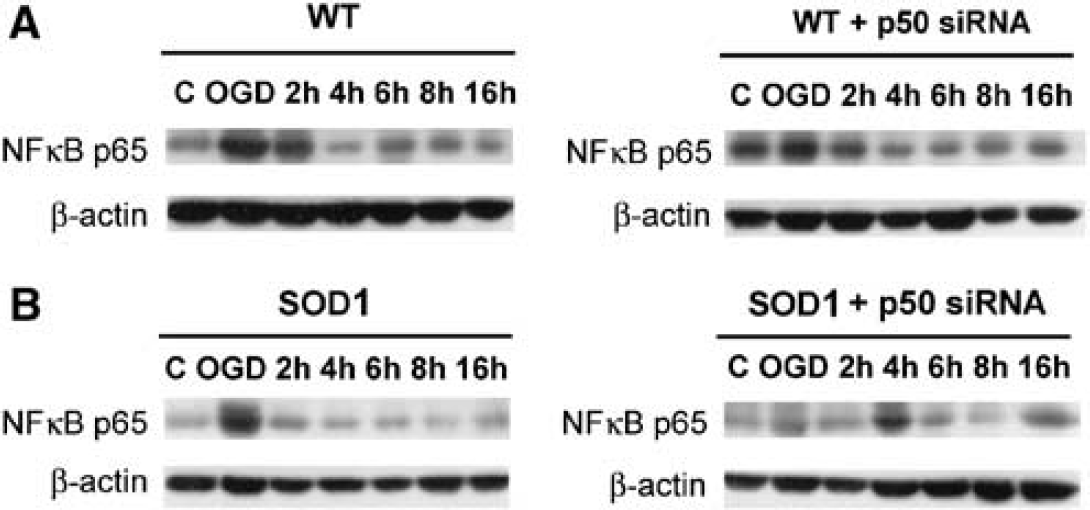
The protein level of NF-kB p65 in WT and SODI Tg astrocytes.
Viability Staining
Most of the WT and SOD1 Tg astrocytes were viable before OGD injury (Figure 5). After 4 h of OGD, viability of the WT astrocytes greatly decreased, while the SOD1 Tg astrocytes were still viable when stained with calcein AM. The SOD1 Tg astrocytes also had less EthD-1-stained cells, suggesting that these astrocytes were more protective against OGD injury (Figure 5, middle panels). When treated with 50 μmol/L of a nitrone-based free radical trapping agent, PBN, for 30 mins, cell death was significantly reduced in both groups of astrocytes after 4 of OGD. In contradistinction, the SOD1 Tg astrocytes treated with PBN showed increased cell viability when stained with calcein AM.
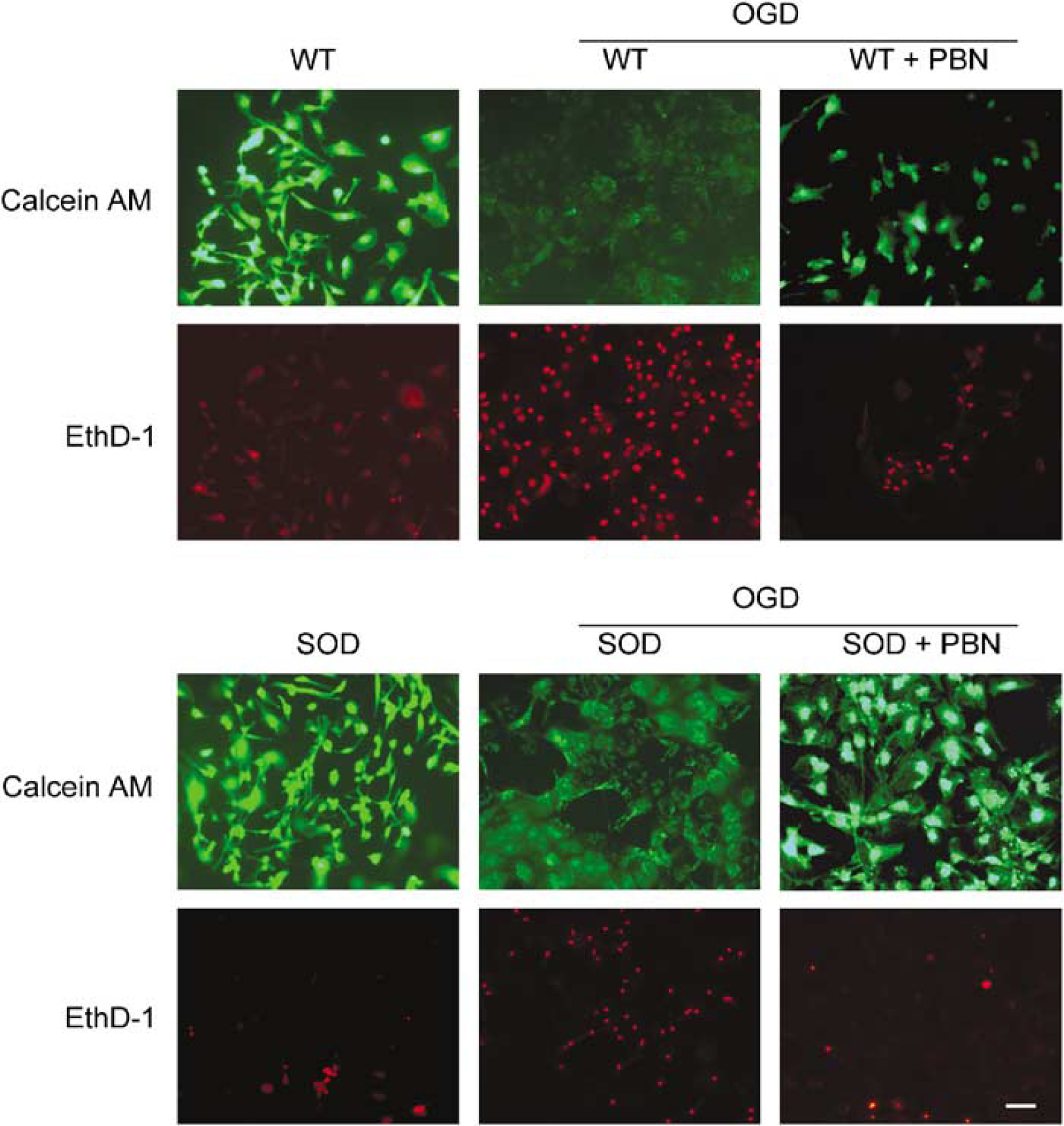
Viability of PBN-treated W Tand SOD1 Tg astrocytes after OGD. Viability of the cells was assessed using calcein AM (green) and EthD-1 (red). Living cells were stained green and dead cells were stained red. Left panels: Most of the WT and SOD1 Tg cells were viable before the injury. Center panels: After 4 h of OGD, dead cells were confirmed by EthD-1 staining. Right panels: PBN-treated cells after 4 h of OGD. Cell death was reduced in both the WT and SOD1 Tg astrocytes. The increase in cell viability was confirmed by the increase in calcein AM cells in the WT astrocytes and by a greater amount in the SOD1 Tg astrocytes. Bar = 50 mm.
Involvement of Nuclear Factor-kB Small Interfering RNA in Oxygen–Glucose Deprivation-Induced Cyclooxygenase-2 Expression
To study the involvement of NF-kB in the upregulation of COX-2 by OGD, the WT and SOD1 Tg astrocytes were transfected with NF-kB siRNA, followed by 4 h of OGD. The expression level of the COX-2 protein was examined by Western blot analysis. As illustrated in Figure 6, there was a 2.8-fold increase in COX-2 levels after OGD. Transfection of NF-kB siRNA reduced COX-2 expression in the WT astrocytes after 4 h of OGD. In the SOD1 Tg astrocytes, there was a 1.25-fold increase in COX-2 levels, but transfection of NF-kB siRNA induced a 2.3-fold increase in COX-2, and this was persistent until 6 h after reperfusion.
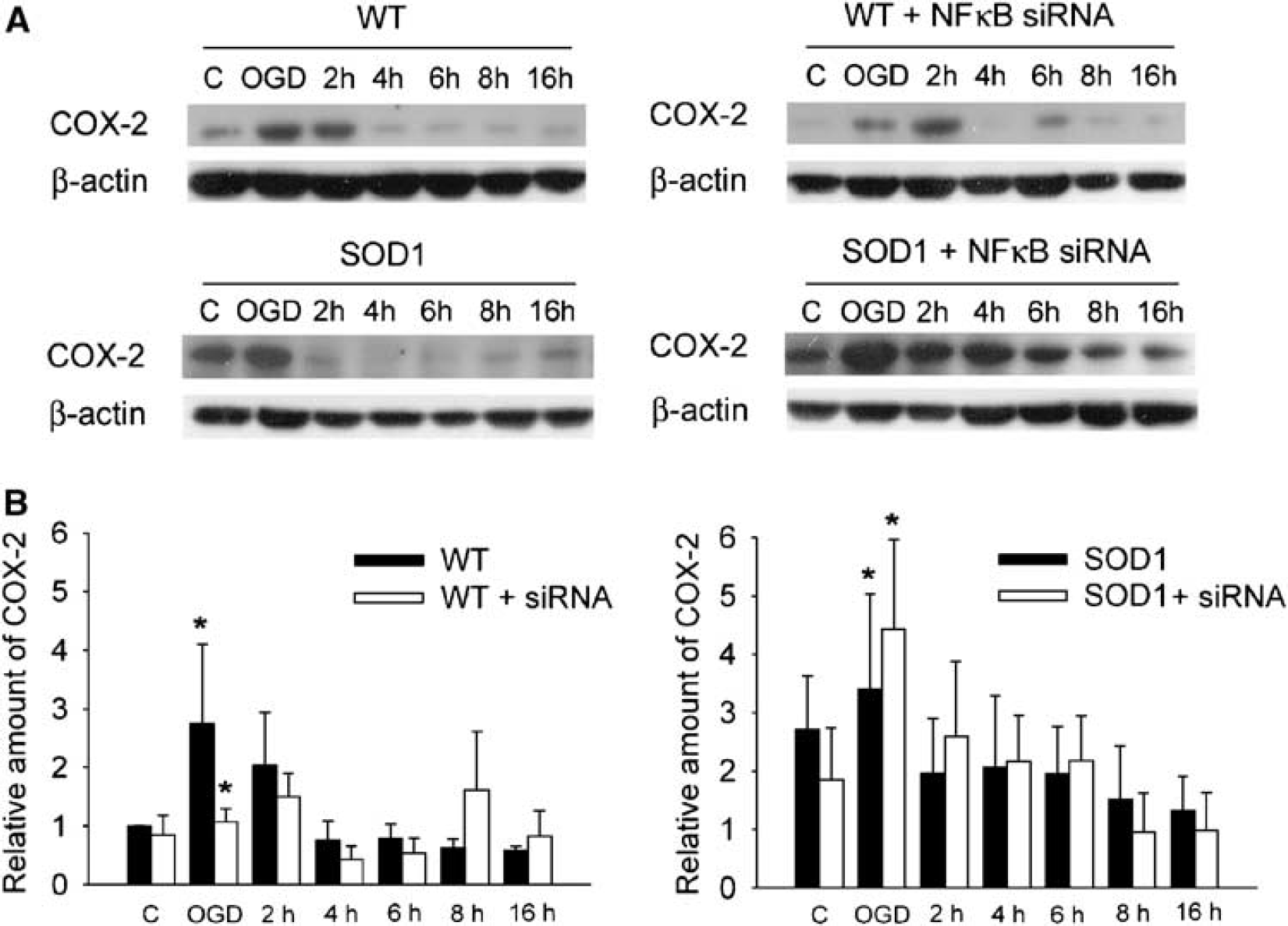
Expression levels of COX-2 protein in WT and SODI Tg astrocytes transfected with siRNA.
These findings were further confirmed by representative immunofluorescent staining of COX-2 in the WT and SOD1 Tg astrocytes and in those transfected with NF-kB siRNA (Figures 7A to 7D). Cyclooxygenase-2 expression was evenly distributed around the cytoplasm in the WT and SOD1 Tg astrocytes after OGD. However, the NF-kB siRNA-transfected SOD1 Tg astrocytes showed a high expression of COX-2 proteins clustered inside the cell after OGD (Figure 7D). Figure 7E shows COX-2 activity from the cytosolic fraction of the WT and SOD1 Tg astrocytes, while eliminating COX-1 activity with SC-560. In normoxia, the SOD1 Tg astrocytes had the least COX-2 activity: 11.073 ± 0.049 nmol/min mL. After OGD, there was an increase of 2.825±0.448 nmol/min mL in COX-2 activity in the WT astrocytes. In the SOD1 Tg astrocytes, there was an increase of 2.76 ± 1.1 nmol/min mL in COX-2 activity. However, when the SOD1 Tg astrocytes were transfected with NF-kB siRNA, there was the highest increase in COX-2 activity at 2 h of reperfusion: 3.224 ± 0.283 nmol/min mL. Adding DuP-697, a potent and time-dependent inhibitor of COX-2 (Kargman et al, 1996), to the assay systems of the WT and SOD1 Tg astrocytes decreased COX-2 activity to the basal level of 2.055 ± 0.324 nmol/min mL.
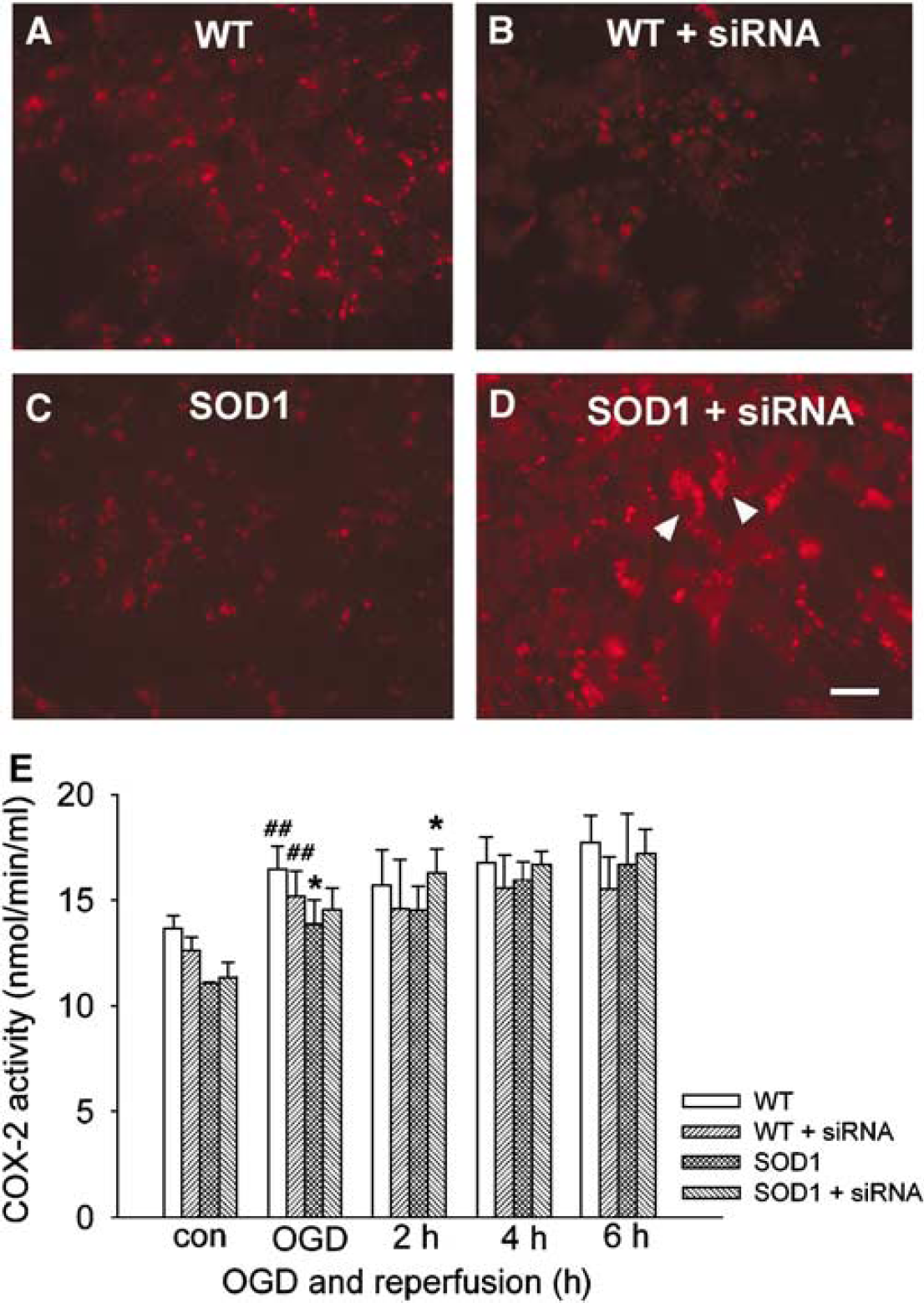
Immunocytochemistry of COX-2 in
Reduction of Nuclear Factor-kB Protein by Small Interfering RNA Induces Cell Death in Superoxide Dismutase Transgenic Astrocytes
We then determined whether inhibition of NF-kB affects OGD-induced cell death. Oxygen–glucose deprivation was terminated by the addition of oxygen and glucose to the medium (reperfusion). The medium was then sampled for LDH release, as a measure of cell injury. In all, 4 h of OGD resulted in 63% cell death in the WT astrocytes (Figure 8). Transfection of NF-kB siRNA significantly protected the WT astrocytes, reducing cell death to 16% after OGD. Oxygen–glucose deprivation in the SOD1 Tg astrocytes resulted in 40% cell death. However, transfection of NF-kB siRNA in these astrocytes resulted in a rapid increase in levels of LDH release, reaching almost 99% of cell death after 4 and 6 h of reperfusion. We examined whether the cell death was necrotic or apoptotic using DNA laddering, which is the hallmark of apoptotic cell death. Results showed that cells underwent necrotic death after OGD and reperfusion injury (data not shown).
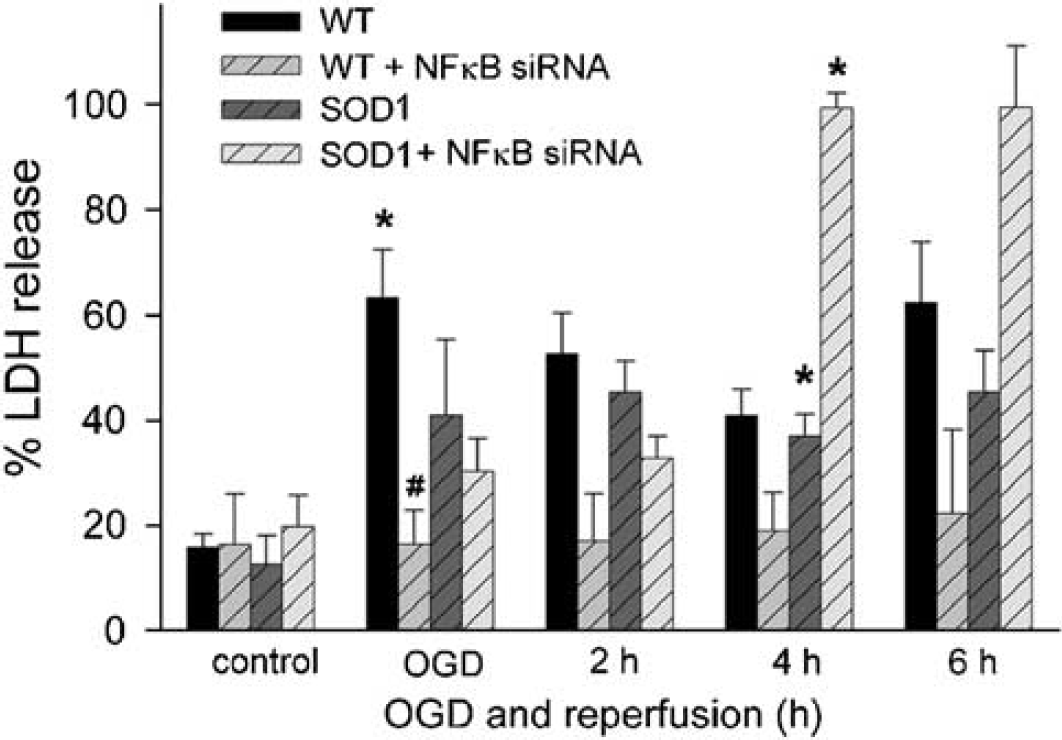
Lactate dehydrogenase release after OGD injury in WT and SOD1 Tg astrocytes transfected with NF-kB p50 siRNA. WT and SOD1 Tg astrocytes were transfected with NF-kB p50 siRNA oligonucleotides as described in Materials and methods. They were then exposed to 4 h of OGD, followed by reperfusion until 6 h. Cell death was assessed by LDH release. Values are expressed as mean ± s.e.m. of three different experiments (n = 6, #P < 0.01, *P < 0.05 indicates statistical significance when compared with control WT or SOD1 Tg astrocytes without NF-kB siRNA transfection).
These results suggest that COX-2 induction might be correlated with NF-kB induction as an early event in ischemic injury in astrocytes. Unlike in the WT astrocytes, inhibiting the expression of NF-kB in the SOD1 Tg astrocytes induced COX-2 expression and aggravated cell death. This suggests that NF-kB might be implicated more in cell survival than in cell death signaling pathways in the SOD1 Tg astrocytes after OGD-induced injury.
Discussion
Oxidative stress is a widespread consequence of a variety of brain insults. Among the prominent and relatively early cellular responses to oxidative stress is the induction of the transcription factor NF-kB (Meyer et al, 1993; Schenk et al, 1994). Our previous studies showed that NF-kB activity increased 60 mins after transient focal cerebral ischemia in mouse brains (Huang et al, 2001), and that oxidative stress downregulated the IKK complex, accompanied by NF-kB activation and IkBα phosphorylation after transient focal cerebral ischemia (Song et al, 2005). Many reports have suggested that ROS regulation of IKK and NF-kB is cell-type restricted, but the molecular mechanisms for the astrocyte-specific NF-kB pathway are not known (Schreck et al, 1991; Meyer et al, 1993; Anderson et al, 1994; Manna et al, 1998; Li and Karin, 1999; Byun et al, 2002). In the present study, we show that OGD injury activates the transcription factor NF-kB through phosphorylation of IKBCX, which finally promotes cell death and COX-2 expression in mouse primary astrocytes. Inhibiting NF-kB activity by siRNA in turn inhibits the expression of COX-2 and increases cell survival.
Astrocytes have a central role in scavenging ROS because they contain high levels of antioxidants and can initially maintain adenosine triphosphate levels via glycolysis (Anderson et al, 2003; Swanson et al, 2004). Superoxide dismutases are known to have a protective role against focal ischemic injury by counteracting the deleterious effects of ROS (Kinouchi et al, 1991; Chan, 1996; Kondo et al, 1997; Murakami et al, 1998). It is well established that astrocyte cultures from SOD1 Tg mice show increased resistance to xanthine oxidase/hypoxanthine and the superoxide generator, menadione (Chen et al, 2001b). Overexpression of SOD1 was protective in some in vivo cerebral ischemia models (Yang et al, 1994; Kondo et al, 1997; Chan et al, 1998) and in astrocyte cultures (Wang et al, 2005).
An important finding of these studies is that NF-kB activation induced by ROS generation was associated with cytoplasmic degradation of IKBCX. Our experiments showed that hypoxia increased phosphorylation of IKBCX at Ser32 and Ser36 in the WT astrocytes, which decreased the level of IKBCX and accelerated NF-kB binding to DNA. Hypoxia followed by reoxygenation has been shown to activate NF-kB p50-p65 heterodimer activity in HeLa cells (Rupec and Baeuerle, 1995). In Jurkat T-cells, hypoxia increased tyrosine phosphorylation of IKBCX, facilitating NF-kB binding to DNA (Koong et al, 1994). Our previous reports showed that, in SOD1 Tg mice that had lower superoxide levels, NF-kB activation and IKBCX degradation were prevented after transient focal cerebral ischemia (Song et al, 2005). Thus, the data from our current study showing lower levels of phosphorylated IKBCX- and NF-kB-binding activity after hypoxic and reoxygenation injury might also be due to lower superoxide levels in the SOD1 Tg astrocytes.
There was an increase in NF-kB p50 proteins in the WT astrocytes and a higher induction of NF-kB p50 in the SOD1 Tg astrocytes at the end of OGD injury. These inductions rapidly decreased after 2 h of reperfusion. Transfection of NF-kB siRNA blocked the transcriptional and translational levels of NF-kB p50 in both the WT and SOD1 Tg astrocytes (Figure 3). However, DNA-binding activity was higher in the WT than in the SOD1 Tg astrocytes after OGD and 6 h of reperfusion (Figure 2). The cellular level of NF-kB p65 was also increased in the WT astrocytes after OGD, which can be supported by our previous findings in neurons (Figure 4A, left panel). Nuclear factor-kB p65 was expressed in the cortex on the ipsilateral side 1 h after focal cerebral ischemia, colocalizing with the neuronal marker, mitogen-activated protein, suggesting that NF-kB p65 is upregulated in certain neurons (Huang et al, 2001). Induction of p65 in the WT astrocytes was not blocked by transfection of NF-kB p50 siRNA (Figure 4B, right panel).
Inhibiting the transcription level of NF-kB by siRNA resulted in protection of the WT astrocytes, whereas it promoted cell death in the SOD1 Tg astrocytes after OGD injury (Figure 8). There are suggestions that NF-kB has a role in the survival of nerve growth factor-dependent neurons by protecting them from apoptosis (Maggirwar et al, 1998). Abrogation of NF-kB, either by deletion of RelA or by overexpression of IkBα, sensitized immune cells to apoptosis in response to tumor necrosis factor a and DNA-damaging agents (Beg and Baltimore, 1996; Liu et al, 1996). There are other reports showing that inhibition of NF-kB in H-Ras-transformed fibroblasts induced apoptosis (Mayo et al, 1997). Overexpression of SOD1 suppressed ischemia-induced activation of NF-kB through a decrease in nuclear translocation, DNA-binding activity, and protein levels, suggesting that superoxide radicals modulate NF-kB activity after focal ischemia in a multistep process (Huang et al, 2001).
Inflammation is a significant source of increased oxidative stress. Cyclooxygenase-2 is believed to contribute to ischemic damage via inflammation and generation of ROS, such as superoxide radicals (Cheung et al, 2002). It has been suggested that superoxide radicals have an important role in the pathogenesis of infarction and edema after focal cerebral ischemia (Kondo et al, 1997). There are reports that COX-2 mRNA and protein expression are upregulated after normoglycemic cerebral ischemia (Kinouchi et al, 1999; Tomimoto et al, 2000). Our study confirmed the increased COX-2 protein expression after OGD and 2 h of reoxygenation in the WT astrocytes.
The NF-kB activation pathway involves proteolytic degradation of the inhibitory protein IKBCX via action of the proteosome complex. Free NF-kB translocates to the nucleus, where it activates the responsive gene (Chen et al, 2001a). There are reports on induction of COX-2 in hypoxia in vascular endothelial cells, and NF-kB control of COX-2 gene transcription (Schmedtje et al, 1997). In human epithelial cells, COX-2 expression was induced by IKKα/β activity and NF-kB activation (Chang et al, 2004). Another report showed that NF-kB activation was mediated via the Ras-dependent signaling pathway, which induces COX-2 transcriptional gene expression in lung epithelial cells (Li et al, 1998). In organotypic hippocampal slice cultures, constitutive NF-kB activity was detected and was correlated with high COX-2 immunoreactivity. The regulation of this COX-2 promoter via NF-kB was solely dependent on the promoter-distal NF-kB 1 site (Kaltschmidt et al, 2002). Our immunostaining results showed expression of COX-2 in the WT astrocytes after OGD. This expression was reduced when the cells were transfected with p50 siRNA (Figure 7A). This result agreed with the decreased expression of the COX-2 protein and decreased COX-2 activity from siRNA-transfected WT astrocytes at the end of OGD, suggesting that NF-kB is responsible for the induction of COX-2 expression in astrocytes after OGD injury (Figures 6 and 7B).
The fact that both NF-kB and COX-2 expressions are induced in WT astrocytes after OGD suggests that hypoxic injury may stimulate COX-2 transcription through NF-kB signaling pathways. From these results, we suggest that an increase in the NF-kB signaling pathway can lead to cell death and COX-2 expression in WT astrocytes.
In the SOD1 Tg astrocytes, the DNA-binding activity of NF-kB was weaker than in the WT cells, which led to less cell death and COX-2 expression in those cells. When siRNAs were transfected to inhibit the transcription activity of NF-kB in the SOD1 Tg astrocytes, the COX-2 level increased and cell death was aggravated. The important protective role of NF-kB in SOD1 Tg astrocytes was further confirmed by enhanced cell death and COX-2 expression, while NF-kB expression was inhibited by siRNA. One possible hypothesis for the marked upregulation of COX-2 and increase in ischemic injury to the SOD1 Tg astrocytes can be supported by a report from Manabe et al (2004). They observed that the COX-2 inhibitor NS-398 provided added neuroprotection when administered with the free radical scavenger superoxide dismutase. The water-and lipid-soluble antioxidant PBN is a ROS scavenger and inhibits NF-kB p50/p65-binding activity (Butterfield et al, 1998). Furthermore, PBN inhibited COX-2 expression (Bazan and Lukiw, 2002) and catalytic activity in a macrophage cell culture (Kotake et al, 1998). Phenylnitrone (PBN) also prevented neoplasia in rat liver not only by its radical scavenging activity but also by inhibiting COX-2 activity at the catalytic level (Nakae et al, 1998). These results coincide very well with our results on increased cell viability after OGD when treated with PBN, suggesting that quenching of free radicals and inhibiting COX-2 activity at the catalytic level during hypoxia in SOD1 Tg and WT astrocytes suppressed cell death.
Induction of NF-kB has been reported in a model of global ischemia (Clemens et al, 1997, 1998). Evidence for NF-kB activation has also been reported in important examples of neurodegeneration, such as Alzheimer's disease (Kaltschmidt et al, 1997; Akama et al, 1998) and Parkinson's disease (Hunot et al, 1997). Zhang et al (2005) recently observed that neuronal activation of NF-kB in cerebral ischemia contributed to ischemic brain damage in vivo. A dual action for NF-kB has been proposed in hippocampal neurons in a global ischemia model, whereby it might be either protective or detrimental depending on the onset and duration of its activation (Clemens et al, 1998). Whether the NF-kB signal transduction pathway, which is highly relevant for inflammatory astrocytic-neuronal signaling in stroke, is protective or detrimental is unknown.
In conclusion, our study showed that the NF-kB signaling pathway is involved in COX-2 expression and cell death in WT astrocytes after OGD injury. Activation of NF-kB in SOD1 Tg astrocytes shifts the balance of cell death-/survival-related proteins in favor of those that induce cell survival. Therefore, NF-kB activation may steer a cell in either of two directions. One induces cell death, and the other induces a cellular defense mechanism, depending on the circumstances and ROS levels in which they are activated. Further studies are needed to address NF-kB regulation in the mediation of the transcription of COX-2 in SOD1 Tg astrocytes and to address the relationship of NF-kB to other survival pathways, such as those involving phosphatidylinositol 3-kinase and AKT (Yao and Cooper, 1995), which are involved in cell survival after ischemic injury.
Footnotes
Acknowledgements
The authors thank Liza Reola and Bernard Calagui for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation.