
Abstract
There are no effective treatments for stroke. The activation of endogenous protective mechanisms is a promising therapeutic approach, which evokes the intrinsic ability of the brain to protect itself. Accumulated evidence strongly suggests that electroacupuncture (EA) pretreatment induces rapid tolerance to cerebral ischemia. With regard to mechanisms underlying ischemic tolerance induced by EA, many molecules and signaling pathways are involved, such as the endocannabinoid system, although the exact mechanisms have not been fully elucidated. In the current study, we employed mutant mice, neuropharmacology, microdialysis, and virus transfection techniques in a middle cerebral artery occlusion (MCAO) model to explore the cell-specific and brain region-specific mechanisms of EA-induced neuroprotection. EA pretreatment resulted in increased ambient endocannabinoid (eCB) levels and subsequent activation of ischemic penumbral astroglial cannabinoid type 1 receptors (CB1R) which led to moderate upregulation of extracellular glutamate that protected neurons from cerebral ischemic injury. These findings provide a novel cellular mechanism of EA and a potential therapeutic target for ischemic stroke.
Introduction
Ischemic stroke is the second most common cause of death worldwide and the leading cause of disability in both the United States and China.1–4 The most-prescribed treatments of ischemic stroke, recommended by current guidelines, are the intravenous thrombolysis of tissue plasminogen activator (tPA) and mechanical thrombectomy. 5 Administration of alteplase between 4.5 and 9.0 hours after stroke onset, or at the time the patient awakes with stroke symptoms, results in a higher percentage of patients with no, or minor, neurologic deficits than after administration of placebo. 6 However, the successful delivery of mechanical thrombectomy requires specialized stroke centers and trained professional staff. 7 Thus, the need for effective stroke prevention and treatment is an urgent medical problem.
It has been suggested that preconditioning methods, such as HBO preconditioning, 8 lipopolysaccharide, 9 brief ischemia 10 and peroxisome proliferator-activated receptor-alpha activation, 11 evoke endogenous protective mechanisms as a potential treatment for stroke. 12 Electroacupuncture (EA), based on traditional acupuncture combined with modern electrotherapy, is a relatively safe, cheap, and straight forward therapy and widely accepted by patients. Animal studies have shown that EA pretreatment may produce neuroprotective effects.13,14 Our previous work has demostrated that EA pretreatment exerts a neuroprotective effect by reducing the ischemic infarct volume, which is accompanied by an increase in neurological scores in rats. 15 Moreover, EA pretreatment elicited the eCBs, which subsequently activated the cannabinoid 1 receptor (CB1R) to produce cerebral ischemia protection.16–18 These studies suggest that EA pretreatment may be a promising clinical application for cerebral ischemia protection and that the eCB system may regulate ischemic tolerance induced by EA. However, the specific neural mechanisms underlying regulation by the eCB system of ischemic tolerance induced by EA pretreatment, such as the specific neurological cells that release eCBs and express the CB1R that modulate the neuroprotective effect, have not yet been determined.
Endocannabinoids, such as 2-arachidonoylglycerol (2-AG) and anandamide, influence a broad range of functions, including learning, memory and neuroprotection.19,20 For example, diacylglycerol lipase alpha (DAGLα) is responsible for the biosynthesis of 2-AG, 21 the most abundant eCB in the brain. Postsynaptic neuron-released 2-AG acts retrogradely on presynaptic CB1R, which is the most abundant G protein-coupled receptor in the central nervous system,22,23 to produce its specific functions. Classically, CB1R was found to be mainly present in presynaptic terminals, 24 mostly in GABAergic interneurons but it is also found at low levels on glutamatergic neurons and astrocytes. 25
Astroglial CB1R play an important role in maintaining many brain functions, such as the memory formation of cannabis addiction, 26 the cannabis-induced impairment of spatial memory 27 and the rapid anti-anxiety of fatty acid amide hydrolase (FAAH) inhibition. 28 Our recent work demonstrated that JZL195 induced long-term depression (LTD) by astroglial CB1R in vivo and produced a neuroprotective effect against ischemic insult. 29 This suggests a crucial role of astroglial CB1R in ischemic tolerance in the brain. However, whether astroglial CB1R are involved in EA has not yet been shown.
Excessive glutamate release plays an important role in the pathophysiology of brain ischemia. 30 Excitatory amino acid transporter 2 (EAAT2) is primarily found on perisynaptic processes of astrocytes 31 and is responsible for approximately 90% of the content of excitatory neurotransmitter glutamate in the synaptic gap. 32 Knockdown of EAAT2 exacerbates the damage of transient middle cerebral artery occlusion-induced ischemic stroke. 33 In contrast, overexpression of EAAT2 enhances neuroprotection following moderate oxygen glucose deprivation in vitro 34 and reduces infarct volume in a middle cerebral artery occlusion (MCAO) model in vivo. 35 Kano et al. demonstrated that activation of astroglial CB1R increased the concentration of extracellular glutamate. 36 Therefore, we hypothesize that an increase of ambient eCB induced by EA pretreatment, as well as the subesequent activation of ischemic penumbral astroglial CB1R, up-regulates EAAT2 and then leads to the moderate upregulation of extracellular glutamate, ultimately producing neuronal protection against lethal ischemic insults, which will benefit those with a high risk of ischemic stroke in the perioperative period.
Materials and methods
Animals
Male C57BL/6J mice at 6-8 weeks old and weighing 22-25 g were used (Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China). They were housed under a 12:12 light-dark cycle with room temperature maintained at 22 ± 1°C and allowed free access to food and water. The experimental protocol used in this study was approved by the Fourth Military Medical University Animal Ethics Committee and was conducted in accordance with the Guidelines for Animal Experimentation of the Fourth Military Medical University (Xi'an, China), and reported according to the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines. 37 All efforts were made to minimize both suffering and the number of mice used.
Generation of mutant mice
CB1R-floxed,29,38 vGLUT1-iCreERT238 and GAD2-iCreERT238 mutant mice were generated by Beijing Biocytogen Co., Ltd., (Beijing, China), as described previously. GFAP-CreERT229,39 mutant mice were purchased from Model Animal Research Center of Nanjing University.
The CB1R-floxed mice were crossed with vGLUT1-iCreERT2, GAD2-iCreERT2 or GFAP-CreERT2 mutant mice to generate vGLUT1-CB1R-KO, GAD2-CB1R-KO and GFAP-CB1R-KO mouse lines using protocols described previously.29,38,39 These seven mouse lines and all iCreERT2 mice received eight daily injections of tamoxifen (Sigma, Missouri, USA, 10 mg/ml, 0.1 ml/d, dissolved in 90% sunflower oil, i.p.) two weeks before experiments.
Electroacupuncture
Mice were anesthetized using 1.0 MAC (1.4%) isoflurane at 1.0 L/min with 100% oxygen and stimulated with EA (G6805-2 EA Instrument, Model No. 227033, Qingdao Xinsheng Ltd., Qingdao, Shandong, China) at the Baihui acupoint (GV 20), which is located at the intersection of the sagittal midline and the line linking the right side of the mouse ears. An intensity of 1 mA and frequency of 2/15 Hz were selected for the EA administration as described in our previous work. 15 A fine needle (0.5 mm diameter) placed at GV 20 was connected to one EA stimulator electrode and to the other electrode on the right ear. Tremor of the ears during administration was regarded as effective EA stimulation. Mice core temperatures of the were maintained at 37.0 ± 0.5°C by surface heating or cooling during anesthesia and EA.
Induction of transient focal cerebral ischemia
Under isoflurane anesthesia (3% for induction and 1.4% for maintenance; maintenance under 100% O2), the right-hand common carotid arteries were exposed, isolated from the adjacent nerve and tissue, and then occluded for 60 minutes using microclips (0.23 mm × 0.126 mm/3 cm, RWD Inc., Shenzhen, China) to induce transient focal cerebral ischemia, which was followed by reperfusion. To prevent the occurrence of hypothermia during surgery, rectal temperature was maintained at 37 ± 0.5°C using a heating pad. Seventy-two hours following reperfusion, all experimental animals were sacrificed for further experimental procedures.
Neurological evaluation and 2, 3, 5-Triphenyltetrazolium chloride staining
Seventy-two hours after reperfusion, a neurological assessment of the mice in different groups was performed by an investigator blind to experimental conditions, using the 18-point scoring system reported by Gracia et al. 40 The system consisted of spontaneous activity, side stroking, vibrissa touch, limb symmetry, and climbing and forelimb walking assessments. After neurological evaluation, mice were decapitated, and brains were rapidly removed and mildly frozen to keep morphology intact during slicing. Infarct volume was measured as described previously.15,41 In brief, the brain was rapidly dissected and sectioned into six coronal blocks in a brain matrix with an approximate thickness of 1 mm and stained with 2% (w/v) 2, 3, 5-triphenyltetrazolium chloride (TTC) (Sigma, Missouri, USA) for 30 minutes at 37°C followed by overnight immersion in 4% (w/v) paraformaldehyde. The infarct tissue area remained unstained (white), whereas normal tissue was stained red. Infarct areas on each slice were demarcated and analyzed using image pixels and Adobe Photoshop CC (2014; Adobe, San Jose, CA, USA). The total infarct volumes were calculated according to the following formula: ((total contralateral hemispheric volume) - (total ipsilateral hemispheric stained volume))/(total contralateral hemispheric volume) × 100%.
Mouse surgery
Male C57BL/6 mice or mutant mice (weighing 22–25 g) underwent surgeries for implantation of a guide cannula for a microinjection needle and microdialysis needle. During pentobarbital anesthesia (50 mg/kg, intraperitoneal), a guide cannula (O.D. 0.48 × I.D. 0.34, RWD Inc., Shenzhen, China) for the microinjection needle (O.D. 0.30 × I.D. 0.14, RWD Inc., Shenzhen, China) was directed stereotaxically at the lateral ventricles. A guide cannula (Eicom Corporation, Kyoto, Japan) for microdialysis (membrane length, 1 mm; OD, 0.22 mm; cut off size, 20 kDA; Eicom Corporation, Kyoto, Japan) was directed stereotaxically at the ischemic penumbra. The tip of the microinjection and microdialysis needle was 1 mm longer than the tip of the guide cannula. The coordinates of the probe tip according to the Paxinos and Watson mouse brain atlas were AP −0.4 mm from bregma, ML +1.0 mm, DV –2.5 mm (lateral ventricles) and AP +0.5 mm, ML –1.2 mm, DV −0.8 mm for the ischemia penumbra. Mice were permitted to recover for at least seven days before any treatment.
Microdialysis
Animals were single-housed following implantation of the microdialysis probe for the remainder of the experiment. During dialysis, aCSF solution (145 mM NaCl, 2.8 mM KCl, 1.2 mM MgCl2, 1.26 mM CaCl2, and 5.4 mM d-glucose (pH 7.4)) was pumped through the microdialysis probe at 1 µL/min. Dialysis samples were collected every 30 minutes and stored on dry ice during the experiment, and then samples were stored at −80°C until analyzed for glutamate content using liquid chromatography coupled with mass spectrometry. After a 2-hour balance period, seven samples were collected per mouse. The first sample collected was used as a baseline. Then, one sample was collected when mice were randomly assigned to an electroacupuncture (EA) group or sham electroacupuncture (sham) group during isoflurane anesthesia for 30 minutes. The subsequent five samples were gathered after the EA/sham treatment when mice were freely moving. Samples were stored at −80°C until detection analysis.
High perforance liquid chromatography analysis of glutamate
The concentrations of glutamate were analyzed using High Performance Liquid Chromatography (HPLC) (Agilent 1100, Agilent, California, USA) with flurescence detection. The microdialysis samples were precolumn derivatized with 2,4-dinitrofluorobenzene for 30 minutes at 60°C, and then 50 mM potassium dihydrogen phosphate was added to each microdialysis sample mixture to stop the reaction. The microdialysis sample mixtures were then filtered using a 0.22-µm filter (Agilent C18; 4.6 × 250 mm, 5 µm). The mobile phase consisted of 99% methanol and 0.1 M sodium acetate buffer (pH 8.0) at a flow rate of 1.0 mL/min, and the column temperature was maintained at 35°C. A UV detector was operated at an excitation wavelength of 250 nm and an emission wavelength of 410 nm to analyze neurotransmitter concentrations in the microdialysis samples. Concentrations were calculated using Aglient Chemstation (B.04.02 SP1) based on standard samples (Sigma-Aldrich, Missouri, USA).
DREADD strategy
We used the Designer Receptors Exclusively Activated by Designed Drugs (DREADD) technique to modulate the activity of ischemic penumbra astrocytes. All DREADD viruses, including AAV2/9-EF1α-DIO-mCherry-WPRE-pA (AAV-mCherry), AAV2/9-EF1α-DIO-hM3Dq-mCherry-WPRE-pA (AAV-hM3Dq-mCherry) and AAV2/9-EF1α-DIO-hM4Di-mCherry-WPRE-pA (AAV-hM4Di-mCherry) were purchased from the Brain VTA company (WuHan, China). Aliquots of virus vectors were stored at −80°C before use in intra-ischemia penumbra microinjections. On the day of surgery, stocks were thawed, cell debris was pelleted by centrifugation at high speed (4000 rpm for 1 minute) in a bench-top centrifuge, and the supernatant was carefully collected for injection. Under isoflurane anesthesia, AAV-mCherry or AAV-hM3Dq-mCherry/AAV-hM4Di-mCherry (5 × 1012 genome copies per mL) was injected into the right primary motor cortex (AP +0.5 mm, ML –1.2 mm, DV −0.8 mm) of GFAP-Cre mice using a Hamilton syringe (7001KH) at a rate of 0.1 µL/min for 3 minutes. Four weeks later, all mice were given clozapine (CNO; Caymen Chemical, Ann Arbor, MI, USA; 3 mg/kg, i.p. dissolved in dimethylsulfoxide (DMSO)) before EA treatment, and the AAV-hM4Di-mCherry and AAV-mCherry groups received EA for 30 minutes. Two hours after EA administration, we induced MCAO in these three mice groups. Seventy-two hours after reperfusion, neurological testing was carried out, followed by brain extraction for TTC staining.
Specific CB1R deletion
The CB1R-floxed mice were used for virus-mediated conditional deletion of astroglail CB1R in the ischemic penumbra by injection of AAV2/9-GFAP-Cre-WPRE-pA (GFAP-Cre) or AAV2/9-GFAP-EYFP-WPRE-pA (GFAP-EYFP) into the right primary motor cortex (AP +0.5 mm, ML −1.2 mm, DV −0.8 mm). Viruses were purchased from the Brain VTA company (WuHan, China). Four weeks later, all mice were administered EA for 30 minutes. Two hours after EA, MCAO was induced. Mice were scored in neurological tests 72 hours after reperfusion, followed by brain extraction for TTC staining.
Procedures and drugs
To investigate the effect of EA treatment after the onset of ischemic stroke, 30 minutes of EA treatment was administered 24 hours after middle cerebral artery occlusion (MCAO) by an intraluminal filament technique as described previously. 42 Reperfusion was accomplished by withdrawing the suture 60 minutes after ischemia. Infarct volume and neurological scores were evaluated 72 hours after reperfusion.
To investigate the role of CB1R in the EA treatment after ischemic stroke, 30 minutes of EA stimuli was performed 24 hours after MCAO, whilst cannabinoid receptor antagonist AM281(Tocris Bioscience, Ellisville, Missouri, USA; 3 mg/kg, i.p, dissolved in 1 × DMSO:1 × Tween 80:18 × physiological saline) or vehicle was injected 15 minutes before EA.38,42 Reperfusion was accomplished by withdrawing the suture 60 minutes after ischemia. Infarct volume and neurological scores were evaluated 72 hours after reperfusion.
To explore the early effect of EA pretreatment, EA stimuli was administered daily for 30 minutes for 5 consecutive days. MCAO surgery was performed 28 days after the 5th consecutive EA treatment. Reperfusion was accomplished by withdrawing the suture 60 minutes after ischemia. Infarct volume and neurological scores were calculated 72 hours following reperfusion.
To identify whether EA pretreatment triggered an increase in the synaptic cleft 2-AG-induced neuroprotective effect, pharmacological interventions were conducted at different times prior to EA, according to specific protocols. This was followed 2 hours later by reperfusion and then another 72 hours later by neurological evaluation. The 2-AG inhibitor RHC80267 (Tocris Bioscience, Ellisville, Missouri, USA; 0.1 µg/5 µL, i.c.v. dissolved in 1 × DMSO: 1 × Tween 80:18 × physiological saline) or vehicle 43 was microinjected into the lateral ventricles 1 hour before EA administration. The monoacylglycerol lipase inhibitor JZL184 (Tocris Bioscience, Ellisville, Missouri, USA; 20 mg/kg, i.p., dissolved in 1 × DMSO:1 × Tween 80:8 × physiological saline) or vehicle 29 was administered by intraperitoneal injection 12 hours before MCAO induction. The FAAH inhibitor PF3845 (Tocris Bioscience, Ellisville, Missouri, USA; 10 mg/kg, i.p., dissolved in 1 × DMSO:1 × Tween 80:18 × physiological saline) 44 was administered 2 hours before MCAO induction.
To investigate the role of CB1R in the neuroprotective effect of EA pretreatment, CB1R antagonists were administered before EA. Either AM281 (Tocris Bioscience, Ellisville, Missouri, USA; 3 mg/kg, i.p, dissolved in 1 × DMSO:1 × Tween 80:18 × physiological saline) or vehicle, and either NESS0327 (Tocris Bioscience, Ellisville, Missouri, USA; 0.3 mg/kg, dissolved in 1 × DMSO:1 × Tween 80:18 × physiological saline) or vehicle 38 was administered by i.p. injection 15 minutes before EA.
To identify the function of EAAT2 in the neuroprotection of the EA pretreatment, pharmacological interventions were conducted to stimulate or inhibit the activity of EAAT2. Either LDN 212,320 (Tocris Bioscience, Ellisville, Missouri, USA; 40 mg/kg i.p., dissolved in 1 × DMSO:1 × Tween 80:18 × physiological saline) or vehicle was administered by i.p. injection 15 minutes before EA, and either Ceftriaxone (Roche, Basel, Switzerland; 200 mg/kg once per day for 5 consecutive days, i.p., dissolved in physiological saline) or saline administered by i.p. injection once a day for five consecutive days before MCAO induction. In addition, dihydrokainic acid (DHKA; Tocris Bioscience, Ellisville, Missouri, USA; 10 mg/kg, i.p., dissolved in 1 × DMSO:1 × Tween 80:8 × physiological saline) or vehicle was administered by i.p. injection 30 minutes before EA.
To clarify the relationship between EAAT2 and astroglail CB1R in the neuroprotective effect of EA pretreatment, the EAAT2 translational activator, LDN 212,320 (Tocris Bioscience, Ellisville, Missouri, USA; 40 mg/kg i.p. dissolved in 1 × DMSO: 1 × Tween 80:18 × physiological saline) or vehicle administered by i.p. injection 15 minutes before EA in GFAP-CB1R-KO mice. Two hours after 30 minutes of EA administration, MCAO was induced. Reperfusion was accomplished by withdrawing the suture 60 minutes after ischemia. Infarct volume and neurological scores were evaluated 72 hours after reperfusion.
Neurological deficit evaluation and cresyl violet staining
Cresyl violet staining was performed to observe morphological changes in cells within the ischemic penumbra following MCAO. Animals were sacrificed 28 days post-stroke. Mice in each group were anesthetized and transcardially perfused with physiologic saline followed by 4% paraformaldehyde (PFA). Brains were subsequently removed, fixed in 4% formaldehyde solution at 4°C post-fixation, then immersed successively in 20% and 30% sucrose until they sank. Brain samples were sectioned with a Leica CM1900 cryostat (Leica Microsystems GmbH, Wetzlar, Germany) into 12-µm coronal sections. For each brain, six sequential sections were taken at 100-µm intervals and processed for cresyl violet staining. Frozen sections were stained with 0.1% cresyl violet (Sigma-Aldrich Corporation, Saint Louis, Mo., USA) solution for 10 minutes at room temperature, rinsed with PBS, dehydrated in a graded series of alcohol, cleared in xylene, and then mounted with neutral gum. The sections were observed using light microscopy. The total number of damaged neurons in the ischemic penumbra was counted in five different fields of view for each section by an observer, blind to the treatment group, using light microscopy at × 400 magnification (BX51; Olympus, Tokyo, Japan). Infarct volume was measured using ImageJ analysis software (version 1.46r; National Institutes of Health, Bethesda, MD, USA).
At 7, 14, and 28 days following reperfusion, a neurological deficit assessment of mice in the different groups was performed by two investigators blind to the group assignment, using the 28-point scoring system reported by Clark. 45 Two investigators blind to the group assignment independently identified and scored individual animals, which were then averaged.
Statistics
All results except the neurological scores are expressed as the mean ± SD. Statistical analysis of the data was performed using a student’s t-test or one-way ANOVA, followed by Bonferronni or LSD post-hoc tests. Neurological scores are expressed as the median (range) and analyzed with the Kruskal–Wallis followed by the Mann–Whitney U tests. Group differences in baseline dialysate glutamate concentrations were evaluated using a one-way ANOVA with EA treatment as the between-subjects factor. Dialysate glutamate levels for each treatment group are expressed as a percentage of mean baseline dialysate concentration (set at 100%) and repeated-measures ANOVA was used to test group differences in dialysate glutamate levels following EA treatment. In the case of significant overall effects, Bonferroni multiple comparison correction was applied for post-hoc comparisons. The level of probability for a statistically significant effects was set at 0.05. Anderson-Darling, D’Agostino-Pearson omnibus normality, Shapiro-Wilk normality, and Kolmogorov-Smirnov normality tests were carried out to conform with journal guidelines, when applicable. All graphs were produced using GraphPad Prism 6 for Mac (GraphPad Software, San Diego).
Results
EA pretreatment-induced eCB activates CB1R and produces cerebral ischemic tolerance
Neuropharmacological agents were used to investigate the role of neuroprotection following EA induced by 2-AG, one of the major endogenous agonists of eCB which acts predominantly on canonical presynaptic CB1R in a retrograde manner to inhibit neurotransmitter release. Following TTC staining and a neurological score evaluation, we observed that i.c.p injection of the 2-AG synthetase inhibitor RHC80267 led to markedly decreased 2‑AG brain levels, similar to inhibition or deletion of CB1R. This resulted in an increase in infarct volume at 72 hours after EA administration and ischemic reperfusion (Figure 1(a) and (b)), as well as a decrease in neurological scores (Figure 1(c)). This suggests that EA-induced neuroprotecion is 2-AG and CB1R dependent. This was further supported by our finding that systemic administration of the 2-AG hydrolase inhibitor JZL184, which increased 2-AG levels in the brain and activated presynaptic CB1R, led to lower infarct volume (Figure 1(d) and (e)) and improved neurological scores (Figure 1(f)) compared to vehicle, which mimicked the neuroprotection of EA.
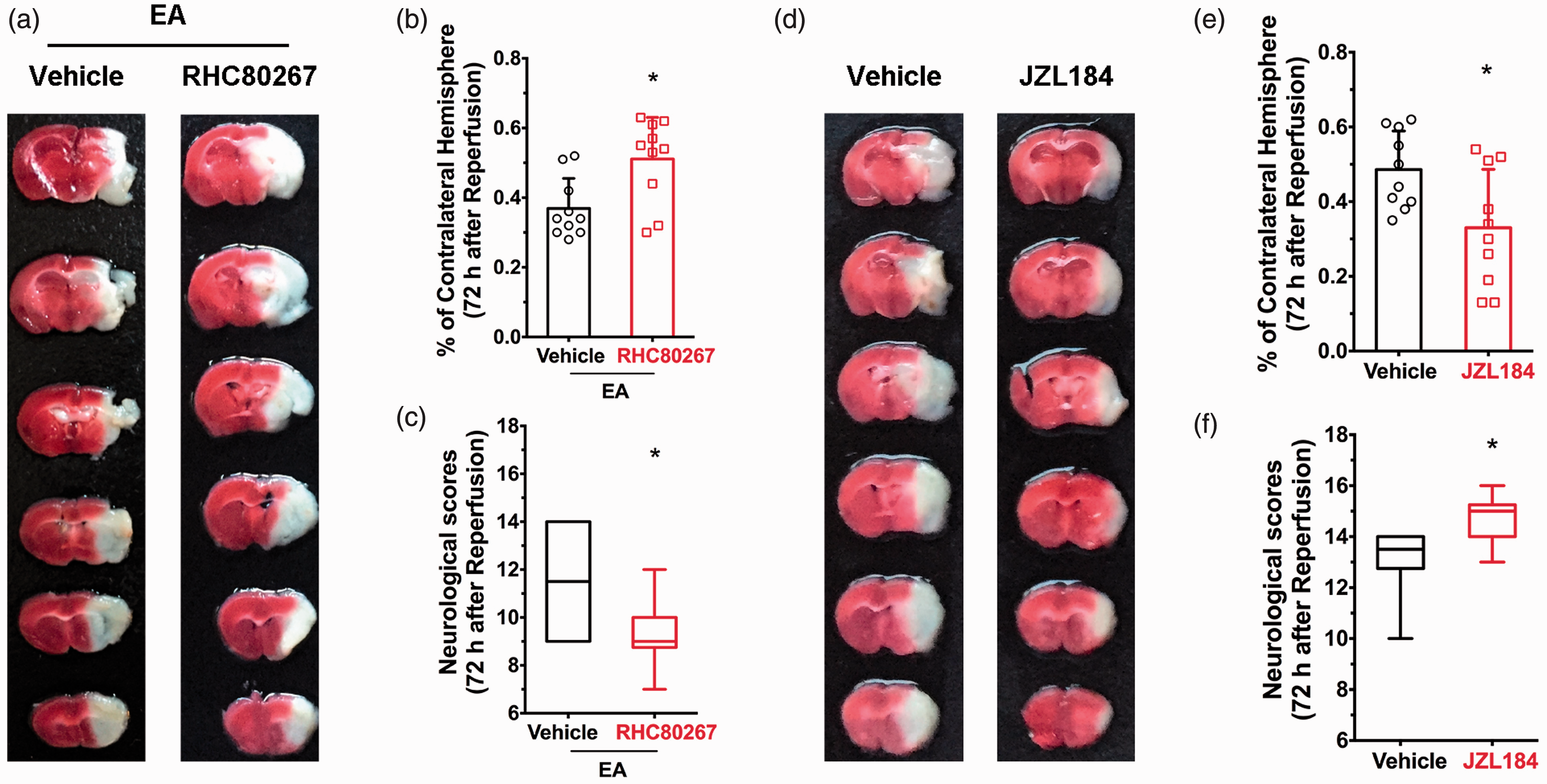
2-AG mediates EA-induced neuroprotection. (a) Representative images showing infarct tissue following pretreatment with RHC80687 and vehicle. (b,c) EA-induced neuroprotection was abolished by pretreatment of RHC80687 with increased infarct volume (b) and lower neurological scores (c) compared with the vehicle group, induced by focal cerebral ischemia for 60 min. (d) Representative images showing infarct tissue following pretreatment with JZL184 and vehicle. (e,f) JZL184 pretreatment protected the ischemic penumbra from 60 min focal cerebral ischemia with smaller infarct volume (e) and higher neurological scores (f). Infarct volume graphs show mean ± SD; n = 10 mice/group. *p < 0.05 vs. vehicle or wild-type littermates, t-test ((b): p = 0.016; (e): p = 0.019). Neurological behavioral scores show median (range) values; group differences tested using the Kruskal–Wallis test followed by the Mann–Whitney U test. *p < 0.05 vs. vehicle, U test ((c): p = 0.0013; (f): p = 0.019).
There was no difference in infarct volume between the EA treatment and sham EA groups after MCAO (sFigure 1(a)), whereras neurological scores were higher in the EA treatment group (sFigure 1b). No significant differences in infarct volume (sFigure 2(a)) or neurological scores (sFigure 2(b)) were observed between the vehicle and the AM281-treated group, suggesting that EA treatment after MCAO does not lead to neuroprotection, and that CB1R were not involved in the process.
Ischemic penumbra astroglial CB1R mediate the neuroprotective effects of EA pretreatment
To investigate the role of presynaptic CB1R in producing neuroprotective effects of EA, two CB1R antagonists (AM281 and NESS0327) were administered systemically 15 minutes before EA initiation. Both AM281 and NESS0327 groups had higher infarct volumes than the vehicle group (Figure 2(b) and (e)) and had lower the neurological scores (Figure 2(c) and (f)), suggesting that CB1R mediate EA-induced neuroprotection. Cell specific CB1R knockout mice were enrolled to determine which cell-specific CB1R were involved in the neuroprotective effect of EA. EA neuroprotection was lower in GFAP-CB1R-KO mice; infarct volume was higher in the knockout group compared to wild-type littermates following MCAO (Figure 2(h) and (i)), and neurological scores were lower (Figure 2(j)). In contrast, EA protection did not differentiate between GABAergic CB1R and wild type, nor between glutamatergic CB1R knockout mice and wild-type littermates (Figure 2(i) and (l)). These results suggest that activation of astroglial CB1R is associated with EA-induced neuroprotection.
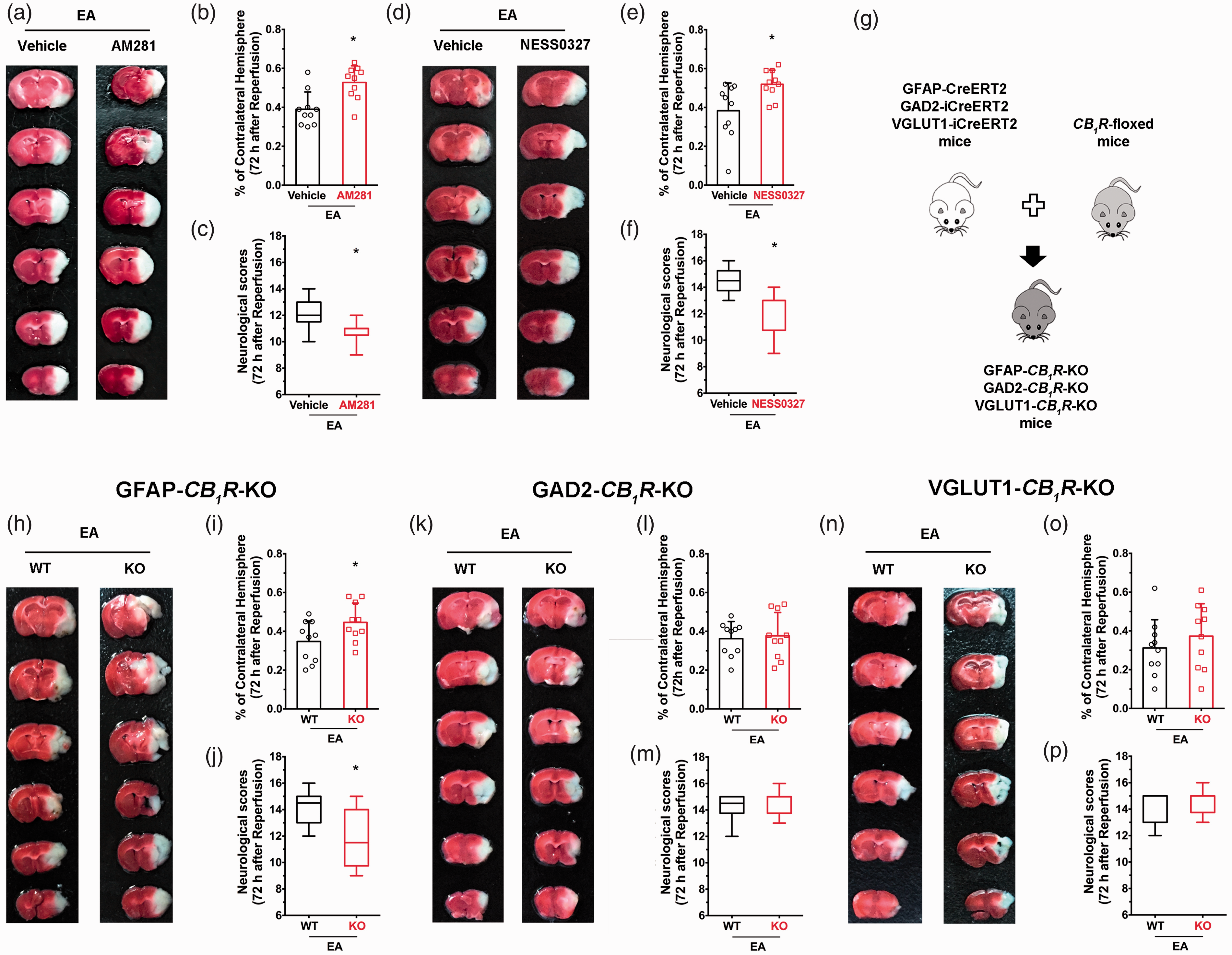
Ischemic penumbra astroglial CB1R mediates the neuroprotective effects of EA pretreatment. (a) Representative images from AM281 and vehicle groups. (b–f) Blocking CB1R with either AM281 or NESS0327 abolished the neuroprotective effect of EA pretreatment. (g) schematic showing generation of cell specific CB1R deletion mice. (h) Representative images from GFAP-CB1R-KO and WT groups. (i,j) CB1R deletion in astrocytes abolished the neuroprotective effect of EA pretreatment. (k–p) EA pretreatment did not produce neuroprotection both in GAD2-CB1R-KO mice and VGLUT1-CB1R-KO mice compared with their wild-type littermates. Infarct volume graphs show mean ± SD, n = 10 mice/group. *p < 0.05 vs. wild-type littermates, t-test ((b): p = 0.0023; (e): p = 0.0069; (i): p = 0.04; (l): p = 0.92; (o): p = 0.39). Neurological behavioral score graphs show median (range) values; group differences were tested with the Kruskal–Wallis test followed by the Mann–Whitney U test. *p < 0.05 vs. vehicle, U test ((c): p = 0.0093; (f): p = 0.01; (j): p = 0.016; (m): p = 0.58; (p): p = 0.68).
To clarify the effect of astroglial CB1R in the ischemic penumbra, the AAV virus was injected into this brain region in CB1R-floxed mice, which resulted in a deletion of astroglial CB1R in the ischemic penumbral (Figure 3(a) and (d)). This resulted in a higher infarct volume than the control virus groupand lower neurological scores (Figure 3(e) to (g)), suggesting that ischemic penumbral astroglial CB1R mediate EA-induced neuroprotection.
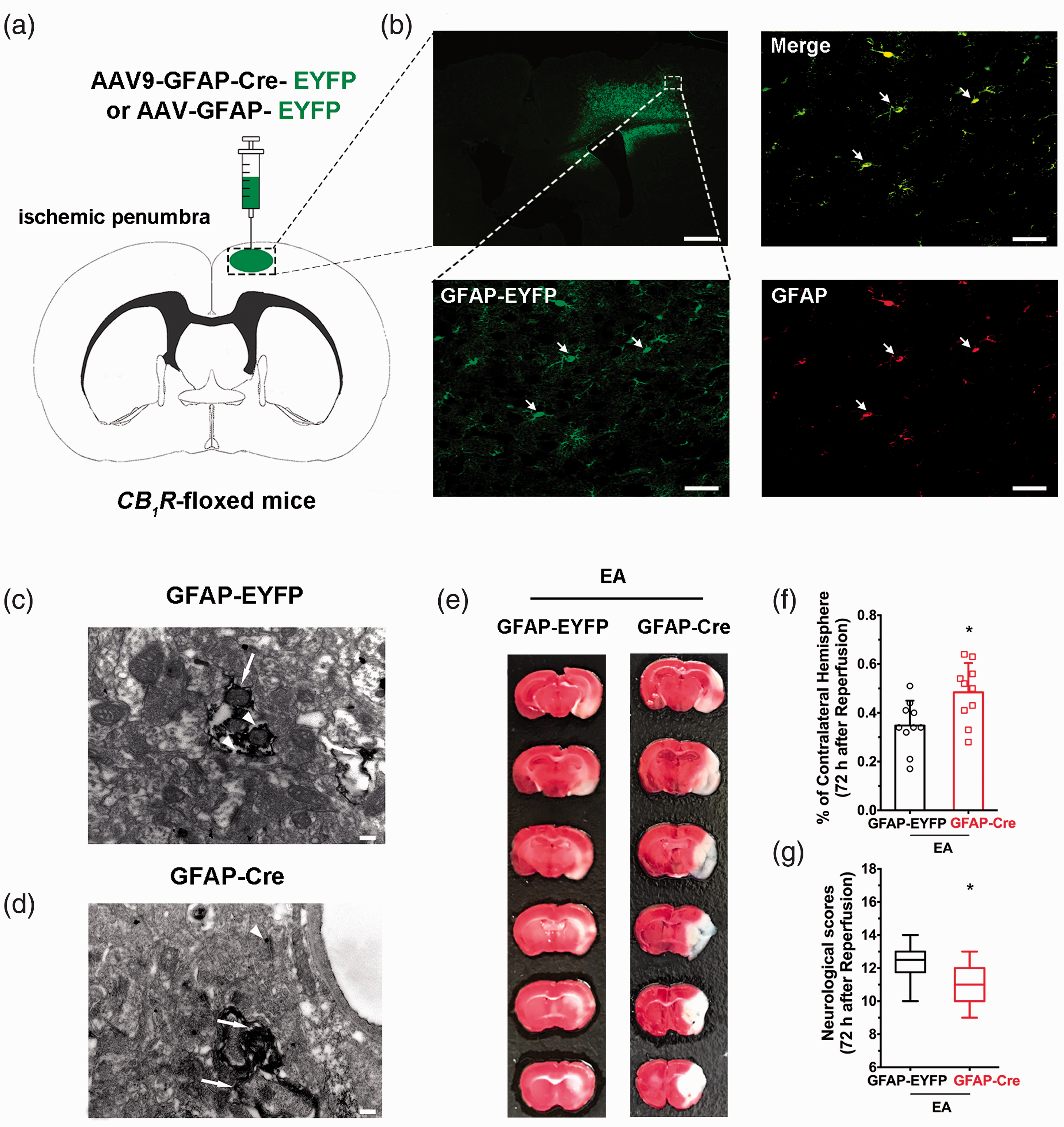
Ischemic penumbral astroglial CB1R mediates the neuroprotective effects of EA pretreatment. (a) Schematic showing intra-ischemic penumbra injection of GFAP-Cre-EYFP or GFAP-EYFP virus in CB1R-floxed mutant mice. (b) Coronal brain section showing the scope with green (GFAP-EYFP) fluorescence in the right ischemic penumbra after microinjection of different viruses. Corresponding magnified images (40×) of the box in showing the virus (green), GFAP (red) and merge (yellow). Scale bar = 50 μm. (c,d) Electron microscopic images used to detect whether astroglial CB1R is knocked out in the ischemic penumbra on CB1R-floxed mice, showing a high density of CB1R immune-positive silver grains (small arrows) in axons/terminals of GFAP-EYFP (c) but not in GFAP-Cre (d) mice. Scale bar = 200 nm. (e) Representative images showing infarct volume in GFAP-EYFP and GFAP-Cre mice. (f,g) Astroglial CB1R deletion in ischemic penumbral abolishes the neuroprotective effect of EA pretreatment induced by focal cerebral ischemia for 60 min. Infarct volume graphs show mean ± SD, n = 10 mice/group. *p < 0.05 vs. GFAP-EYFP group, t-test ((f): p = 0.013). Neurological behavioral score graphs show median (range) values; group differences were tested with the Kruskal–Wallis test followed by the Mann–Whitney U test. *p < 0.05 vs. GFAP-EYFP group, U test ((g): p = 0.02).
Activation of ischemic penumbra astrocytes mediate the neuroprotective effects of EA pretreatment
Activation of astroglail CB1R mediate the neuroprotection of EA. The DREADD strategy was used to illuminate the function of ischemic penumbral astrocytes on the neuroprotective effect of EA pretreatment (Figure 4(a) and (b)). We assessed the severity of brain injury and found that there was no difference in infarct volume (Figure 4(c) and (d)) or neurological scores (Figure 4(e)) between the hM3Dq group and the mCherry group. However, 72 hours after ischemic perfusion, infarct volume (Figure 4(c) and (d)) was higher and neurological scores (Figure 4(e)) were lower in the hM4Di group than in the mCherry group, suggesting that ischemic penumbral astrocytes are necessary for the EA-induced neuroprotection.
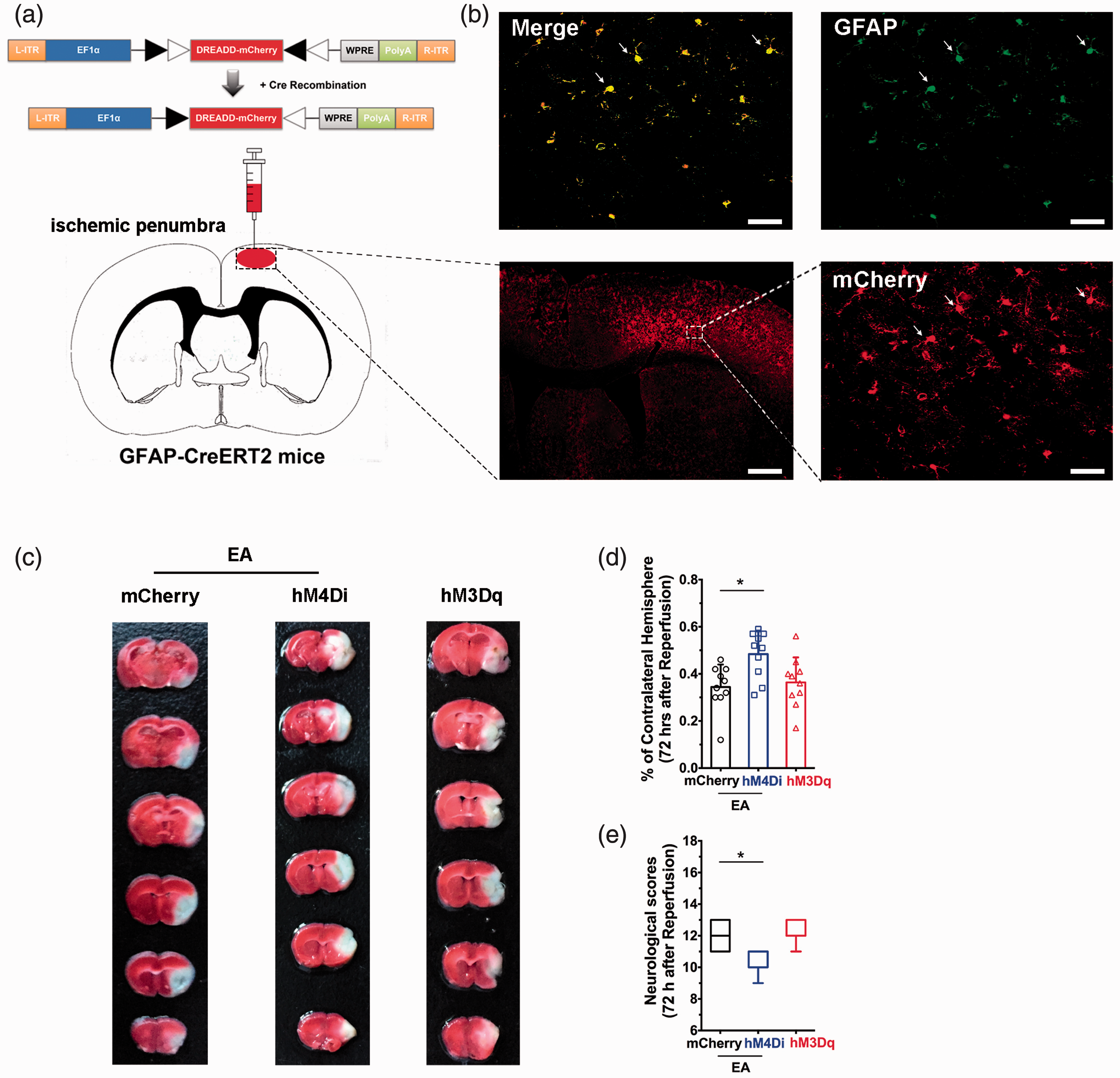
Ischemic penumbral astrocytes mediate the neuroprotective effects of EA pretreatment. (a) Schematic showing intra-ischemic penumbra injection of DREADD virus in GFAP-Cre mice. (b) Coronal brain section shows the scope with red (mCherry) fluorescence in the right ischemic penumbra after microinjection of different viruses. (c–e) corresponding magnified images (40×) of the box in (b) showing mCherry (red), GFAP, and merge (yellow). Scale bars = 50 µm. (c) Representative images comparing infarct volume in mCherry, hM4Di, and hM3Dq groups. (d,e) Activation of ischemic penumbral astrocytes decreased infarct volume (d) and improved neurological scores (e) induced by focal cerebral ischemia for 60 min. By contrast, inhibition of ischemic penumbral astrocytes decreased infarct volume (d) and decreased neurological scores (e) after EA pretreatment. Infarct volume graphs show means ± SD, n = 10 mice/group, *p < 0.05 vs. mCherry group, one-way ANOVA ((d): p = 0.0023; hM4Di vs mCherry; p > 0.999; hM3Dq vs. mCherry). Neurological behavioral score graphs show median (range) values; group differences were tested with the Kruskal–Wallis test followed by the Mann–Whitney U test. *p < 0.05 vs. mCherry group, U test ((e): p = 0.0092; hM4Di vs. mCherry; p > 0.999; hM3Dq vs. mCherry).
Ischemic penumbra astroglial CB1R mediate the long-term neuroprotective effect of EA pretreatment
To determine whether the long-term neuroprotection of EA is via astroglial CB1R, a CB1R antagonist was tested in GFAP-CB1R-KO mice. Systemic administration of the CB1R antagonist AM281 led to higher infarct volume after MCAO than the vehicle group (Figure 5(a) and (d)) in addition to lower neurological test scores (Figure 5(e)). This result suggests that the EA-induced neuroprotection lasted for 28 days. Compared to wild-type littermates, infarct volume was higher in GFAP-CB1R-KO mice after MCAO (Figure 5(f) to (i)), and neurological scores were lower (Figure 5(j)). This suggests that the astroglial CB1R also mediate the long-term neuroprotection of EA pretreatment for 28 days. We then investigated the effect of early EA administration and found no significant difference in infarct volume or neurological scores between the EA-pretreated group and sham EA group 28 days after MCAO (sFigure 3), suggesting no benefit from early EA pretreatment following an interval of this length.
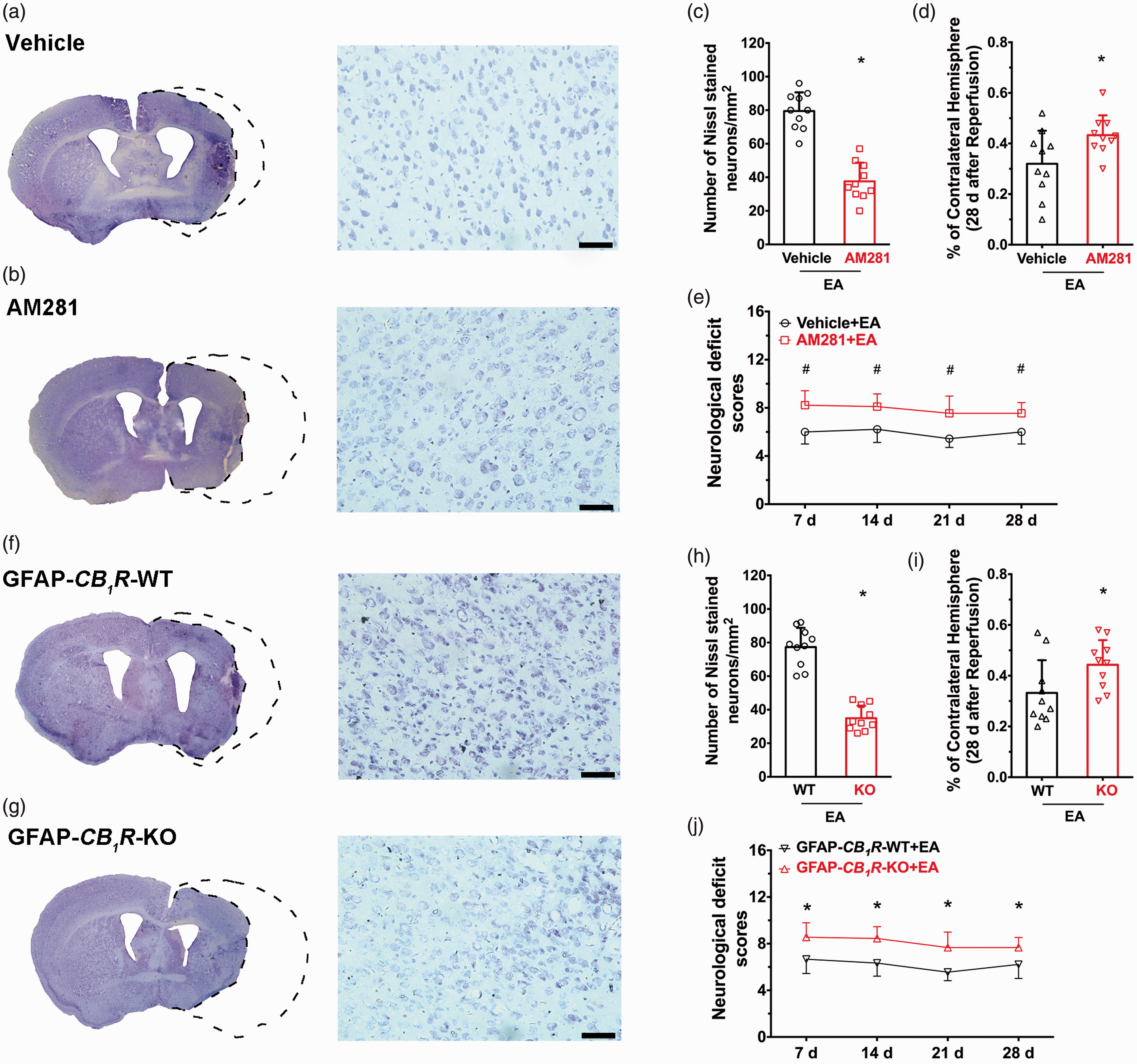
Ischemic penumbra astroglial CB1R mediates the long-term neuroprotective effect of EA pretreatment. (a,b) Left images represent coronal sections of Nissl staining in (a) vehicle or (b) AM281 groups; Right images show higher magnifications (20×) of the images on the left. (c) Neuronal density ratio in the left/right cerebral cortex. EA-induced long-term neuroprotection is abolished by pretreatment with AM281 compared to the vehicle group, showing decreased infarct volume (d) and neurological deficit scores (e) induced by focal cerebral ischemia for 60 min. (f,g) Left images represent coronal sections of Nissl staining in the GFAP-CB1R-WT (f) or GFAP-CB1R-KO (g) groups; Right images show a higher magnification (20×) of the images on the left. (h) Neuronal density ratio in the left/right cerebral cortex. EA-induced long-term neuroprotection is abolished by pretreatment with GFAP-CB1R-KO mice compared to wild-type littermates, showing decreased infarct volume (i) and neurological deficit scores (j) induced by focal cerebral ischemia for 60 min. Infarct volume graphs show mean ± SD, n = 10 mice/group. *p < 0.05 vs. vehicle or wild-type littermates, t-test ((c): p < 0.0001; (d): p = 0.03 (h): p < 0.0001; (i): p = 0.04). Neurological deficit scores graphs show median (range) values; group differences were tested using the Kruskal–Wallis test followed by the Mann–Whitney U test. *p < 0.05 vs. wild-type littermates, U test ((e): p = 0.004; (i): p = 0.005).
EA pretreatment increases extracellular glutamate levels in the ischemic penumbra, which is neuroprotective
Astrocytes regulate the concentration of interstitial glutamate. To evaluate the impact of EA-induced activation of astrocytes on neurotransmitter release, in vivo microdialysis (Figure 6(a) and (b)) was employed. Extracellular fluid samples were detected and analyzed, and the concentration of extracellular glutamate in the ischemic penumbra was higher 0.5 hours and 2 hours after EA pretreatment than the sham group (Figure 6(c)), which suggests that EA-induced activation of astroglail CB1R elevates glutamate levels.
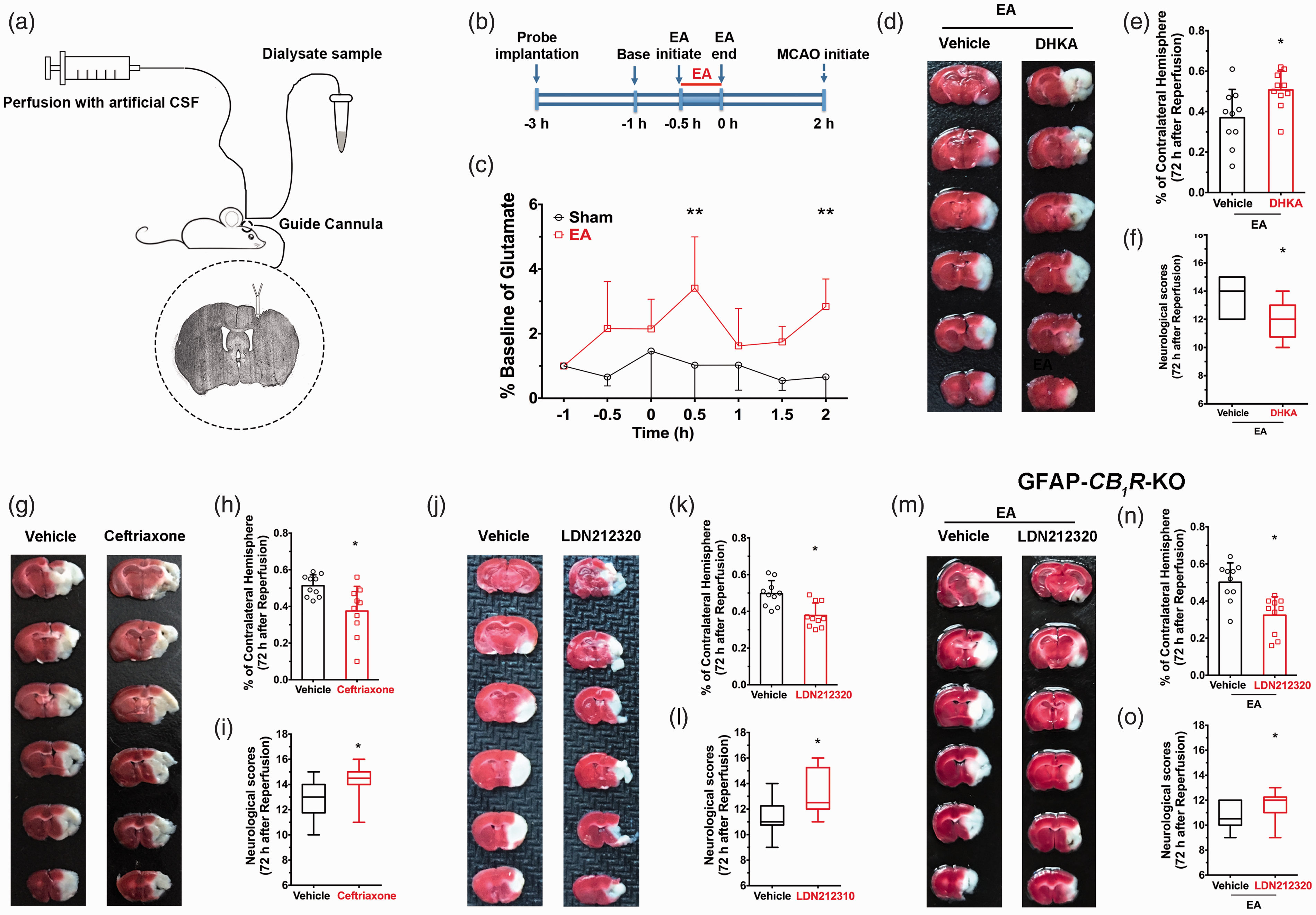
Pharmacological activation of EAAT2 restores EA-induced neuroprotective effect in astroglial CB1R deficient mice by mediating the ambient glutamate. (a) Photomicrograph of the representative location of the tip of microdialysis probe in the ischemic penumbra. The vertical black arrow indicates the guide cannula and the lower black arrow represents the tip of microdialysis probe (microdialysis probe is 1 mm longer than guide cannula). (b) Timeline showing microdialysis experimental procedure. (c) EA pretreatment raised extracellular glutamate level in the ischemic penumbra 0.5 h and 2 h after administration of EA compared with the sham group (*p 0.5 h= 0.025 vs. sham, *p 2 h< 0.007 vs. sham, (c). (d–f) Pharmacological approaches were used to detect the role of EAAT2 in EA-induced neuroprotection. (d) Representative images comparing infarct volume in vehicle and DHKA (e,f) Inhibition of EAAT2 abolished the neuroprotective effect of EA pretreatment. (g,j) Representative images comparing infarct volume in vehicle and ceftriaxone (g) and in vehicle and LDN212320 (j). Activation of EAAT2 either by LDN212320 or Ceftrixone decreased infarct volume (h,k) and improved neurological scores (i,l). (m–o) LDN212320 restored EA-induced neuroprotection in the GFAP-CB1R-KO mice showing decreased infarct volume (n) and improved neurological scores (o) after MCAO compared with the vehicle group. Group differences in baseline dialysate glutamate concentrations were evaluated using one-way ANOVA with EA treatment as the between-subjects factor. Subsequently, per treatment dialysate glutamate levels were transformed to percentages of mean baseline dialysate concentration (set at 100%) for evaluation of changes in dialysate glutamate content following EA treatment as performed by ANOVA with repeated measures over time. In the case of significant overall effects, a Bonferroni multiple comparison test was used for post-hoc comparisons. Infarct volume graphs are shown as mean ± SD, n = 10 mice/group. *p < 0.05 vs. vehicle, t-test ((e): p = 0.001; (h): p = 0.0083; (k): p = 0.01; (n): p = 0.001). Neurological behavioral scores graphs show median (range) values; group differences were tested using the Kruskal–Wallis test followed by the Mann–Whitney U test. *p < 0.05 vs. vehicle, U test ((f): p = 0.019; (i): p = 0.02; (l): p = 0.01; (o): p = 0.04).
Astroglail EAAT2 is the principal transporter that removes the excitatory neurotransmitter glutamate from the extracellular space. 29 We used a neuropharmacological approach to detect the role of EAAT2 in EA-induced neuroprotection. When the EAAT2 inhibitor DHKA was injected systemically prior to EA, there was a higher infarct volume (Figure 6(e)) and lower neurological scores than the vehicle group (Figure 6(f)) 72 hours after EA administration and ischemic reperfusion. These results suggest that inhibition of EAAT2 reversed the neuroprotective effect of EA. In contrast, systemic injection of either the EAAT2 translational activator, LDN212320, or the EAAT2 transcriptional activator, Ceftrixone, led to lower infarct volume after MCAO (Figure 6(g), (h), (j) and (k)) and higher neurological scores (Figure 6(i) and (l)) than the vehicle group suggesting that the activation of EAAT2 mimicked the neuroprotective effect of EA. These neuropharmacological results demonstrate that EAAT2 mediates the neuroprotective effect of EA.
Pharmacological activation of EAAT2 restores EA-induced neuroprotective effect in astroglial CB1R-deficient mice
Both activation of astroglial CB1R and EAAT2 mediate EA-induced neuroprotection. Nevertheless, the relationship between astroglial CB1R and EAAT2 in EA-induced neuroprotection is unclear. To evaluate this issue, the EAAT2 activator LDN212320 was administered in GFAP-CB1R-KO mice. After neurological scoring and TTC staining, an effect of administering LDN212320 was observed: seventy-two hour after reperfusion, infarct volume was higher (Figure 6(m) and (n)) and neurological scores were lower (Figure 6(o)) in GFAP-CB1R-KO mice than in the GFAP-CB1R-WT group. These results suggest that activation of EAAT2 led to moderate upregulation of extracellular glutamate, which rescued EA neuroprotection in astroglial CB1R knockout mice.
Discussion
In this study, we investigated the mechanism underlying the neuroprotective effect of EA pretreatment. First, we demonstrated that EA pretreatment elicited eCBs, which subsequently activated ischemic penumbra astroglial CB1R, which to led to cerebral ischemic protection. It has been shown that activation of astroglial CB1R increases extracellular glutamate. Using microdialysis, we found that EA pretreatment increased interstitial glutamate in the ischemic penumbra by up-regulation of EAAT2. Our findings suggest that EA pretreatment induced an increase of ambient eCBs and subesequent activation of ischemic penumbral astroglial CB1R led to the moderate upregulation of extracellular glutamate, which protected neurons from cerebral ischemic injury. These findings provide clear evidence for the cell-specific and brain region-specific mechanism underlying EA-induced neuroprotection.
Stroke is one of the leading causes of mortality and morbidity worldwide and results in cognitive, affective, and sensorimotor dysfunction. 4 However, there are currently no efficacious interventions. Evoking endogenous protective mechanisms is a promising therapeutic approach for stroke patients. Our previous studies demostrate that EA pretreatment develops a neuroprotective effect by reducing ischemic infarct volume, accompanied with higher neurological scores in rats. 15 Subsequent studies16,18,46–50 show that several transmitters, modulators and molecular pathways, such as certain neurochemicals (for example, opioid peptides and 5- hydroxytryptamine),47,51 cerebral blood flow, 46 apoptosis (cholecystokinin octapeptide (CCK-8)),47,48 inflammation48–50 and eCBs,16,18 regulate the ischemic tolerance effect of EA. Although Wang et al. 18 and others demonstrated that eCBs are involved in the neuroprotective effect of EA,16,17 the detailed mechanism had not yet been described.
In recent years, neuroprotective effects of EA have been demonstrated in both animal studies and clinical trials. An accumulation of evidence indicates that EA can attenuate ischemic brain injury via specific mechanisms, such as regulating apoptosis or inflammation-related factors, which in turn regulate glutamate release, etc. Meanwhile, EA greatly enhances cerebral blood flow and promotes daily-life activity for stroke patients in clinics. Our results strongly indicate that the neuroprotective effect of EA pretreatment on cerebral ischemic injury occurs through the regulation of astroglial eCBs in the ischemic penumbra. This is a newly described neuroprotective mechanism that underlies EA and further confirms the value of the broad application of this approach in neurology.
Pharmacological intervention was used to up- and down-regulate the concentration of 2-AG before EA administration and it was found that when elevated, infarct volume increased, and when reduced, neurological outcomes following cerebral ischemia were better. Conversely, we noted that the neuroprotective effects of EA were reversed following inhibition of 2-AG. These results suggest that 2-AG mediates the neuroprotective effect of EA pretreatment. Two different CB1R antagonists, AM281 and NESS0327, reversed the neuroprotective effect of EA. By employing mutant mice with knockout of CB1R in either glutamergic neurons, GABAergic neurons or astrocytes, we found that the neuroprotective effect of EA was reversed by deletion of astroglial CB1R, which indicated that astroglial CB1R mediated the neuroprotective effect of EA. However, EA did not provide neuroprotection after ischemic stroke. As reported, monoacylglycerol lipase (MAGL) inhibitors such as JZL184 or MJN110, administered 60 minutes after stroke, produced neuroprotection against permanent focal cerebral ischemic injury. 52 However, to apply this kind of intervention strategy as a better alternative to EA treatment in such a narrow therapeutic time window, further evidence is necessary to evaluate safety with regards to cerebral hemorrhage. Otherwise, widespread application of MAGL inhibitors remain limited by the narrow treatment time windows and the related risks of cerebral hemorrhage.
Several studies have demonstrated that interactions between astrocytes and surrounding cells can influence neuronal activity and brain injury outcomes.53–58 However, the function of astrocytes in EA-induced neuroprotection is unclear. Our previous finding demonstrated that long-term depression induced by endogenous cannabinoids produced neuroprotective effects via astroglial CB1R after stroke in rodents, 29 which also indicates that astroglia are of great importance in neuroprotection. To determine whether astroglia participated in the EA-induced neuroprotective effects, we used a DREADD strategy. Excitation or inhibition of astrocytes in the ischemic penumbra by microinjection of the DREADD virus showed that excition of astrocytes decreased infarct volume and improved neurological scores, while inhibition of astrocytes in the ischemic penumbra reversed the neuroprotective effect of EA, suggesting that activation of astrocytes protect neurons from ischemic injury.
When a sudden interruption of cerebral blood supply to a specific region of the brain happens within minutes, especially a reduction in the blood supply to under 15∼20% of baseline levels, an irreversibly damaged infarct core with rapidly evolving necrotic cell death may occur. The ischemic penumbra offers a target for therapies in the treatment of stroke.59,60 By crossing CB1R-floxed mice with GFAP-CreERT2 mutant mice to generate a GFAP-CB1R-KO mouse line, we found that astroglial CB1R is associated with EA-induced neuroprotection. Moreover, in order to eliminate side effects due to widespread astroglial CB1R knockout in the whole brain, we specifically knocked out astroglial CB1R in the primary motor cortexand found that deletion of ischemic penumbral astroglial CB1R reversed the neuroprotective effect of EA.
We also found that the EA-induced neuroprotective effect lasted for 28 days. It has been demonstrated that endogenous protective mechanisms could be induced to the development of the neuroprotection by preconditioning.8–10 Therefore, we suppose that EA preconditioning also activates an endogenous protective mechanism, which then reduces brain damage produced during ischemia-reperfusion and contributes to brain function restoration, which subsequently leads to protracted protection (about 28 days). However, early EA pretreatment 28 days before MCAO showed no neuroprotective effect on ischemic stroke. The reason might be due to preconditioning windows of neuroprotection. It has been demonstrated that there are two specific time windows in which preconditioning can occur. The first window (rapid tolerance) appearring immediately after the preconditioning maneuver lasts no longer than 3 hours, and then the neuroprotective effect wears off, whereas a second window of neuroprotection (delayed tolerance) reappears 24 hours after preconditioning and lasts more than 12 hours.61–64 Combined with the previous findings, an interval of 28 days may be too long to maintain the neuroprotective effect against ischemic stroke.
CB1R are expressed at the membranes of neuronal presynaptic terminals responsible for the inhibition of neurotransmitter release.36,59 Postsynaptic depolarization leads to release of eCBs and then activation of presynaptic CB1R and transient suppression of the release of inhibitory and excitatory neurotransmitters. The same eCB feedback mechanism operates at astroglial CB1R and leads to the increase of extracellular glutamate. Recently, Wang et al. 29 demonstrated that acute elevation of extracellular eCB following eCB clearance inhibition results in neuroprotection through induction of long-term depression following sequential activation of astroglial CB1R and postsynaptic glutamate receptors. We speculated that the EA produced mild long-term depression via activation of astroglail CB1R, which increased glutamate. We employed in vivo microdialysis to detect the concentration of glutamate following EA administration in the ischemic penumbra. We found that the concentration of glutamate was increased in the ischemic penumbra after EA pretreatment, which suggests that EA-induced activation of astroglail CB1R increased glutamate levels.
Glutamate clearance is necessary for proper synaptic activation and to prevent neuronal damage from excessive activation of glutamate receptors. 60 EAAT2 is the principal transporter that clears the excitatory neurotransmitter glutamate from the extracellular space at synapses in the central nervous system, and is responsible for over 90% of glutamate reuptake within the brain.65,66 The neuropharmacological results from the currect study illustrate that activation of EAAT2 mimicked the neuroprotective effect of EA, whereas inhibition of EAAT2 reversed the neuroprotective effect of EA. Administration of the EAAT2 translational activator, LDN212320, in GFAP-CB1R-KO mice rescued the neuroprotective effect of EA. This suggests that activation of EAAT2 leads to moderate upregulation of extracellular glutamate, which rescued EA neuroprotection in astroglial CB1R knockout mice.
This study also has limitations. We used pharmacological methods to augment or reduce the concentration of 2-AG before administration of EA. The results showed that 2-AG mediated the neuroprotective effect of EA pretreatment. Nevertheless, we have not determined the cellular origin of EA-induced 2-AG. DAGLα and DAGLβ both contribute substantially to the regulation of steady-state levels of 2-AG in the brain and other tissues. 59 Levels of 2-AG are reduced by up to 80% in the brain and spinal cord, and by ∼90% in the liver in mice lacking DAGLα and DAGLβ, respectively. 67 It follows that the concentration of 2-AG is dominated by DAGLα. Further study should employ cell-specifi DAGLα knockout mice to illuminate the origins of EA-induced 2-AG. In addition, we have not explained the mechanism by which activation of EAAT2 leads to moderate upregulation of extracellular glutamate in astroglial CB1R knockout mice, which mediates the neuroprotective effect of EA. As we know, glutamate is an important excitatory neurotransmitter in the CNS. Under pathological conditions, such as ischemic stroke, the accumulation of extracellular glutamate stimulates NMDA receptors either on the neuron or the glial cells, leading to cell death through multiple pathways. EA pretreatment involves a variety of endogenous protective mechanisms. Further research should explore the underlying mechanism by which EAAT2 mediates glutamate-related neuroprotective effects of EA pretreatment.
In summary, the present study provides robust evidence for the cell-specific and brain region-specific mechanism of EA-induced neuroprotection. It is suggested that EA pretreatment induced an increase of ambient eCB and subesequent activation of ischemic penumbral astroglial CB1R, which led to a moderate upregulation of extracellular glutamate to protect neurons from cerebral ischemic injury. These findings provide a more complete understanding of the neurobiological mechanism underlying the neuroprotective effects of EA pre-treatment and suggest a potential therapeutic target for ischemic stroke. EA pretreatment may also benefit the patients undergoing intracranial aneurysm procedure, which otherwise could result in cerebral ischemic injury.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X21994395 - Supplemental material for Activation of astroglial CB1R mediates cerebral ischemic tolerance induced by electroacupuncture
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X21994395 for Activation of astroglial CB1R mediates cerebral ischemic tolerance induced by electroacupuncture by Cen Yang, Jingjing Liu, Jingyi Wang, Anqi Yin, Zhenhua Jiang, Shuwei Ye, Xue Liu, Xia Zhang, Feng Wang and Lize Xiong in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the International Cooperation and Exchange of the National Natural Science Foundation of China (No. 81420108013 to L.X.), National Natural Science Foundation of China (No. 81701362 to Y.C.), Guangdong Provincial Key Laboratory of Brain Connectome and Behavior (No. 2017B030301017), CAS Key Laboratory of Brain Connectome and Manipulation (No. 2019DP173024).
Acknowledgments
We thank Shiquan Wang for technical assistance. We thank David Bett for linguistic assistance during the preparation of this manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
CY, JL, ZJ, JW, and AY conducted experiments and analyzed data, and contributed to writing the manuscript. XL, SY and XZ were involved in partial data analysis and manuscript writing. FW and LX designed the project and wrote the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.