
Abstract
Nuclear factor-kappa B (NF-kB) is a multisubunit transcription factor that when activated induces the expression of genes encoding acute-phase proteins, cell adhesion molecules, cell surface receptors, and cytokines. NF-kB is composed of a variety of protein subunits of which p50-and p65-kDa (RelA) are the most widely studied. Under resting conditions, these subunits reside in the cytoplasm as an inactive complex bound by inhibitor proteins, IkBα and IkBβ. On activation, IkB is phosphorylated by IkB kinase and ubiquitinated and degraded by the proteasome; simultaneously, the active heterodimer translocates to the nucleus where it can initiate gene transcription. In the periphery, NF-kB is involved in inflammation through stimulation of the production of inflammatory mediators. The role of NF-kB in the brain is unclear. In vitro, NF-kB activation can be either protective or deleterious. The role of NF-kB in ischemic neuronal cell death in vivo was investigated. Adult male rats were subjected to 2 hours of focal ischemia induced by middle cerebral artery occlusion (MCAO). At 2, 6, and 12 hours after reperfusion, the expression and transactivation of NF-kB in ischemic versus nonischemic cortex and striatum were determined by immunocytochemistry and by electrophoretic mobility gel-shift analysis. At all time points studied, p50 and p65 immunoreactivity was found exclusively in the nuclei of cortical and striatal neurons in the ischemic hemisphere. The contralateral nonischemic hemisphere showed no evidence of nuclear NF-kB immunoreactivity. Double immunofluorescence confirmed expression of p50 in nuclei of neurons. Increased NF-kB DNA-binding activity in nuclear extracts prepared from the ischemic hemisphere was further substantiated by electrophoretic mobility gel-shift analysis. Because the activation of NF-kB by many stimuli can be blocked by antioxidants in vitro, the effect of the antioxidant, LY341122, previously shown to be neuroprotective, on NF-kB activation in the MCAO model was evaluated. No significant activation of NF-kB was found by electrophoretic mobility gel-shift analysis in animals treated with LY341122. These results demonstrate that transient focal cerebral ischemia results in activation of NF-kB in neurons and supports previous observations that neuroprotective antioxidants may inhibit neuronal death by preventing the activation of NF-kB.
The transcription factor NF-kB, originally described for its role in B lymphocyte activation, is now known to be present in a wide variety of cell types including those in the central nervous system (Thanos and Maniatis, 1995; O'Neill and Kaltschmidt, 1997). NF-kB is an oxidative responsive transcription factor that can be activated by reactive oxygen species, cytokines, or viruses. When activated, it induces a number of target genes including those encoding cytokines, cell adhesion molecules, oxidative-stress related enzymes, cell surface receptors, and acute-phase proteins. The mechanism of activation has been well characterized in cell culture systems (Lenardo and Baltimore, 1989; Schreck et al., 1992; Baeuerle and Henkel, 1994; Siebenlist et al., 1994). NF-kB is present in the cytoplasm as an inactive multisubunit complex associated with an inhibitory subunit IkB. Stimulation of the cell induces dissociation of the NF-kB/IkB complex by degrading IkB and allowing the nuclear translocation of free NF-kB. Dissociation of IkB from the heterotrimeric complex involves phosphorylation of the IkB subunit which is then degraded by the proteasome. In the nucleus, NF-kB binds to a specific DNA motif and regulates transcription of target genes containing NF-kB consensus sequences in their promoter region. Because activation does not require new protein synthesis, it represents an attractive intracellular mechanism to generate rapid responses.
In the periphery, NF-kB activation has been shown to contribute to immunologically mediated diseases. What role NF-kB plays in the nervous system is not clear. NF-kB in neurons may play a role in synaptic activity (Guerrini et al., 1995; Meberg et al, 1996). Furthermore, neuronal NF-kB is developmentally regulated (Bakalkin et al., 1993). Both neurons and glial cells in vitro can express NF-kB (Sparacio et al., 1992; Moynagh et al., 1993; Rattner et al., 1993; Kaltschmidt et al., 1994a, b , 1995; Guerrini et al., 1995; Carter et al., 1996). In primary cultured neurons derived from embryonic mouse cortex and hippocampus, NF-kB appears to be constitutively activated (Kaltschmidt et al., 1994b). NF-kB has been shown to be activated in multiple types of central nervous system lesions including kainate toxicity (Perez-Otano et al., 1996), convulsant-induced seizures (Prasad et al., 1994), spinal cord injury (Bethea et al., 1998), cerebral infarction (Terai et al., 1996b), and HIV encephalopathy (Rattner et al., 1993) as well as in chronic neurodegenerative conditions such as Alzheimer's disease (Terai et al., 1996a; Kaltschmidt et al., 1997), multiple sclerosis (Gveric et al., 1998), and Parkinson's disease (Hunot et al., 1997).
NF-kB can be activated by numerous factors, many of which are known to be induced after ischemia—reperfusion. These include inflammatory cytokines such as TNFα and IL-1β (Krasnow et al., 1991; Moynagh et al., 1993; Siebenlist et al., 1994), hydrogen peroxide/reactive oxygen species (Schreck et al., 1991; Meyer et al., 1993), increased intracellular calcium (Sen et al., 1996; Pahl and Baeuerle, 1997), and glutamate (Kaltschmidt et al., 1995; Guerrini et al., 1997). After experimental myocardial ischemia-reperfusion, NFkB is activated and inhibition of this activation confers protection from injury (Morishita et al., 1997).
Our previous work demonstrated activation of NF-kB in a rat model of global ischemia (Clemens et al., 1997). The present study was undertaken to determine whether NF-kB is activated in rats subjected to focal ischemia—reperfusion induced by MCAO. Several time points after reperfusion were evaluated to determine a putative time course of activation. Using a combination of gel shift analysis and immunocytochemistry, we sought to determine the identity of the cell type that expressed active NF-kB. Finally, we evaluated the effect of the neuroprotective antioxidant, LY341122 [2-(3, 5-di-t-butyl-4-hydroxyphenyl)-4-(2-(4-methylethylaminomethyl-phenyloxy)ethyl)oxazole], on NF-kB activation in this model.
MATERIALS AND METHODS
Focal ischemia—reperfusion
A total of 37 male Sprague Dawley rats (Harlan, Indianapolis, IN, U.S.A.) weighing 275 to 310 g each were subjected to transient MCAO by the intraluminal filament technique of Zea Longa et al. (1989). Before surgery, the animals were housed in groups of 3 to 4 with food and water available ad libitum. Animal procedures were performed in accordance with the Lilly Research Laboratories animal care and use committee and the guidelines of the National Institutes of Health for the care and use of laboratory animals for experimental procedures. Body temperature was maintained at 37°C with the use of a homeothermic temperature system (Harvard Apparatus, Holliston, MS, U.S.A.). Postsurgery, rats were housed individually. After the initiation of reperfusion, rats were killed at one of the following time points: 2 hours (n = 8), 6 hours (n = 8), and 12 hours (n = 8). Brain tissue from rats killed at each time point was pooled and processed for electrophoretic mobility gel shift analysis (EMSA) (n = 4 per time point) or processed separately for immunocytochemistry (n = 4).
For MCAO, a 3-0 nylon monofilament suture (Harvard Apparatus, Holliston, MS, U.S.A.) was prepared by rounding the tip by heating near burning embers, dipping in poly-
An additional 5 rats were treated with the antioxidant LY341122 [2-(3, 5-di-t-butyl-4-hydroxyphenyl)-4-(2-(4-methylethylaminomethyl-phenyloxy)ethyl)oxazole]. Drug treatment was administered by means of a jugular cannula which was inserted before MCAO surgery. LY341122 was prepared by dissolving in physiologic saline (30 mg/mL), sonicating, and adjusting the pH to 6.0. At 1 hour after occlusion of the MCA, a 10-mg/kg intravenous bolus dose was administered slowly over a period of 2 to 5 minutes followed by constant infusion at the same dose. Infusion (0.1 mL/hour) was carried out for 17 hours at which time the animals were killed and prepared for EMSA. This dosing regime was chosen because Huh et al. (1999) had shown it confers maximal neuroprotection (∼85% reduction in infarct volume) in this model. The time point for termination of the study was chosen because we were interested in evaluating early events that may be related to ischemia/neuroprotection. As a control, rats (n = 4) were treated under the same conditions with the same volume and infusion rate of saline (pH 6.0). At the termination of the study, rats were rapidly decapitated and the ischemic or the contralateral striatum and cortex were frozen separately in liquid nitrogen. In these experiments, rats were processed individually for EMSA because previous studies with chemically related antioxidants have shown that individual rats can vary slightly in their response to drug treatment (Clemens et al., 1998).
Immunocytochemistry. Polyclonal antisera generated against specific regions of NF-kB were used for immunocytochemistry. The antisera Ab392 and Ab567 were kindly supplied by Dr. Warner Greene (Gladstone Institute, UCSF). Ab392 was made against the N-terminal peptide of kBF-1, the p50 homodimer; Ab567 was raised against the N-terminal peptide of p65. These antibodies are highly specific and have been characterized previously (Walker et al., 1992; Doerre et al., 1993). Monoclonal antibodies directed against specific cellular markers were used. MAP-2 (Sigma Immunochemicals, St Louis, MO, U.S.A.) was used at 1:200, GFAP (concentrated antibody, Biogenex labs, San Ramon, CA, U.S.A.) was used at 1:75, and OX42 (Harlan Bioproducts for Science, Indianapolis, IN, U.S.A.) was used at 1:50.
At specific time points after reperfusion, rats for immunocytochemistry were perfused with phosphate-buffered saline followed by ice-cold periodate lysine paraformaldehyde (2%). Excised brains were postfixed by immersion in the same fixative for 24 hours and cryoprotected in 30% sucrose until the tissue was equilibrated. Tissues were rapidly frozen in isopentane chilled with dry ice. Cryosections (16-μm) were thaw mounted onto polylysine coated slides (Fisher Scientific), 3 sections per slide in the coronal plane, throughout the rostro-caudal extent of the MCA territory. For anatomic localization of tissue integrity, every fifth slide was stained with cresyl violet for Nissl substance. Immunocytochemistry was performed using the avidin-biotin-peroxidase system (ABC kit, Vector Labs, Burlingame, CA, U.S.A.). Briefly, tissue sections were incubated with 10% goat serum to block nonspecific staining followed by treatment overnight with primary anti-NF-kB p50 or p65 antiserum (1:500 dilution). Sections were stained with the ABC immunoperoxidase system according to the recommendations of the manufacturer. The reaction product was visualized by development with 3, 3'-diaminobenzidine and H2O2. Positive and negative controls were conducted in parallel with NF-kB stained sections in each experiment. Staining of sections with commercially available antibodies served as the positive control. Negative controls included staining tissue sections with omission of the primary antibody. Stained sections were dehydrated, mounted in Permount, and examined. Photomicrographs were taken on a Nikon Microphot equipped with Nomarsky optics.
Double immunofluorescence. For colocalization experiments on the same tissue section, double immunofluorescence techniques were used. Double staining was performed sequentially as follows: sections were stained first with NF-kB p50 antiserum which was localized using biotin antirabbit followed by avidin-Texas red (Vector Labs) and then with one of the monoclonal antibodies: MAP2 for neurons, GFAP for astrocytes, or OX42 for microglia. The monoclonal antibody was localized using goat antimouse immunoglobulins (high fluorescent conjugate; Antibodies Inc., Davis, CA, U.S.A.). Slides were mounted with fluorescence mounting medium containing DAPI (Vectashield, Vector Labs). Sections were examined using a Nikon Microphot equipped with epifluorescence and Nomarsky optics.
EMSA. For gel shift analysis, animals were rapidly decapitated at specific time points after reperfusion, and tissue was microdissected on ice. In nontreated animals, the cortex and striatum were isolated from each hemisphere and frozen separately in liquid nitrogen. Subsequently, striatum or cortex was pooled from each of the 4 rats killed at each time point and nuclear extracts prepared. For animals treated with LY341122 or vehicle, each side of the brain including both cortex and striatum was processed separately from each animal. Nuclear extracts were prepared from each tissue homogenate. NF-kB DNA binding probes were generated by 5' end-labeling of double-stranded cDNA encoding 5′-CAACGGCAGGGGAA-TTCCCCTCTCCTT-35' with τ-[32P]ATP and T4 polynucleotide kinase. Binding reactions were prepared in a final volume of 20 μL containing 10 mmol/L HEPES, pH 7.5, 50 mmol/L KCl, 1 mmol/L EDTA, 50 μg/mL poly(dI-dC), 0.05% NP-40, 5% glycerol, and 0.1 mmol/L dithiothreotol. Nuclear extracts (20 μg), buffer, and 1 × 105 cpm of radiolabeled NF-kB probe were incubated at room temperature for 15 minutes. Competition reactions were prepared as described, except that 0.1 μg/mL of cold double-strand DNA oligonucleotides was added 15 minutes before the radiolabeled probe. Bound complexes were separated from free probe by electrophoresis in 5% Trisbuffered EDTA nondenaturing polyacrylamide gels and then visualized by autoradiography. The same amount of each sample was loaded based on the protein concentration (Bradford assay, BioRad, Richmond, CA, U.S.A.).
Semiquantitation of the EMSA was performed with densitometry. The X-ray film containing gel shift bands was scanned with Hpscanjet 6200C. The relative intensity of each band was then determined with Kodak digital science 1D image analysis system. The background intensity level was subtracted from the absolute optical density and plotted. Statistical analysis was performed using ANOVA followed by Dunnett's post-hoc test for significance.
RESULTS
Occlusion of the middle cerebral artery (MCAO) in rats produces a large focal cerebral infarct that exhibits a well-characterized histopathologic profile (Osborne et al., 1987; Chiamulera et al., 1993; Clark et al., 1993; Zhang et al., 1994). At 24 hours after MCAO, a circumscribed region of infarction is observed that is characterized by a central core of necrotic tissue and a peripheral area termed the penumbra that is thought to undergo delayed cell death. There is breakdown of the blood-brain barrier and trafficking of peripheral inflammatory cells into the parenchyma (Clark et al., 1993; Garcia et al., 1994; Zhang et al., 1994). The contralateral side is spared from injury and serves as a useful internal negative control. We evaluated NF-kB by immunocytochemistry in the forebrain at early time points after transient MCAO. Nissl staining confirmed the presence of ischemic neurons in the cortex and striatum of rats after transient MCAO (Fig. 1). Immunocytochemistry of NF-kB produced a distinctive pattern of immunoreactivity confined to the region of infarction at 2, 6, and 12 hours after reperfusion (Figs. 2 and 3). Within the infarcted area as well as in the periinfarct region, specific cells expressed specific NF-kB p50 immunoreaction product (Fig. 2). p65 immunoreactivity revealed a staining pattern that was very similar to that observed with p50 antiserum (Fig. 3). Cells scattered throughout the ischemic cortex and striatum expressed p50 and p65 immunoreactivity, and the stain appeared to be distributed in the nucleus of the cell (Figs. 2B, 2D, 3B, and 3D). In the cortex, both pyramidal (Fig. 3D, lamina V) and nonpyramidal neurons (Fig. 2D, lamina II) expressed p50 and p65 immunoreactivity. The contralateral noninfarcted side showed no detectable immunoreactivity with p50 antiserum (Figs. 2A and 2C). A diffuse, cytosolic distribution of stain was observed in the nonischemic cortex with anti-p65 (Fig. 3C). The abundance and distribution of staining with both p50 and p65 antibodies did not appear to differ qualitatively when comparing sections from animals evaluated at the different time points postreperfusion. Moreover, the ischemic striatum and the ischemic cortex appeared very similar with respect to the intensity, distribution, and abundance of immunopositive cells. However, quantitative analysis of immunoreactivity was not carried out in the present study.
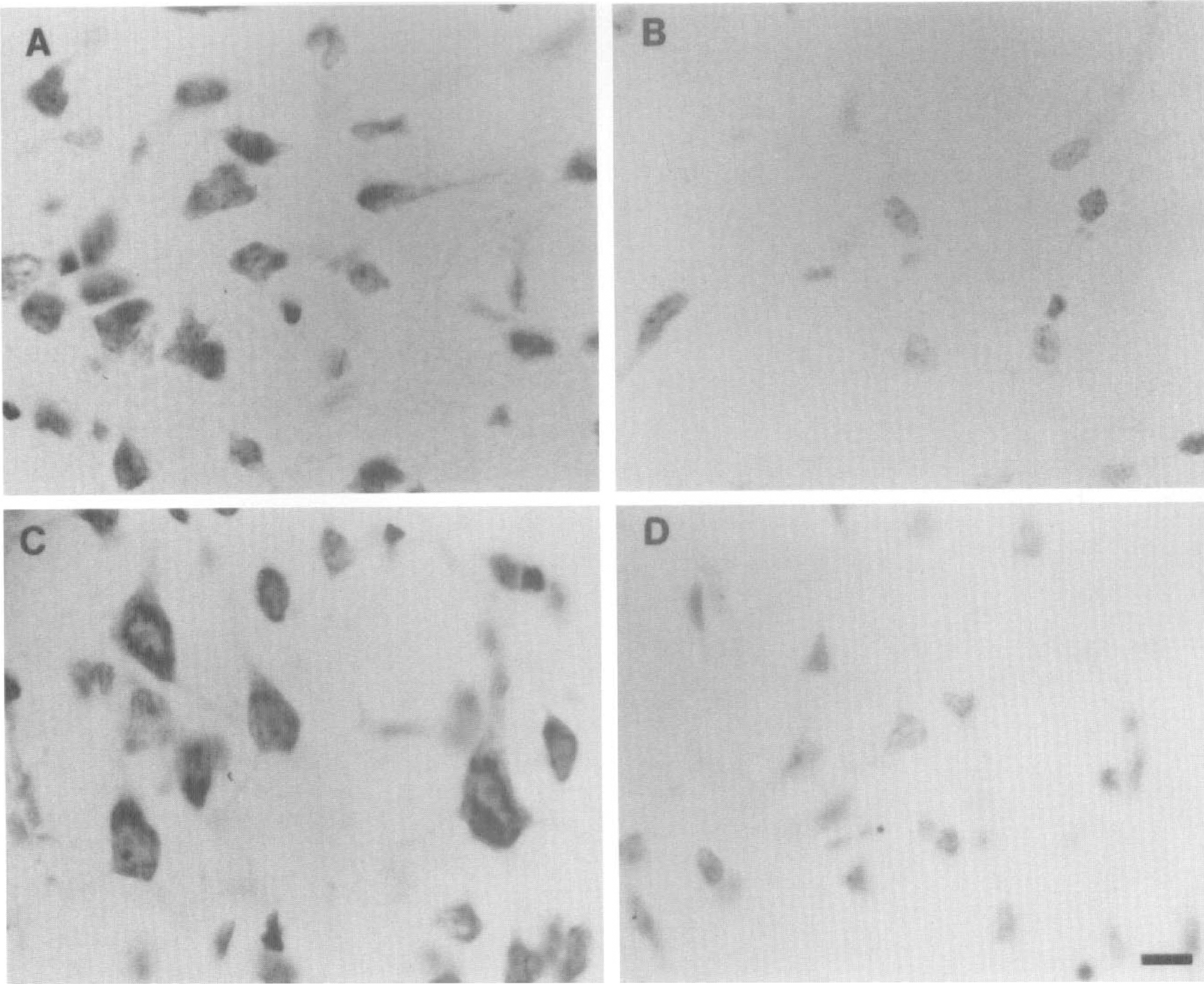
Transient middle cerebral artery occlusion (MCAO) results in ischemic cell change within the infarcted hemisphere. (
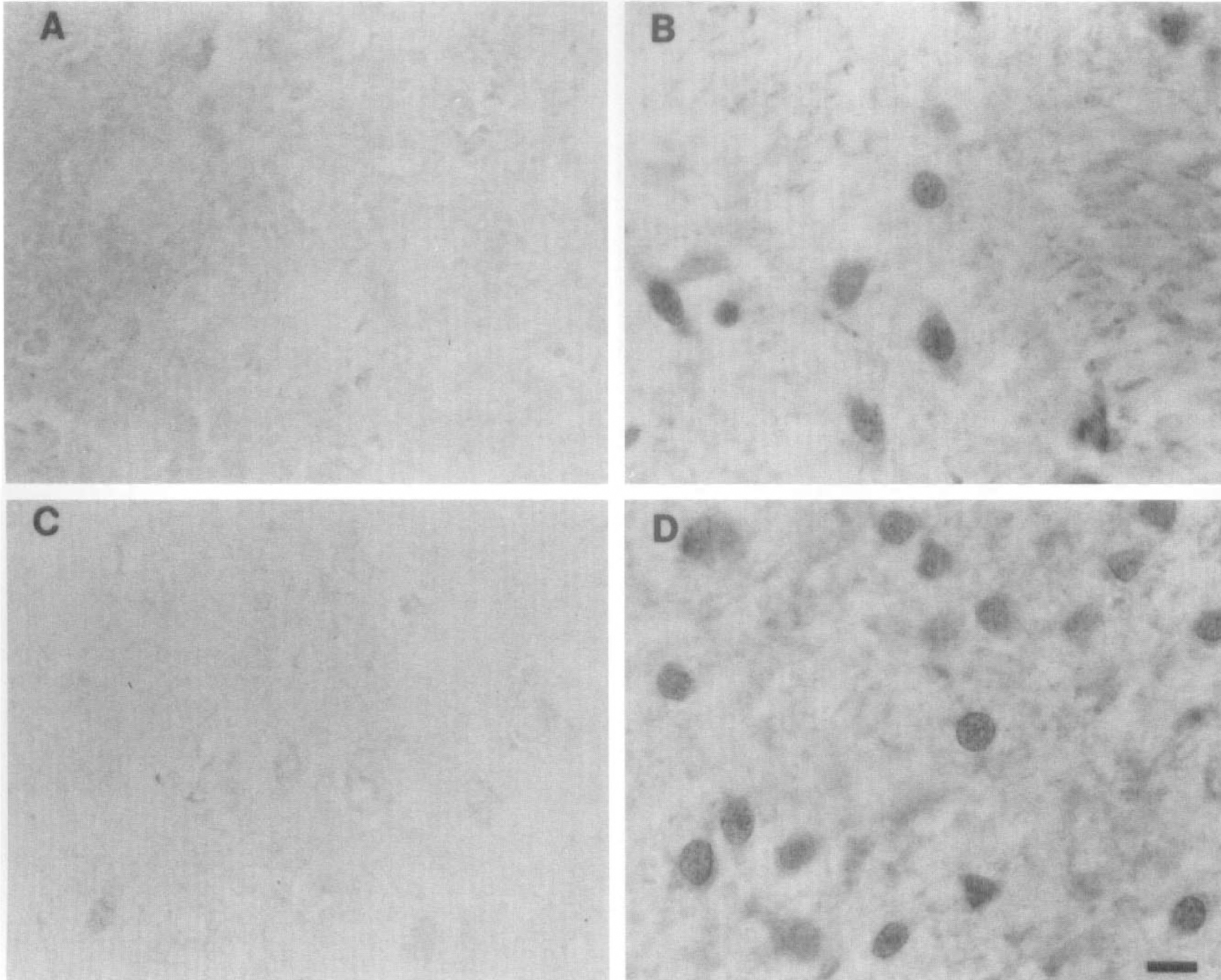
The p50 NF-kB subunit is detected by immunocytochemistry in cells scattered throughout the ischemic cortex and striatum. Immunocytochemistry of p50 subunit in the striatum (
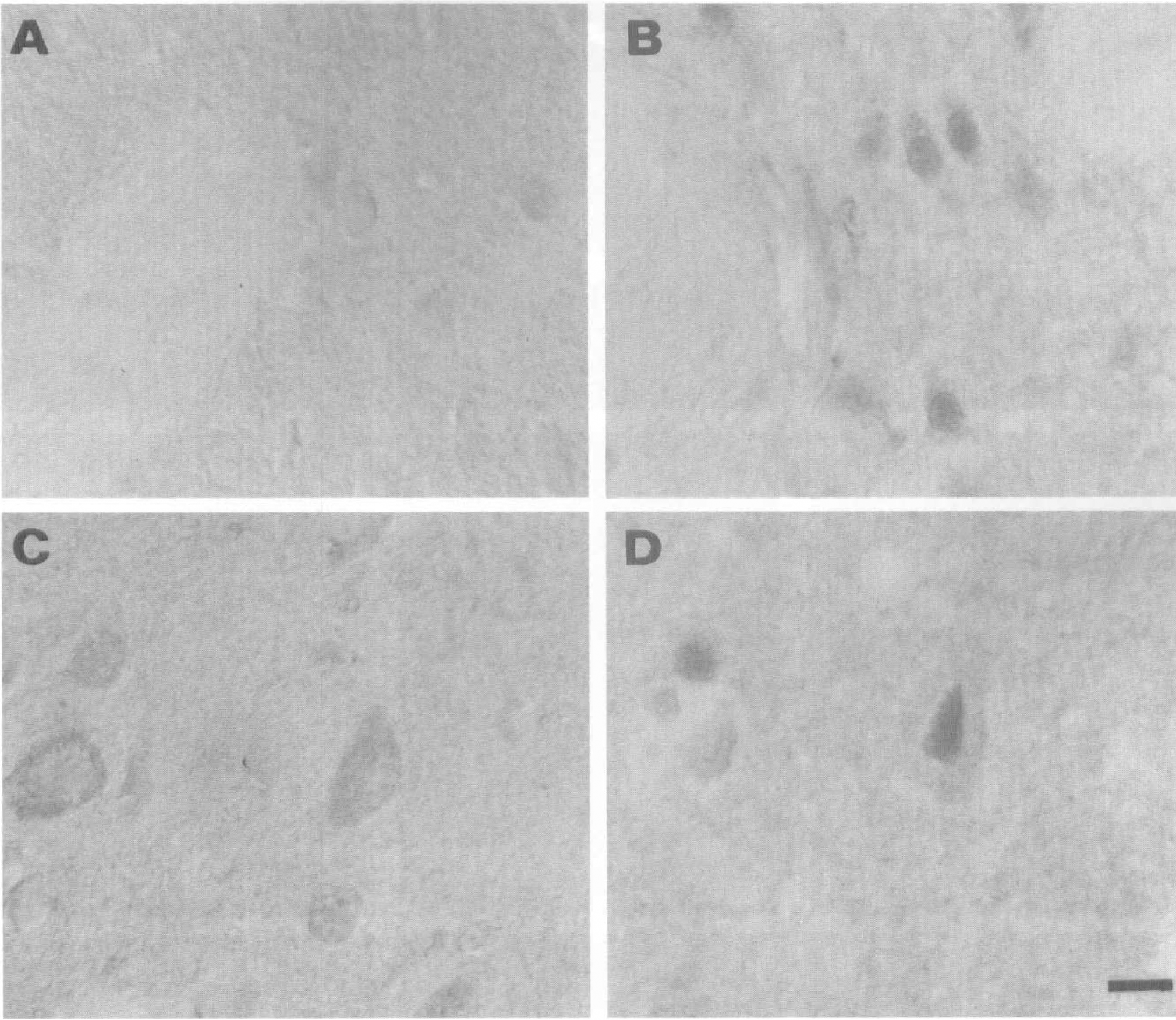
Photomicrographs of p65 immunoreactivity in the brain of a rat at 6 hours after reperfusion. Immunoperoxidase reaction product is localized to presumptive nuclei of scattered cells present in the ischemic striatum (
The pattern of immunoreactivity appeared to be localized to the nucleus of presumptive ischemic neurons, the precise domain of activated transcription factors. Double immunofluorescence techniques were used to determine the cell type and precise cellular location of NF-kB immunoreactivity. The DNA fluorescent dye 4', 6-diamidino-2-phenylindole was used to label nuclei. The choice of which marker to use for double staining was based on the known expression pattern of markers after focal ischemia. Because ischemic neurons stop expressing markers of neuronal integrity (e.g., MAP-2) very early on after ischemia (Dawson and Hallenbeck, 1996), colocalization with a neuronal marker was directed at analysis of the periinfarct region. Within the ischemic core, nonneuronal cells predominate, even though astrocytes degenerate in this region as well (Li et al., 1995). However, at early time points after ischemia, GFAP immunoreactivity can still be observed. Neurons at the border of the MCA territory were found to coexpress both p50 and MAP 2 (Figs. 4A and 4B, arrows). Within the MCA territory, p50 immunoreactivity did not colocalize with the astrocyte marker, GFAP (Figs. 4D and 4E), or the microglial marker OX42 (Figs. 4G and 4H). In all fields, p50 immunofluorescence was found to colocalize precisely with 4', 6-diamidino-2-phenylindole-stained nuclei (Figs. 4A, 4C, 4D, 4F, 4G, and 4I, arrows). p50 immunoreactive nuclei were approximately 10 μm and some, particularly in the striatum, showed evidence of compromised integrity, i.e., they appeared shrunken and deformed (Figs. 4F and 4I, arrows). The morphology and size of immunopositive cells combined with the colocalization data strongly suggest that NF-kB immunoreactivity is present in the nuclei of neurons.

Immunofluorescence colocalization illustrates p50 immunoreactivity is present in the nucleus of neurons. Photomicrographs are taken from a rat at 12 hours after reperfusion. Fields illustrated on the left represent p50 localized using Texas Red conjugated detection system. The middle panel shows staining with monoclonal antibody to either MAP-2 (
We then determined NF-kB transcriptional activation in ischemic tissues by EMSA. A low level of constitutive NF-kB activity was found in the nonischemic hemisphere, suggesting EMSA is a more sensitive measure of transcription factor activation than immunocytochemistry. Increased NF-kB DNA binding activity was observed in the left ischemic striatum and cortex at 2, 6, and 12 hours after reperfusion (Fig. 5A, lanes A and B) with the most dramatic induction observed at 12 hours postreperfusion. At 6 hours, both ischemic (Figs. 5 and 6H, lanes A and B) and nonischemic hemispheres (Figs. 5 and 6H, lanes C and D) showed elevated NF-kB. Two bands were observed by EMSA. Because our previous studies have shown that the upper band consists of NF-kB p65-p50 heterodimers (Clemens et al., 1997), this band was used for quantitative densitometry. Quantitation of the band (Fig. 5A, arrow) showed that there was an approximate 3-fold induction at 2 hours, 1.4-fold induction at 6 hours, and 7-fold induction at 12 hours postreperfusion when comparing the ischemic versus nonischemic hemispheres (Fig. 5B). There was no significant difference between the extent of activation in the cortex as compared to that of the striatum at any of the time points evaluated (Figs. 5A and 5B).
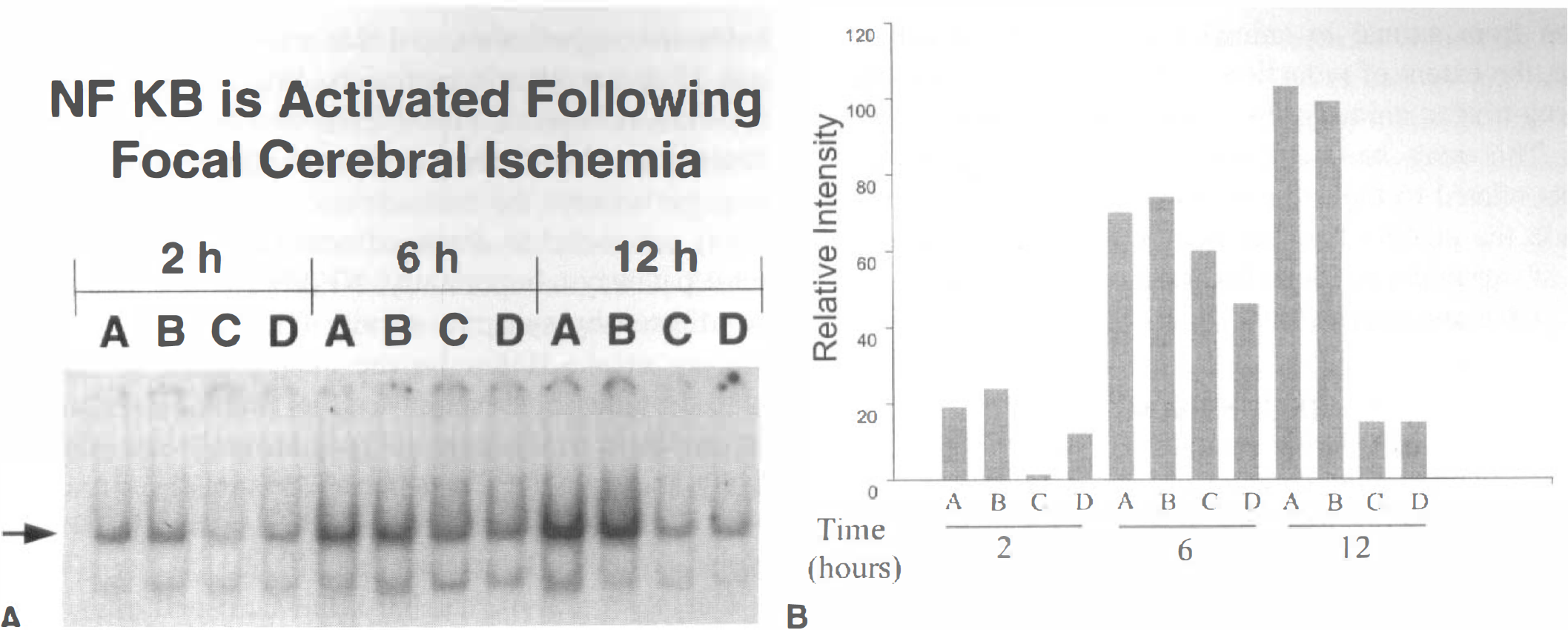
NF-kB DNA-binding activity is increased in the ischemic cortex and striatum after transient middle cerebral artery occlusion (MCAO). DNA binding activity of NF-kB is present at 2, 6, and 12 hours after ischemia-reperfusion. (
The antioxidant, LY341122, is potently neuroprotective in rat models of global as well as focal cerebral ischemia (Clemens et al., 1999; Huh et al., 1999). A structural analog, LY231617, also neuroprotective (Clemens et al., 1993; Block et al., 1995), has been shown to attenuate persistent activation of NF-kB in global ischemia (Clemens et al., 1997). To gain insight into the possible function of NF-kB in focal ischemia, we tested the effect of the antioxidant LY341122 on NF-kB in rats subjected to MCAO. The dosing conditions chosen for these experiments were based on those shown previously to exert the most potent neuroprotection (Huh et al., 1999). Figures 6A and 6B shows that tissues from vehicle-treated animals exhibit a significant activation of NF-kB in the ischemic versus contralateral hemisphere. However, tissues from LY341122-treated rats showed a significant reduction in NF-kB DNA- binding activity in the ischemic hemisphere compared with controls (Fig. 6B). For optimal dosing conditions and detection of early changes after ischemia—reperfusion, animals in this experiment were killed at 16 hours after reperfusion. Densitometric analysis of each band in Fig. 6A shows a statistically significant (P < 0.01) increase in DNA binding activity in the ischemic compared to nonischemic hemispheres in the vehicle control group of animals when pooled (Fig. 6B). In contrast, no significant activation of NF-kB was observed in the ischemic compared to control hemisphere from rats treated with LY341122 (Fig. 6B). The degree of NF-kB activation in the vehicle-treated group of animals varied from animal to animal (Fig. 6A). This is not unexpected given the observation that normotensive rats subjected to MCAO show a considerable degree of variation with respect to the size of lesion from animal to animal (Coyle, 1986). Furthermore, the extent of reduction in NF-kB-binding activity in drug-treated animals also varied from animal to animal. This may be attributable to pharmacodynamic events related to the drug because our previous experience in the model of global ischemia showed some degree of variability in drug efficacy among individual animals (Clemens et al., 1999).
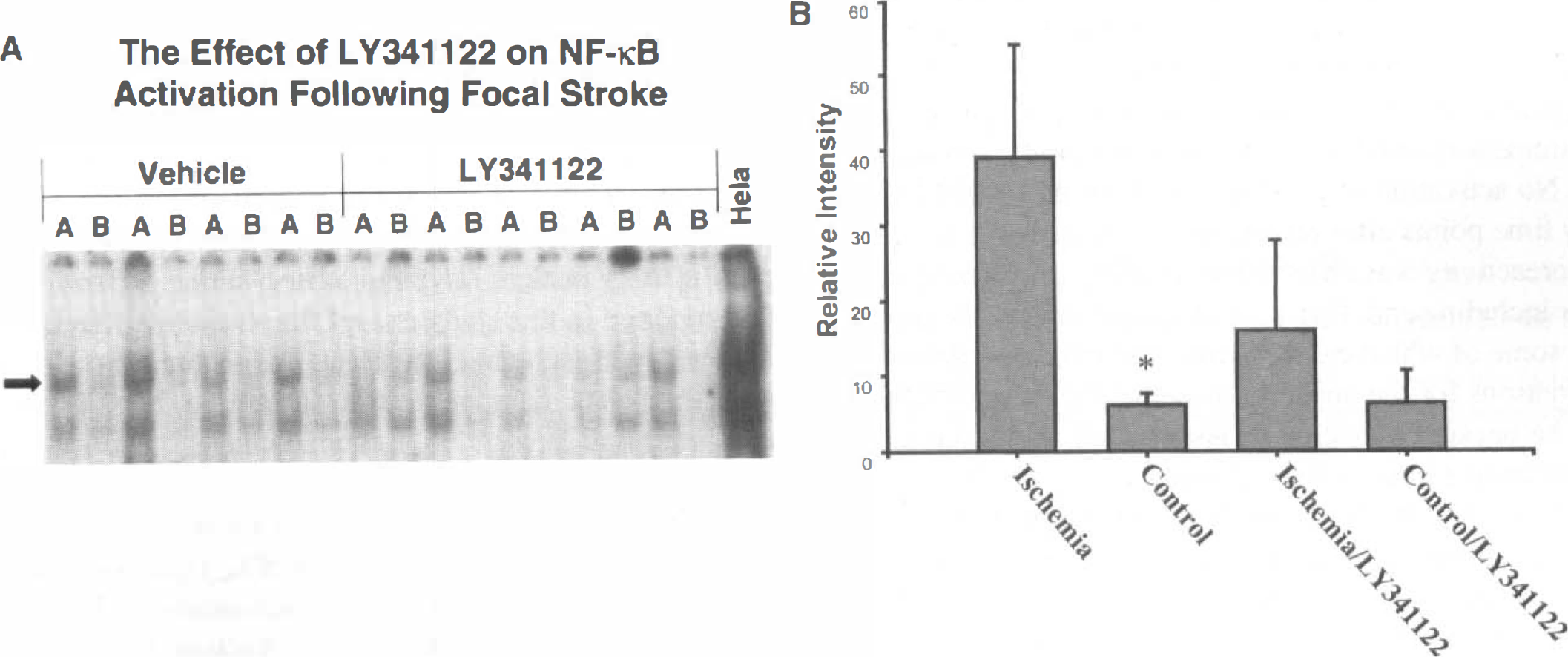
DNA binding activity of NF-kB is not increased in animals subjected to middle cerebral artery occlusion (MCAO) and treated with antioxidant LY341122. (
DISCUSSION
We investigated the changes in NF-kB activity during the early hours after transient focal ischemia. This is an important time window because this is the time during which neuronal death takes place. In the present model, 2 hours of MCAO produces a large infarct in the cortex and striatum. Immunocytochemistry of NF-kB p50 and p65 subunits at 2, 6, and 12 hours after reperfusion demonstrated that NF-kB immunoreactivity was localized to the nuclei of neurons in the ischemic hemisphere. At these time points, NF-kB immunoreactivity was not observed in other cell types. Importantly, the presence of p50 and p65 in the nucleus corresponded to active NF-kB at all the time points because we observed DNA binding activity by gel shift analysis. Electrophoretic mobility gel-shift analysis appears to be a more sensitive index of transcription factor activation compared to immunocytochemistry because constitutively active NF-kB was observed by means of EMSA and not immunocytochemistry, consistent with our earlier observation in global ischemia (Clemens et al., 1997). Our results differ from the study of Gabriel et al. (1999) who observed enhancement of NF-kB binding activity only at 1, 4, and 7 days after the injury, time points during which nuclear p65 immunoreactivity could be observed only in reactive glia. No activation of NF-kB was observed by EMSA at early time points after reperfusion. Furthermore, p65 immunoreactivity was observed in a variety of different cell types including endothelial cells, glia, and some neurons, only some of which exhibited nuclear immunoreactivity. The reasons for the apparent discrepancy are unclear but may be because of technical issues such as the duration of ischemia (1 hour in Gabriel study versus 2 hours in our study), use of different antibodies, or most likely because of the different time points evaluated after ischemia.
EMSA analysis showed a low level of constitutive NF-kB activity in the nonischemic hemisphere which is in agreement with in vitro and in vivo experiments demonstrating constitutively active NF-kB in neurons (Kaltschmidt et al., 1994b). In our study, constitutive NF-kB expression in the nonischemic hemisphere could not be observed as nuclear NF-kB immunoreactivity probably because this level of activation was below the level of detection of the assay. NF-kB DNA binding activity was observed in the ischemic hemisphere at 2 hours after reperfusion, and this activity intensified at 6 and 12 hours after reperfusion. We also observed an apparent increase in NF-kB DNA-binding activity in the nonischemic hemisphere at 6 hours after occlusion. This may be because the nonischemic hemisphere is extensively connected to damaged neurons through commissural pathways. Importantly, NF-kB has been shown to be affected by synaptic activity (Guerrini et al., 1995; Meberg et al., 1996). So the altered synaptic activity caused by the injury might lead to alterations in NF-kB activation in neurons on the contralateral side. Alternatively, there may be a transient widespread activation of NF-kB at an early time point similar to that observed in global ischemia (Clemens et al., 1997).
Our observations that NF-kB is activated after focal cerebral ischemia is consistent with previous studies. However, our observations differ markedly with respect to time course of activation and cell type. Salminen et al (1995) and Gabriel et al. (1999) detected no increase in NF-kB binding activity during the first 24 hours after ischemia. Both studies observed an increase in activation several days after ischemia likely attributable to infiltration of inflammatory cells. Peripheral inflammatory cells as well as glial cells have been shown to possess active NF-kB (Sparacio et al., 1992; Moynagh et al., 1993; Rattner et al., 1993; Baeuerle and Henkel, 1994; Frankenberger et al., 1994; Carter et al., 1996). Our studies at the early time points after ischemia did not observe NF-kB in nonneuronal cells. Thus, at the present time there is no evidence that glia contribute to active NF-kB at early time points after focal stroke. Very early activation of NF-kB was observed by Carroll et al (1998) after 2 hours of MCAO. In that study, the activation of NF-kB increased transiently in the infarcted hemisphere at 15 and 30 minutes after reperfusion. However, by 60 minutes after reperfusion, NF-kB activation returned to baseline and remained at this level for the next 120 hours. At present it is unclear why our observations differ from that reported in this study except for possible differences in severity of ischemia, rat strain, and techniques. However, our data demonstrating that active NF-kB heterodimers are present in ischemic nuclear extracts as well as neuronal nuclei up to 12 hours after reperfusion provide strong evidence to support a longer lasting activation than that reported by Carroll et al. (1998) because the present data use two different measurements for NF-kB activation. Consistent with our observations, activation of NF-kB in neurons has recently been reported at 20 hours after reperfusion in mice subjected to transient MCAO (Schneider et al., 1999).
The localization of p50 and p65 in the nuclei of neurons in the present study bears similarities to our previous findings in the model of global ischemia where NF-kB was localized to nuclei of neurons in the hippocampal CA1 layer after four-vessel occlusion (Clemens et al., 1997). The time course of cell death in this model differs markedly from that which occurs after global ischemia. In the four-vessel occlusion model, it takes several days for histologic evidence of cell death to occur (Pulsinelli et al., 1982) whereas after focal stroke neurons begin to degenerate within hours after the insult (Osborne et al., 1987; Chiamulera et al., 1993; Clark et al., 1993; Zhang et al., 1994). After four-vessel occlusion, we found that only the neurons that were destined to die had persistently activated NF-kB. The remainder of the neurons in the brain that survived exhibited transient NF-kB activation. In the model of global ischemia, cell death could be blocked by treatment with the antioxidant LY231617 (Clemens et al., 1998), a structural analog of LY341122. Antioxidant treatment was found to prevent the persistent rise in NF-kB. However, the transient rise in NF-kB could not be prevented, suggesting that these two rises in NF-kB activation occur by different mechanisms. In the present study using the model of focal stroke, NF-kB activation occurred earlier than that observed after global ischemia likely because of the more rapid time course of cell death. In global ischemia (Clemens et al., 1998), as well as in this study, rats treated with the antioxidant did not show significant NF-kB DNA-binding activity. Other studies have shown that the antioxidant LY341122 is neuroprotective in models of global ischemia (Clemens et al. 1999), traumatic brain injury (Dietrich et al., 1997), and focal stroke (Huh et al., 1999). Because the above models are all associated with long-lasting increases in NF-kB (Clemens et al., 1997, Bethea et al., 1998; Gabriel et al., 1999), the mechanism of neuroprotection of LY341122 may be mediated by attenuation of persistent NF-kB activation. In vitro studies support this idea because antioxidants that block activation of NF-kB (Schreck et al., 1991; Meyer et al., 1993) can protect neurons against oxidative stress-induced cell death (Behl et al., 1994; Kaltschmidt et al., 1997).
It is interesting that some studies performed in cultured cells report that NF-kB activation can exert a protective effect (Barger et al., 1995; Beg and Baltimore, 1996; Liu et al., 1996; Van Antwerp et al., 1996; Mattson et al., 1997; Tagliartela et al., 1997; Lezoualc'h et al., 1998; Tamatani et al., 1999), but other in vitro studies report that NF-kB activation is necessary for cell death to occur (Lin et al., 1995; Grilli et al., 1996; Grimm et al., 1996). Few studies have addressed the putative function of NF-kB activation in vivo. Morishita et al. (1997) reported that inhibition of NF-kB binding decreases damage from myocardial infarction. Qin et al. (1998) and Nakai et al. (1999) reported that NF-kB contributes to excitotoxin-induced apoptosis in rat striatum. Phillips et al. (1998) reported that inhibition of NF-kB activation by proteasome inhibition dramatically reduced infarct size after MCAO. Most recently, a study published by Schneider et al. (1999) demonstrated that mice with targeted deletion of p50 exhibit reduced infarction when subjected to focal ischemia (Schneider et al., 1999). These in vivo studies provide evidence that NF-kB activation contributes to neuronal degeneration. Interestingly, the in vivo conditions where NF-kB seems to contribute to neuronal cell death seem to be conditions where reactive oxygen species have been proposed to be the initiators of the cascade of events that ultimately results in cell death. Substantial evidence exists for a link between reactive oxygen species, apoptosis, and cell death (Buttke and Sandstrom, 1994; Gorman 1996; Jacobson et al., 1996).
In conclusion, the results of our study demonstrate that NF-kB is progressively activated at 2, 6, and 12 hours after reperfusion in a model of MCAO. The EMSA data which demonstrates active NF-kB heterodimers in nuclear extracts prepared from the ischemic hemisphere support our immunocytochemical data showing localization of p50 and p65 subunits in the nucleus. The cell type which demonstrates nuclear NF-kB subunits is the neuron. Induction of nuclear p50 and p65 was seen in neurons confined to the region of ischemia. Double-labeling experiments showed that nuclear NF-kB subunits were present in neurons that exhibited compromised nuclear morphology. Importantly, the antioxidant LY341122, which has been shown to reduce ischemic neuronal death resulting from global (Clemens et al., 1993) and focal ischemia (Huh et al., 1999), can also reduce the activation of NF-kB in this model. This study, in combination with the recent p50 knockout data (Schneider et al., 1999), suggests that NF-kB plays a deleterious role in the pathology of cerebral ischemia.
Footnotes
Acknowledgments
The authors thank Rosa Simmons for assistance with data analysis and Dr. Marcelle Bergeron for review of the manuscript.