
Abstract
Several studies have suggested that cyclooxygenase-2 (COX-2) plays a role in ischemic neuronal death. Genetic disruption of COX-2 has been shown to reduce susceptibility to focal ischemic injury and N-methyl-D-aspartate-mediated neurotoxicity. The purpose of this study was to examine the effects of COX-2 deficiency on neuronal vulnerability after transient forebrain ischemia. Marked upregulation of COX-2 immunostaining in neurons was observed at the early stage and prominent COX-2 staining persisted in the CA1 medial sector and CA2 sector over 3 days after ischemia. The immunohistologic pattern of COX-2 staining in these sectors gradually condensed to a perinuclear location. The degree of hippocampal neuronal injury produced by global ischemia in COX-2–deficient mice was less than that in wild-type mice, coincident with attenuation of DNA fragmentation in the hippocampus. Also, treatment with a selective COX-2 inhibitor, nimesulide, after ischemia decreased hippocampal neuronal damages. These results of genetic disruption and chemical inhibition of cyclooxygenase-2 show that inhibition of COX-2 ameliorates selective neuronal death after transient forebrain ischemia in mice.
Cyclooxygenase (COX) is a key enzyme in the synthesis of prostaglandins. Two isoforms, COX-1 and COX-2, have been identified, and COX-2 is expressed constitutively at relatively high levels in brain and kidney (Yamagata et al., 1993). COX-2 expression is induced immediately after global (Koistinaho et al., 1999; Ohtsuki et al., 1996) or focal ischemia (Collaco-Moraes et al., 1996). Prolonged COX-2 expression has been reported in vulnerable neurons after global ischemia (Matsuoka et al., 1999) and in neurons of the ischemic penumbra region after focal ischemia (Nogawa et al., 1997). The effects of genetic manipulation of COX-2 on focal ischemia have recently been reported. The genetic disruption of COX-2 reduced cerebral infarction after middle cerebral artery occlusion (Iadecola et al., 2001), and neuronal overexpression of COX-2 enhanced cerebral infarction (Dore et al., 2003). COX-2 has also been shown to promote neuronal apoptosis (Andreasson et al., 2001; Mirjany et al., 2002). Apoptosis is one of the crucial mechanisms underlying ischemic brain injury in response to global ischemia (Kitagawa et al., 1998a; Nitatori et al., 1995; Okamoto et al., 1993). Chemical inhibitors of COX-2 reduce ischemic brain injury in transient forebrain ischemia (Candelario-Jalil et al., 2002b; Nakayama et al., 1998). It remains unclear whether genetic manipulation of COX-2 expression mitigates selective neuronal vulnerability after transient global ischemia. In this study, we investigated the effect of genetic disruption of COX-2 on neuronal vulnerability to transient forebrain ischemia.
MATERIALS AND METHODS
Cyclooxygenase-2-deficient mice were obtained from Jackson laboratories (Dinchuk et al., 1995). Mice were backcrossed with C57Black/6 mice five to seven times and studied at 10 to 12 weeks of age. Experiments were performed with age-matched littermates to minimize any effects deriving from genetic background. These included wild-type mice (+/+) and those homozygous for knockout (–/–). The genotypes of all COX-2–knockout mice were determined by polymerase chain reaction (PCR) analysis. The PCR primers were 5′-ATCTCAGCACTGCATCCT-3′, 5′-CACCATAGAATCCAGTCCGG-3′, and 5′-CTTGGGTGGAGAGGCTATTC-3′, amplifying a 922-bp product in the wild-type allele and a 1,200-bp product in the disrupted allele. For subsequent experiments, the genotype was not identified during the experiment and was confirmed postmortem by PCR amplification of tail genomic DNA and by immunoblotting of cerebral homogenates with a polyclonal antibody against murine COX-2 (Cayman Chemical, Ann Arbor, MI, U.S.A.). The experimental protocol was approved by the Institutional Animal Care and Use Committee of Osaka University Graduate School of Medicine. The animals were fed standard laboratory chow and had free access to water before and after all procedures. Animal care was given according to the guidelines of Animal Center of Osaka University Graduate School of Medicine.
General anesthesia was maintained with 1% halothane by means of an open facemask. A polyacrylamide column for measurement of cortical microperfusion by laser-Doppler flowmetry (LDF; Advanced Laser Flowmetry) was attached to the skull, 3 mm lateral to bregma on the right side, with dental cement. Body and skull temperatures were monitored and maintained at 36.5°C to 37.5°C with a heat lamp and mat. Both common carotid arteries (CCAs) were occluded for 12 minutes with microaneurysm clips and then reperfused. As described previously, only mice that showed less than 13% of baseline control microperfusion during the first minute of occlusion were used in subsequent experiments (Kitagawa et al. 1998b). Twenty-five mice met the criteria during the first minute and were subjected to extended bilateral CCA occlusion for an additional 11 minutes without interruption. After discontinuation of halothane anesthesia, each mouse was allowed to recover for 12 hrs in a 37°C chamber to prevent hypothermia, and then each mouse was kept at room temperature.
Samples of the hippocampus of both COX-2–deficient mice and wild-type littermates in control and 24 hours after ischemia were isolated. Proteins were separated by sodium dodecyl sulfate – polyacrylamide gel electrophoresis, and electrophoretically transferred to polyvinylidene difluoride sheet (Immunobilon P; Millipore, Bedfold, MA, U.S.A.). Blots were probed with a monoclonal COX-1 antibody (Cayman Chemical) and a polyclonal COX-2 antibody, then detected using either sheep antirabbit or antimouse horseradish peroxidase-conjugated secondary antibody (Amersham Pharmacia Biotech, Buckinghamshire, UK) followed by enhanced chemoluminescence (ECL; Amersham Pharmacia Biotech).
Mice were killed for COX-2 immunohistochemistry 1, 3, and 7 days after ischemia, and the brains were removed quickly and frozen at −80°C. Ten-micron coronal cryostat sections were fixed for 2 minutes in absolute methanol at −20°C. After a washing, sections were incubated with a 1:200 dilution of rabbit polyclonal anti-COX-2 antibody (Cayman Chemical) diluted with TBS/0.1% TritonX-100 containing 1.5% normal donkey serum at 4°C overnight, and then incubated with appropriate anti-immunoglobulin G secondary donkey antibodies conjugated to fluorescein isothiocyanate for 90 minutes at room temperature. After rinsing with Tris buffer, sections were mounted with Vector-shield (Vector Laboratories, Burlingame, CA, U.S.A.) and visualized or photographed with a confocal microscopy system (Zeiss LSM-410, Oberkochen, Germany).
At 7 days after ischemia, mice were subjected to deep pentobarbital anesthesia, perfused transcardially with 4% paraformaldehyde, and the brains were removed and fixed in 4% paraformaldehyde at 4°C overnight. For staining with cresyl violet and in situ terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end-labeling (TUNEL) of fragmented DNA, each tissue block was dehydrated after fixation and embedded in paraffin. Tissue sections encompassing the dorsal hippocampus, 1.9 mm caudal to bregma, were examined after the staining with cresyl violet. For TUNEL labeling of fragmented DNA, sections were treated with 10-μg/mL proteinase K in Tris/edetic acid buffer for 10 minutes at 37°C and then with 2% hydrogen peroxide for 5 minutes. The sections were immersed in TdT buffer for 15 minutes at 37°C and then incubated with fresh TdT buffer containing 0.3-U/μL TdT and 0.01-mmol/L biotin-11-dUTP for 60 minutes at 37°C. The reaction was terminated by transferring the sections to buffer consisting of 300-mmol/L NaCl and 30-mmol/L of sodium citrate for 10 minutes. After being washed, the sections were incubated with avidin-biotin complex (Vector Laboratories) for 45 minutes at room temperature and then reacted with 0.05% 3′3-diaminobenzidine in the presence of 0.01% H2O2.
Mice were killed 7 days after ischemia and semiquantitative evaluation of the degree of ischemic damage in the hippocampus was performed as described previously (Buchan and Pulsinelli, 1990; Kitagawa et al., 1998a). Briefly, the degree of damage was assessed in the CA1 to CA3 sector on the basis of the percentage of damaged cells as follows: grade 0, no cell damage visible; grade 1, less than 10% of cells damaged; grade 2, 10% to 50% of cells damaged; grade 3, more than 50% of cells damaged. The distance from the CA1 to CA3 sector was measured with each grade, and mean histologic score was obtained for each mouse by dividing the integration of each grade and its length by the total length from the CA1 to CA3 sector. The grading of neuronal damages was undertaken with an observer who was blinded to the COX-2 genotypes and the treatment group.
Mean histologic score was determined by the following equation:
(1 × length with grade 1) + (2 × length with grade 2) + (3 × length with grade 3)/ total length of the CA1 to CA3 sector
To investigate the effect of a COX-2 inhibitor on neuronal damage after global ischemia, both right and left common carotid arteries were occluded for 12 minutes as described above. Ischemic mice were divided randomly into two groups. Nimesulide (N- (4-nitro-2-phenoxyphenyl)-methanesulfonamide; Cayman Chemical) at a dose of 6 mg/kg (n = 8) or vehicle alone was given intraperitoneally at 1, 12, 24, 48, and 72 hours of reperfusion. Nimesulide, a highly selective COX-2 inhibitor, has been widely used to treat inflammatory conditions and fever (Candelario-Jalil et al., 2002b) and known to cross the blood-brain barrier in both humans and rodents (Taniguchi et al., 1997). The dose and treatment schedule were based on previous reports (Candelario-Jalil et al., 2002b; Nakayama et al., 1998). At 7 days after ischemia, mice were killed and histologic analyses were undertaken as described above.
Data are reported as mean ± SD. To analyze differences between proportions of histologic damage in each hippocampal sector, we used the Fisher exact test. The difference in mean histologic score between wild-type and COX-2–deficient mice was evaluated statistically by the Mann-Whitney U test and considered significant at P < 0.05.
RESULTS
There were no significant differences in any of the parameters, including CBF and rectal and skull temperatures, between COX-2–deficient mice and their wild-type littermates (Figs. 1A and 1B) during and after transient forebrain ischemia. Western blot analysis of hippocampal homogenate showed a major band at 72-kd corresponding to the size of COX-2 protein (Fig. 1C). The level of COX-2 was low in the normal hippocampus; however, COX-2 proteins were markedly upregulated 24 hours after ischemia in wild-type mice. COX-2 protein expression was not detectable in COX-2–deficient mice (Fig. 1C). In contrast, there was no significant difference in COX-1 expression between COX-2–deficient mice and wild-type littermates (Fig. 1D). Also, the levels of COX-1 protein were similar in control and ischemic hippocampus (Fig. 1D). These data showed that there was no compensatory upregulation of COX-1 protein in COX-2–deficient mice.
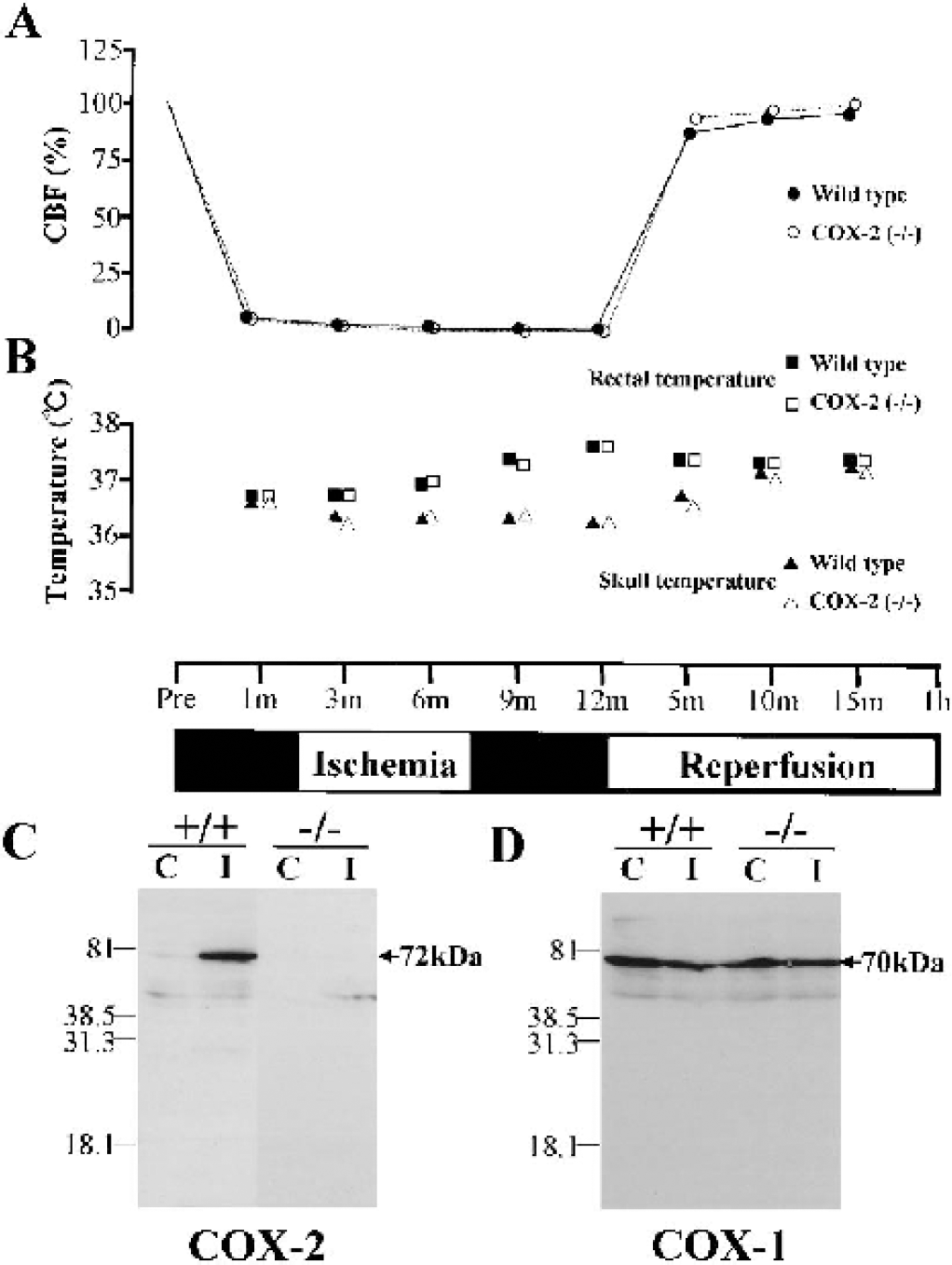
Physiologic parameters during and after transient forebrain ischemia and analysis of COX-2 and COX-1 expression in hippocampus of COX-2–deficient mice and wild-type littermates. Effect of bilateral common carotid artery (CCA) occlusion on CBF (
In control animals, neuronal localization of COX-2 immunoreactivity was evident and was relatively strong in the CA2 and CA3 sector of the hippocampus (Figs. 2D and 2G). Only a faint immunoreactivity was observed in the CA1 subregion (Fig. 2 A). The immunoreactivity showed a characteristically perinuclear localization, as reported previously (Morita et al., 1995). At 24 hours after ischemia, COX-2 staining was dramatically increased in the whole hippocampus (Figs. 2B, 2E, and 2H) including CA1 to CA3 sector and dentate gyrus. At 72 hours after ischemia, COX-2 immunoreactivity was persistently increased in scattered neurons of CA1 sector and in the CA2 sector (Figs. 2C and 2F), whereas the level of COX-2 immunoreactivity returned to the basal level in CA3 hippocampal sector and dentate gyrus. COX-2 immunoreactivity in CA1 and CA2 sectors was morphologically pyknotic and condensed to a perinuclear location (Figs. 2C and 2F). At 7 days after ischemia, marked and long-lasting COX-2 immunoreactivity was still observed in a few pyknotic cells of both the CA1 medial sector and the CA2 sector (data not shown).
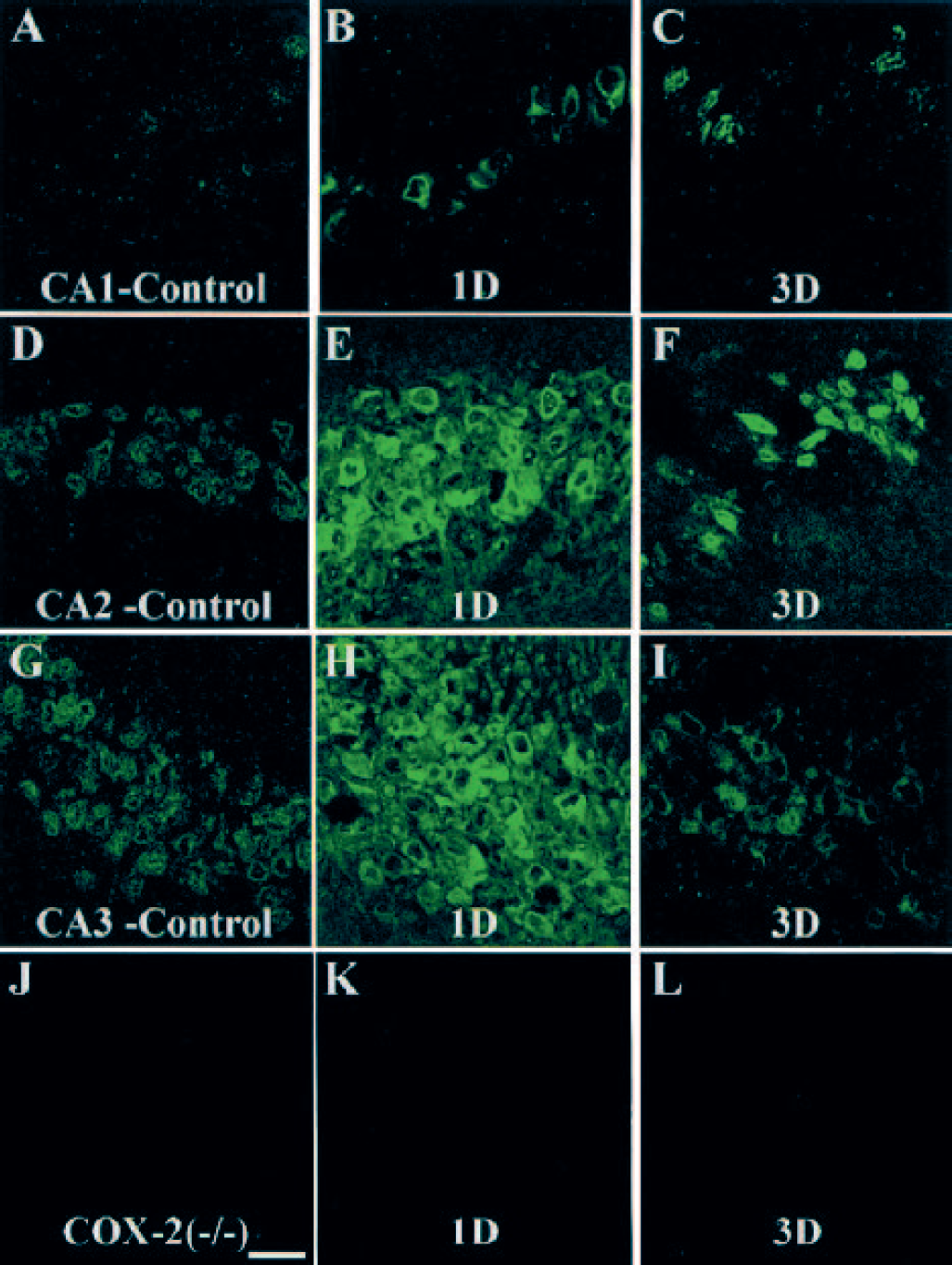
Distribution of COX-2 immunoreactivity in the CA1, CA2, and CA3 sector of the hippocampus after global ischemia (
Ischemic neuronal damage in hippocampal neurons in COX-2–deficient mice, as evaluated by Nissl staining, was less than that in wild-type littermates (Figs. 3A and 3B). In addition, neuronal damage in the hippocampus was identified concomitantly with DNA fragmentation in both strains of mice. Fewer scattered damaged cells labeled with TUNEL were observed in COX-2–deficient mice than in wild-type littermates (Figs. 3C and 3D).
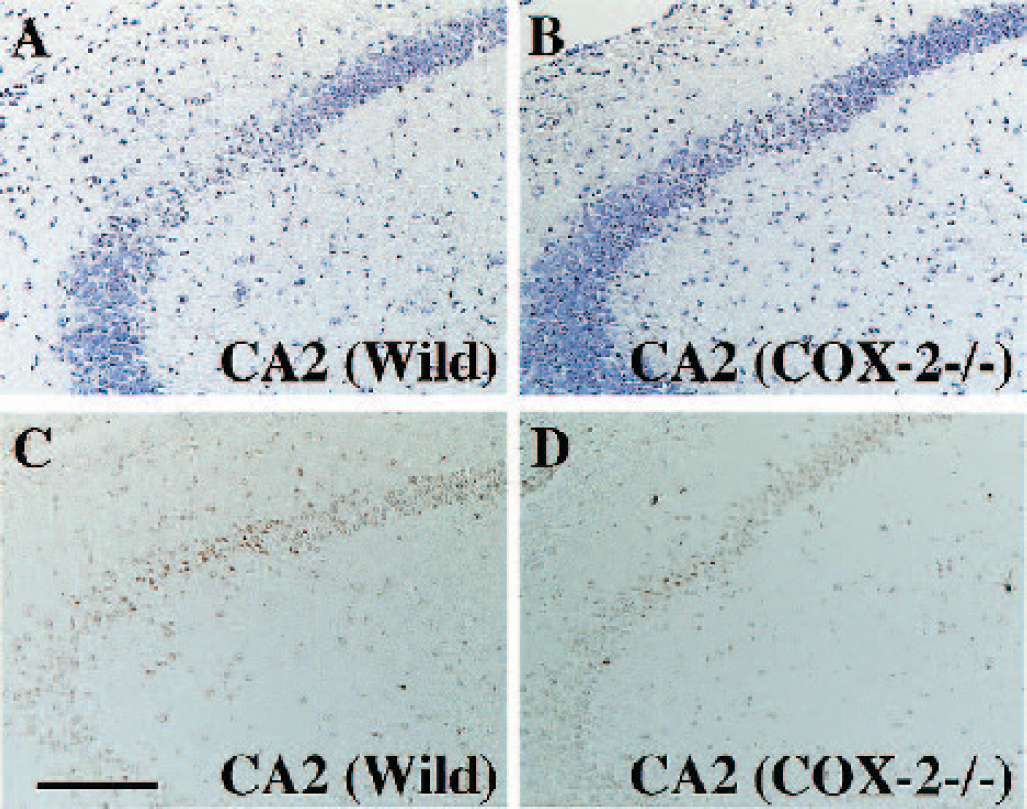
Histologic assessment of hippocampal pyramidal neurons after transient forebrain ischemia. Cresyl-violet staining showed less hippocampal neuronal damage in COX-2–deficient mice (COX-2 –/–) (
The frequency of neuronal damage observed in the CA1 and CA2 sector in COX-2–deficient mice was less than that observed in wild-type mice. The difference in the CA2 sector was significant (Fig. 4). By semiquantitative analysis in the CA1 to CA3 sector as evaluated by Nissl staining, expressed as mean histologic score, neuronal injury in COX-2–deficient mice was significantly reduced compared to that in wild-type littermates (Fig. 5).
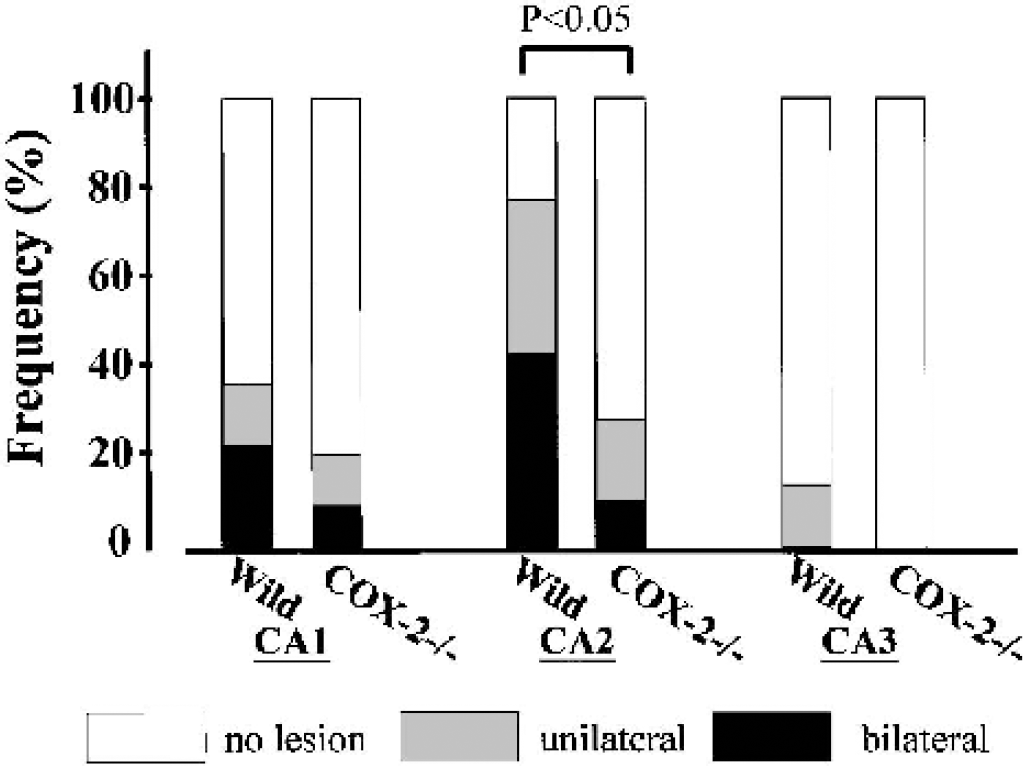
Frequency of neuronal damage after transient forebrain ischemia in the CA1 to CA3 sector of the hippocampus. The frequency of ischemic neuronal damage identified unilaterally and bilaterally is shown. The frequency of neuronal damage in the CA1 and CA2 sector of COX-2–deficient mice (COX-2 –/–) was lower than in wild-type mice (Wild). The difference in the CA2 sector was statistically significant.
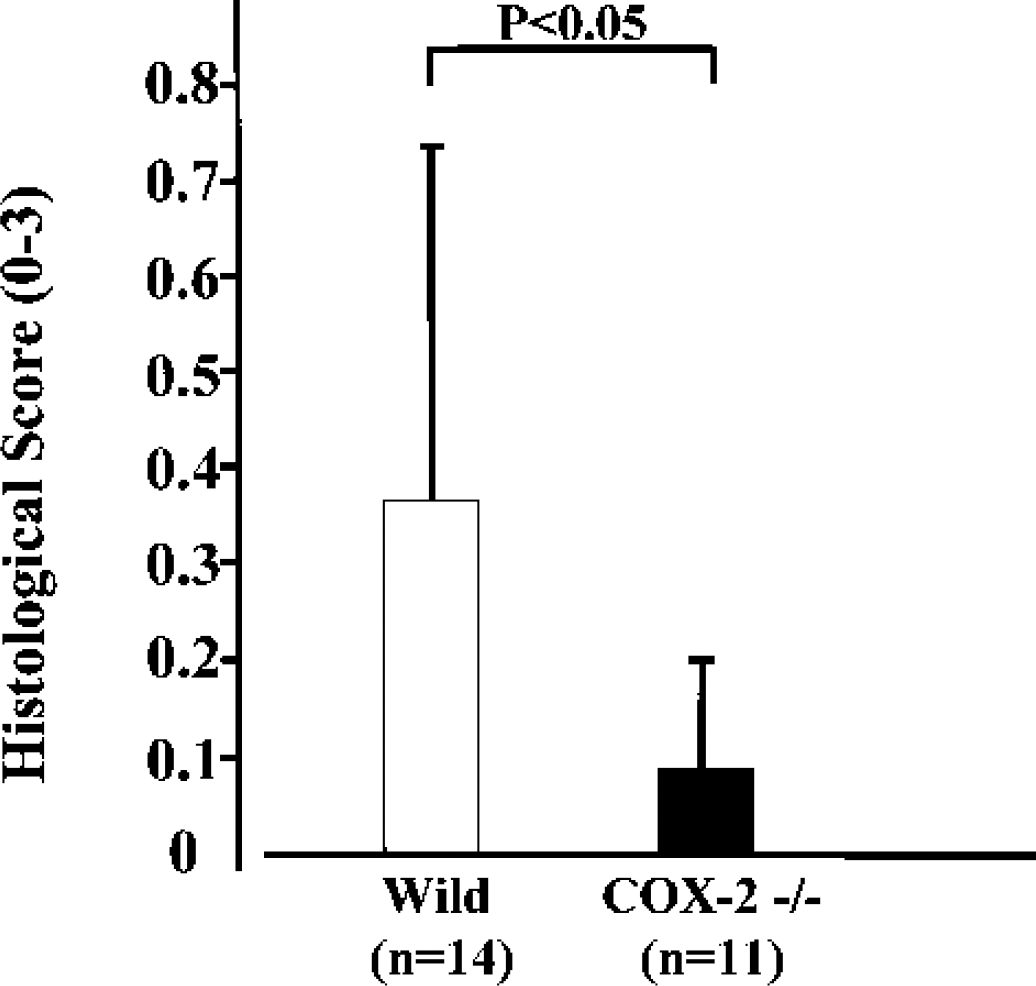
Semiquantitative assessment of ischemic neuronal damage in the hippocampus after transient forebrain ischemia, as evaluated by Nissl staining. The histologic score for each mouse was obtained by dividing the integration of each grade and its length by the total length of the CA1 to CA3 sector. The mean histologic score was significantly lower in COX-2–deficient mice (COX-2 –/–, n = 11) than in wild-type mice (Wild, n = 14) (007 ± 0.14 vs. 0.36 ± 0.42).
The treatment with nimesulide after ischemia significantly attenuated neuronal damage in the CA1 to CA3 sector by semiquantitative analysis as described above (Fig. 6). There were no significant differences in any of the parameters, including CBF and rectal and skull temperatures, between vehicle-treated and nimesulidetreated groups (data not shown).
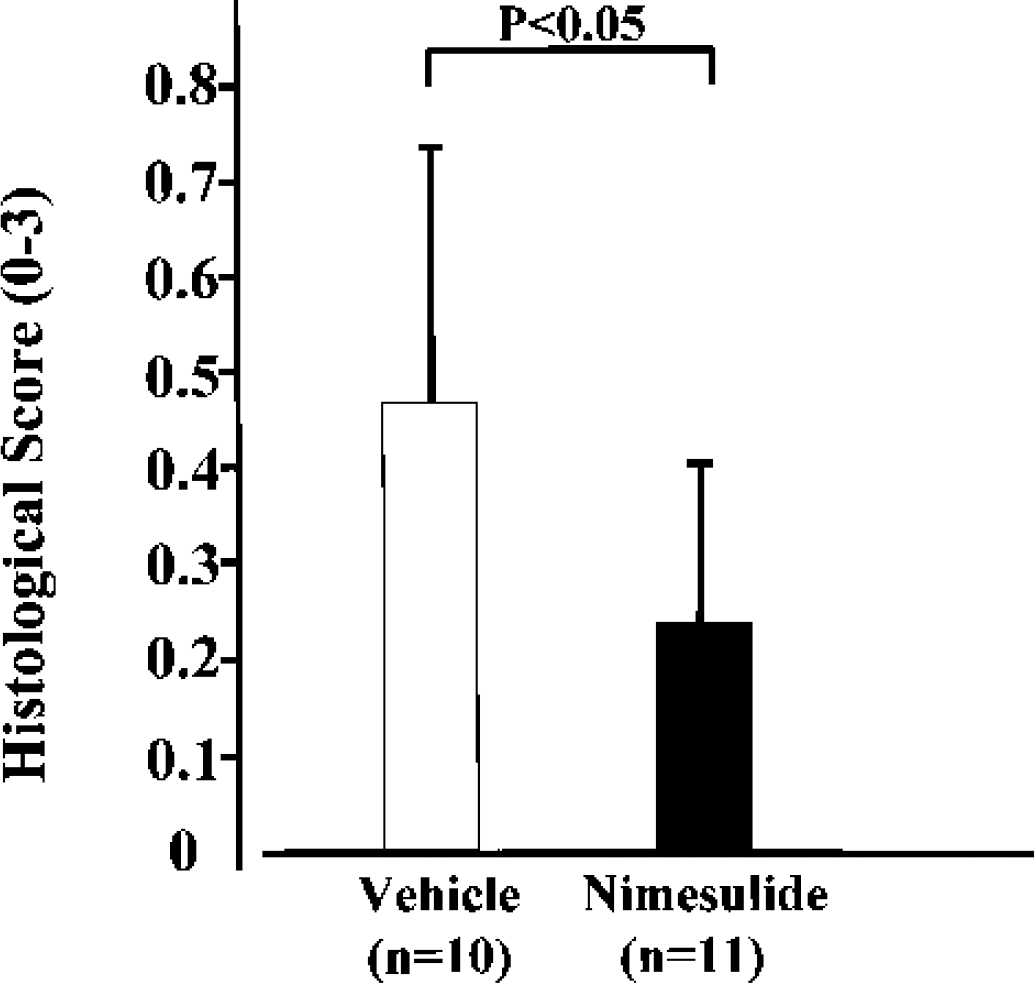
Treatment with the COX-2–specific inhibitor nimesulide decreased hippocampal neuronal damage after transient forebrain ischemia. Nimesulide (6 mg/kg) (n = 11) or vehicle (n = 10) was administered intraperitoneally at 1, 12, 24, 48, and 72 hours of reperfusion. At 7 days after ischemia, mice were killed for histologic analysis. Semiquantitative assessment of ischemic neuronal damage in the hippocampus after transient forebrain ischemia was evaluated by Nissl staining. The mean histologic score in the nimesulide group (n = 11) was significantly lower than that in vehicle group (n = 10) (0.25 ± 0.17 vs. 0.47 ± 0.28, P < 0.05).
DISCUSSION
Here, we showed that genetic disruption of COX-2 and the use of a COX-2–selective inhibitor ameliorated hippocampal neuronal damage after transient forebrain ischemia. No significant difference in COX-1 protein expression after brain ischemia between wild-type and COX-2–deficient mice was found, consistent with the similar COX-1 mRNA expression in COX-2–deficient mice (Iadecola et al., 2001). These data show that there is no compensatory upregulation of COX-1 protein in COX-2–deficient mice. We previously established the C57BL/6 strain to be the best strain for producing reproducible forebrain ischemia by bilateral CCA occlusion due to the lack of patency of the posterior communicating artery (Kitagawa et al., 1998a), as previously reported (Fujii et al., 1997; Murakami et al., 1997). Importantly, Iadecola et al. (2001) reported no alteration in the anatomy of large cerebral vessels in COX-2–deficient mice. Because of variable neuronal damage observed after global ischemia in mice, semiquantitative evaluations of damaged neurons have been performed in the mouse brain (Kitagawa et al., 1998a; Murakami et al., 1997). Also, our previous study suggested the possibility that neurons in the CA2 hippocampal subregion were more vulnerable than those in the CA1 sector (Yang et al., 2000). In this study, the levels in increased COX-2 immunoreactivity after ischemia were more prominent in the CA2 sector than in the CA1 sector. These findings suggest that COX-2 protein activities are closely correlated with selective neuronal death after transient forebrain ischemia in mice.
The reduction and recovery of CBF in LDF did not differ between COX-2–deficient and wild-type mice. Because the CBF value measured in LDF provides relative changes in CBF but does not indicate absolute CBF value, the CBF measurements obtained with LDF should be evaluated carefully. Particularly, it has been shown that COX-1, COX-2, or prostaglandins can impact CBF (Niwa et al. 2000; Niwa et al., 2001). Recently, neuron-to-astrocyte signaling via COX product was shown to be centrally involved in dynamic control of brain microcirculation (Zonta et al., 2003). However, we have previously shown that CBF changes in cortical perfusion detected by LDF at 1 minute after bilateral CCA occlusion were closely correlated with functional anastomosis at the circle of Willis, ATP depletion after vessel occlusion, and ischemic neuronal death 7 days after ischemia (Kitagawa et al., 1998a). Additionally, Iadecola et al. (2001) also showed that the genetic disruption of COX-2 did not impact on blood flow regulation. Therefore, it is unlikely that the severity of ischemic insults or altered vascular function contribute to the neuronal protection observed in COX-2–deficient mice.
Among the few ischemia-induced genes that reached the protein expression level because of the inhibition of protein translation, COX-2 was induced at an early stage and translated into protein. COX-2 was expressed before the death of neurons, and the induction was persistent increased over 3 days, when neurons died. Thus, the COX-2 expression pattern overlapped with the temporal profile of neuronal death after global ischemia. Therefore, these findings support the notion that COX-2 activity is closely correlated with selective neuronal vulnerability in mice after transient forebrain ischemia. In addition, their marked COX-2 immunostaining pattern was morphologically pyknotic and was condensed to a perinuclear location. The present study also showed a decrease in the number of TUNEL-positive cells in COX-2–deficient mice, suggesting the involvement of COX-2 with apoptosis in neuronal death after global ischemia. Mirjany et al. (2002) recently showed that COX-2 overexpression potentiated glutamate-mediated apoptotic neuronal damage and that the COX-2–selective inhibitor nimesulide significantly attenuated apoptotic damage. However, TUNEL labeling simply showed DNA damage, and neuronal death in rat hippocampus after transient global ischemia was shown to be caused by both apoptosis and necrosis (Sugawara et al., 1999). Therefore, the evaluation of TUNEL labeling in cell death after transient forebrain ischemia must be done carefully.
In focal ischemia, inflammation and microcirculatory disturbances play important roles in the expansion of cerebral ischemia (Barone and Feuerstein, 1999; Kochanek and Hallenbeck, 1992; Vogel et al., 1999). Cellular responses of astrocytes, microglia, and endothelial cells to ischemia could play substantial roles in determining infarct size. COX-2 immunoreactivity has been noted in astrocytes (Hirst et al. 1999) and in microglia (Basu et al., 2002) after brain injury, and it has also been detected in brain vascular cells (Schiltz and Sawchenko, 2002; Yamagata et al. 2001). Although the neuroprotective effects of COX-2 inhibitors are more beneficial when administered early after ischemic insult, COX-2–selective inhibitors show a wide therapeutic window for the prevention of neuronal death in both focal and global ischemia (Candelario-Jalil et al., 2002a,b; Nakayama et al., 1998; Sugimoto and Iadecola, 2003). Considering that microglial and astroglial activation occurs after transient forebrain ischemia (Morioka et al., 1991; Tsuda et al., 1996), COX-2 expression in these cells may be also involved in tissue injury.
In recent studies, COX-2–deficient mice showed attenuated glutamate-mediated neurotoxicity (Iadecola et al., 2001), and transgenic mice overexpressing neuronal COX-2 showed increased infarct volume after transient focal ischemia (Dore et al., 2003) or enhanced glutamate-mediated cytotoxity (Kelley et al., 1999), suggesting a cause-effect relationship between neuronal COX-2 expression and excitotoxicity. A recent study indicated that neuronal COX-2 promoted glutamate-mediated excitotoxicity through cell cycle activity (Mirjany et al., 2002). Our present results with transient forebrain ischemia support the notion that COX-2 is directly involved in neuronal injury, suggesting the use of targeting of neuronal COX-2 in brain ischemia therapy.
The present study is the first to show an effect of COX-2 gene disruption on neuronal injury after transient forebrain ischemia, and provides additional evidence that COX-2 is closely involved in ischemic neuronal death. Further studies are necessary to determine the time window and site of action of COX-2 for therapeutic benefit.
Footnotes
Acknowledgements
The authors thank R. Morimoto and S. Imoto for secretarial assistance.