
Abstract
Bid is a proapoptotic member of the Bcl-2 family that mediates cell death by caspase-dependent and-independent pathways. We tested mice genetically deficient in Bid in a controlled cortical impact (CCI) model to examine the hypothesis that Bid contributes to cell death and functional outcome after traumatic brain injury. After CCI, truncated Bid (15 kDa) was robustly detected in cortical brain homogenates of wild-type mice. Bid –/– mice had decreased numbers of cortical cells with acute plasmalemma injury at 6 h (wild type (WT), 1721 ± 124; Bid –/–, 1173 ± 129 cells/ × 200 field; P < 0.01), decreased numbers of cells expressing cleaved caspase-3 in the dentate gyrus at 48 h (WT, 113 ± 15; Bid–/–, 65 ± 9 cells/ × 200 field; P < 0.05), and reduced lesion volume at 12 days (Bid –/–, 5.9 ± 0.4 mm3; WT, 8.4±0.4 mm3; P < 0.001), but did not differ from WT mice at later times after injury regarding lesion size (30 days) or brain tissue atrophy (40 days). Compared with naïve mice, injured mice in both groups performed significantly worse on motor and Morris water maze (MWM) tests; however, mice deficient in Bid did not differ from WT in postinjury motor and MWM performance. The data show that Bid deficiency decreases early posttraumatic brain cell death and tissue damage, but does not reduce functional outcome deficits after CCI in mice.
Introduction
Distinct modes of cell death contribute to delayed, progressive cell loss, and secondary damage after traumatic brain injury (TBI) (Faden, 1996; McIntosh et al, 1998; Raghupathi et al, 2000). Necrosis likely predominates (Faden et al, 1989), but caspase-dependent and -independent programmed death mechanisms have also been described (Yakovlev et al, 1997; Zhang et al, 2002). Programmed cell death can be initiated by activation of death receptors of the tumor necrosis factor (TNF) receptor family via at least two apoptotic pathways (Ashkenazi and Dixit, 1998). Type I cell death involves rapid assembly of a death-inducing signalling complex (DISC) (composed of activated death receptor(s), cytosolic adapter protein(s), and initiator caspases), and robust activation of caspase-8, which directly cleaves and activates caspase-3. Type II cell death is characterized by less efficient DISC assembly and a requirement for amplification of death receptor signalling via the mitochondrial (intrinsic) death pathway (Green and Reed, 1998).
Bid is a 22-kDa ‘BH3 domain-only’ proapoptotic Bcl-2 family protein that links death receptor activation to the intrinsic death pathway. After oligomerization of death receptors and assembly of a DISC, activated caspase 8 cleaves cytosolic Bid to its truncated (active) form (tBid) (Li et al, 1998). Truncated Bid targets BAK and BAX in the outer mitochondrial membrane and induces release of cytochrome c (Wei et al, 2000; Zha et al, 2000). In the cytosol, cytochrome c, Apaf-1, and caspase-9 assemble an apoptosome, which activates caspase-3, and other effector caspases that induce cell death (Zou et al, 1999). Bid can also induce necrosis associated with oxidative injury, although the mechanisms involved remain to be clarified (Wang et al, 2003).
Bid is essential for Fas-mediated liver cell death in vivo (Yin et al, 1999), and Bid knockout mice have decreased infarct size after transient focal ischemia (Plesnila et al, 2001; Yin et al, 2002). Consistent with a role in posttraumatic cell death, Bid is cleaved in the injured cortex at 6 h to 3 days after controlled cortical impact (CCI) in rats, and tBid-immunoreactive neurons and astrocytes exhibit apoptotic-like nuclear morphology (Franz et al, 2002). To directly examine the role of Bid in the pathogenesis of TBI, we subjected Bid-deficient and wild-type mice to CCI, and tested the hypotheses that: (1) Bid knockout decreases overall cell death, (2) Bid is essential for caspase-3 activation, and (3) Bid knockout is associated with reduced functional deficits after injury.
Materials and methods
For all studies, investigators were masked to mouse genotype during surgery, data acquisition, and analysis. Mouse genotype was masked during surgery by an investigator other than the operator. The tails of the mice were marked before surgery so that only the independent investigator could identify the genotype of the mice before or after surgery.
Controlled Cortical Impact
The CCI model was used as described previously, with minor modifications (Qiu et al, 2002). The trauma protocol was approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals. Mice (8 to 12 weeks of age) were anesthetized with 4% isoflurane (Anaquest, Memphis, TN, USA) in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH, USA) and positioned in a stereotaxic frame. Anesthesia was maintained using 2% to 3% isoflurane. A 5 mm craniotomy was made using a portable drill and trephine over the left parietotemporal cortex, and the bone flap was removed. Mice were then subjected to CCI using a pneumatic cylinder with a 3-mm flat-tip impounder, velocity 6 m/s, depth 0.6 mm, and impact duration 100 ms. The bone flap was discarded and the scalp was sutured closed. The mice were returned to their cages to recover from anesthesia, and were able to ambulate within 15 mins.
Bid Knockout and Wild-Type Mice
Mice deficient in Bid (Bid–/–) were generated as described previously (Yin et al, 1999) and backcrossed 7 to 8 generations into a C57Bl/6 background. Bid–/– mice show no apparent developmental brain abnormalities, and do not differ from C57Bl/6 mice regarding baseline cerebral blood flow, cerebrovascular anatomy, baseline blood pressure, and baseline arterial blood gas values under general anesthesia (Plesnila et al, 2001; Yin et al, 2002). C57Bl/6 mice from the same vendor as those used to construct the Bid–/– mice (Jackson Laboratories, Bar Harbor, ME, USA) were used as controls. Only male mice matched for age (8 to 12 weeks) and weight (20 to 25 g) were used for experiments.
Immunoblots
Brain tissue dissected from contused cortex within the injured hemisphere or from the left hemisphere of naive (uninjured) control mice was homogenized on ice in a modified RIPA buffer (50 mmol/L Tris-HCl, pH 7.5, 50 mmol/L NaCl, 4 mol/L urea, 0.5% sodium dodecyl sulfate (SDS), 0.5% NP-40, 0.5% Na-deoxycholate, 5 mmol/L phenylmethylsulfonylfluoride (PMSF), 1 mmol/L ethylene diamine tetraacetate (EDTA), 5 mmol/L ethyleneglycol tetraacetate (EGTA), 10 mmol/L dithiothreitol, and protease inhibitor cocktail (1:100, Sigma). Homogenates were cleared by centrifugation at 12,000 g for 15 min at 4°C. Protein content of the supernatant was assayed (Bio-Rad laboratories, Hercules, CA, USA) and aliquots of protein were boiled in denaturing sample buffer (62.5 mmol/L Tris, pH 6.8, 2% SDS, 5 mmol/L EDTA, 10% glycerol, 0.25% 2-mercaptoethanol, 0.01% bromophenol blue). Brain samples were loaded at 100 μg/lane. Denatured protein was size fractionated on 12% SDS-polyacrylamide gels (Invitrogen, Carlsbad, CA, USA) and blotted onto Immobilon 0.45 μm polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). Membranes were blocked for 1 h in 5% milk in Trisbuffered saline, pH 7.4, containing 0.1% Tween 20 (TBST), then incubated overnight at 4°C with rabbit anti-Bid antibodies that recognize the proform (22 kDa) (AR53) or the cleaved form (15 kDa) (AR54) of mouse Bid (Krajewska et al, 2002). Primary antibodies were diluted (1:1000) in TBST. Membranes were washed in TBST, and then incubated for 1 h with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:20,000 in TBST) at room temperature. Bid was detected using the enhanced chemiluminescence Western blotting detection system kit (ECL Plus, Amersham, Buckinghamshire, UK) and Hyperfilm (Amersham, Oakville, Ontario, Canada). Three to five mice per time point were examined.
Administration of Propidium Iodide and Detection of Propidium Iodide-Positive Cells
Propidium iodide (PI; 10 mg/ml, Sigma, St Louis, MO, USA) was diluted in 0.9% NaCl and 1 mg/kg was administered 1 h before killing by intraperitoneal injection in a total volume of not more than 100 μL. Mice (n = 8 to 9/group) were killed at 6 h after CCI, the brains frozen in nitrogen vapor, and cryostat brain sections (12 μm) were cut at 150- to 200-μm intervals from the anterior to posterior hippocampus. Cryostat sections were placed on poly-L-lysine slides and stored at −80°C. For detection of PI-labelled cells, brain sections were fixed in 100% ethanol for 10 min at room temperature, coverslipped with Permount (Biomeda, Foster City, CA, USA) and photographed on a Nikon Eclipse T300 fluorescence microscope (Tokyo, Japan) using excitation/emission filters 568/585 for PI.
Caspase-3 Immunohistochemistry
At 48 h after CCI, mice (n = 10 to 11/group) were anesthetized with isoflurane and the brain rapidly removed intact and frozen at −80°C. Coronal brain sections (10 μm) were placed on poly-L-lysine-coated slides and were fixed in 100% ethanol at room temperature for 10 min. Slides were washed in phosphate-buffered saline (PBS; pH 7.4). Sections were blocked for 30 mins in PBS with 3% normal goat serum and then incubated overnight at 4°C with rabbit polyclonal anti-caspase-3 antibody (conditioned medium (CM)-1, Idun Pharmaceuticals, La Jolla, CA, USA). The CM-1 antibody reacts specifically with the p18 fragment of cleaved caspase-3, but not with full-length caspase-3 (Srinivasen et al, 1998). Sections were then washed in PBS and incubated with goat anti-rabbit IgG–Cy-3 conjugate (1:300, Jackson ImmunoResearch, West Grove, PA, USA) for 60 mins. After washing in PBS, sections were incubated with mouse monoclonal anti-mouse neuronal nuclear protein (NeuN) (Chemicon, Temecules, CA, USA, 1:300) for 30 mins and then incubated with goat-anti-mouse IgG-bodipy for 30 mins (1:300, Molecular Probes, Eugene, OR, USA). Sections were washed in PBS and dehydrated in ascending alcohol series, immersed in xylene, and coverslipped with Permount. Sections were analyzed on a Nikon Eclipse T300 fluorescence microscope. Excitation/emission filters were 488/522 nm for bodipy and 568/585 for Cy3.
Assessment of Propidium Iodide-Positive and Caspase-3-Positive Cell Counts
The regions of interest in injured cortex (× 200 microscopic fields, 1100 × 1100 μm) for PI and caspase-3-positive cell counts were a cortical field at the medial edge of the impact site, a cortical field at the lateral edge, and a cortical field directly under the impact site (Figures 2 and 3). Propidium iodide-positive and caspase-3-positive cells in the cortex were counted in a total of three brain sections located within the center of the contusion (bregma —1.90 to —2.70) and separated by at least 150 μm, for a total of nine × 200 cortical fields assessed per animal [n = 8 to 11 mice/group). For caspase-3 cell counts in the dentate gyrus, an × 200 field (1100 × 100 μm) encompassing the upper and lower blades of the dentate gyrus was evaluated in two brain sections located in the center of the contusion and separated by at least 150 μm. These predetermined regions of interest were chosen based on the consistent pattern of prominent in vivo PI labelling and caspase-3 staining produced by our CCI model. Cell count data for each mouse were reported as the average of the nine cortical brain fields or the two hippocampal fields.
Assessment of Dentate Gyrus Cell Loss After Controlled Cortical Impact
As prominent cell loss occurs in dentate gyrus by 48 h after CCI, we assessed for possible differences in cell loss in this region in Bid–/– versus wild type (WT) mice. Two brain sections at least 150 μm apart that contained regions of dentate gyrus with overt cell loss were chosen for analysis. The number of cells remaining in a portion of the upper blade of the dentate gyrus (located near the most medial part of the dentate) was counted in an × 400 (450 × 450 μm) field in each of the two brain sections (Figure 3). The cell counts from the two sections were averaged and reported as a single data point for each animal (n = 5 mice/group).
Assessment of Lesion Volume and Brain Tissue Loss
Morphometric image analysis (MCID, Imaging Research Inc., St Catherines, Ontario, Canada) was used to determine lesion volume (n = 20 to 21/group for 14 days and 10/group for 28 days) and brain tissue loss (n = 9 to 10/group) after CCI. Mice were deeply anesthetized with isoflurane, decapitated, and brains were removed and frozen in nitrogen vapor. Coronal brain sections (12 μm) were cut on a cryostat at 0.5-mm intervals from the anterior to the posterior and mounted on poly-L-lysine-coated slides. Sections were stained with hematoxylin and the area of the lesion was quantitated using image analysis. Lesion volume was expressed in mm3. Brain tissue loss was calculated by obtaining the total volume of remaining tissue in the right (uninjured) and left (injured) hemispheres. Percent tissue loss was calculated as volume of the ([Right hemisphere–Left hemisphere]/Right hemisphere) × 100%.
Evaluation of Motor and Morris Water Maze Performance
Vestibulomotor function was assessed using a wire-grip test (Hall, 1989). Mice (n = 7/group) were placed on a metal wire (45 cm long) suspended 45 cm above a foam pad and were allowed to traverse the wire for 60 secs. The latency that a mouse remained on the wire within a 60-sec interval was measured, and wire grip scores were quantitated using a 5-point scale (Hall, 1989). A score of one point was given if the mouse failed to hold on to the wire with both sets of fore paws and hind paws together; two points were given if the mice held on to the wire with both forepaws and hind paws but not the tail; three points were given if the mouse used its tail along with both forepaws and both hindpaws; four points were given if the mouse moved along the wire on all four paws plus tail; and five points were given if mice that scored four points also ambulated down one of the posts used to support the wire. Mice that were unable to remain on the wire for less than 30 secs were given a score of zero. The wire grip test was performed in triplicate and an average value calculated for each mouse on each day of testing.
The Morris water maze (MWM) task was used to evaluate spatial memory performance as described previously (Sheibani et al, 2004). The apparatus consisted of a white pool (83 cm diameter, 60 cm deep) filled with water to 29 cm depth, with several highly visible cues located on the walls of each of the four quadrants. Water temperature was maintained at 21°C to 25°C. A clear plexiglass goal platform 5 cm in diameter was positioned 0.5 cm below the water's surface approximately 15 cm from the southwest wall. Each mouse was subjected to a series of four trials per day. For each trial, mice were randomized to one of four starting locations (north, south, east, or west) and placed in the pool facing the wall. Mice were given a maximum of 60 secs to find the submerged platform. If the mouse failed to reach the platform by 60 secs, it was placed on the platform by the experimenter and allowed to remain there for 10 secs. Mice were placed in a warming chamber for at least 4 mins between trials. To control for possible differences in visual acuity or sensorimotor function between groups, two trials were performed using a visible platform raised 1 cm above the surface of the water. Performance in the MWM was quantitated by latency to find the platform. For injury studies, n = 20 mice/group were used. For naïve MWM studies, n = 6 to 10 mice/group were used.
Statistical Analyses
Data are mean ± s.e.m. except caspase-3-positive cell counts, which are graphed as individual data points because the cortical counts were not normally distributed. Propidium iodide-positive and caspase-3 cell count data, and dentate gyrus cell counts, were analyzed by rank sum test. Lesion volume and brain tissue loss data were analyzed by t-test. Motor and MWM test data were analyzed by two-factor repeated-measures analysis of variance (ANOVA) (group × time). For all comparisons, P < 0.05 was regarded as significant.
Results
All mice survived CCI and appeared healthy during the experimental period. The histopathology of the lesion produced by our CCI model has been described previously (Qiu et al, 2002). The contusion produced by CCI in Bid–/– and WT mice had similar histopathologic characteristics, including edema (24 and 48 h), acute cell death in cortical and subcortical brain regions (6 to 48 h), and robust leukocyte accumulation (24 h) in the injured cortex and hippocampus (data not shown). We assessed histopathology at 12 days after CCI to allow for resolution of tissue edema and delayed cell death in the acute postinjury period.
Immunoblot analysis of Bid–/– and WT mice showed that Bid–/– mice lacked detectable Bid, whereas a prominent band at 22 kDa was observed on Western blots of naïve wild-type mouse brain homogenates (Figure 1). Truncated Bid was not detected in naïve mouse brain; however a band with a molecular weight of 15 kDa, corresponding to tBid, was detected in the injured brain of wild-type mice as early as 6 h (not shown) and robustly at 48 h after CCI (Figure 1). The 15-kDa band was not detected in the brain homogenates of injured Bid knockout mice, confirming the identity of the 15-kDa band as tBid (data not shown).
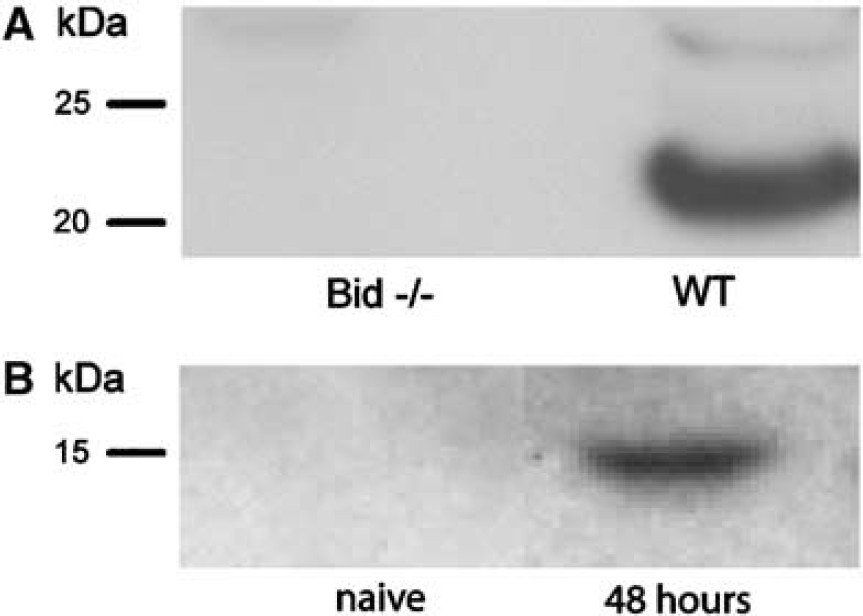
Expression of Bid in naïve mouse brain and after CCI. (
To assess the effect of Bid knockout on acute cellular injury, we used in vivo PI labelling as a marker of plasmalemma permeability that accompanies cell death in our CCI model. Propidium iodide-labelled cells were detected at 6 h after CCI in injured cortex (Figures 2A and 2B) and hippocampal brain regions including dentate gyrus (not shown) in Bid–/– and WT mice. Compared with WT, Bid–/– mice had significantly decreased numbers of PI-positive cells in the injured cortex at 6 h after CCI (P < 0.01; Figure 3C).
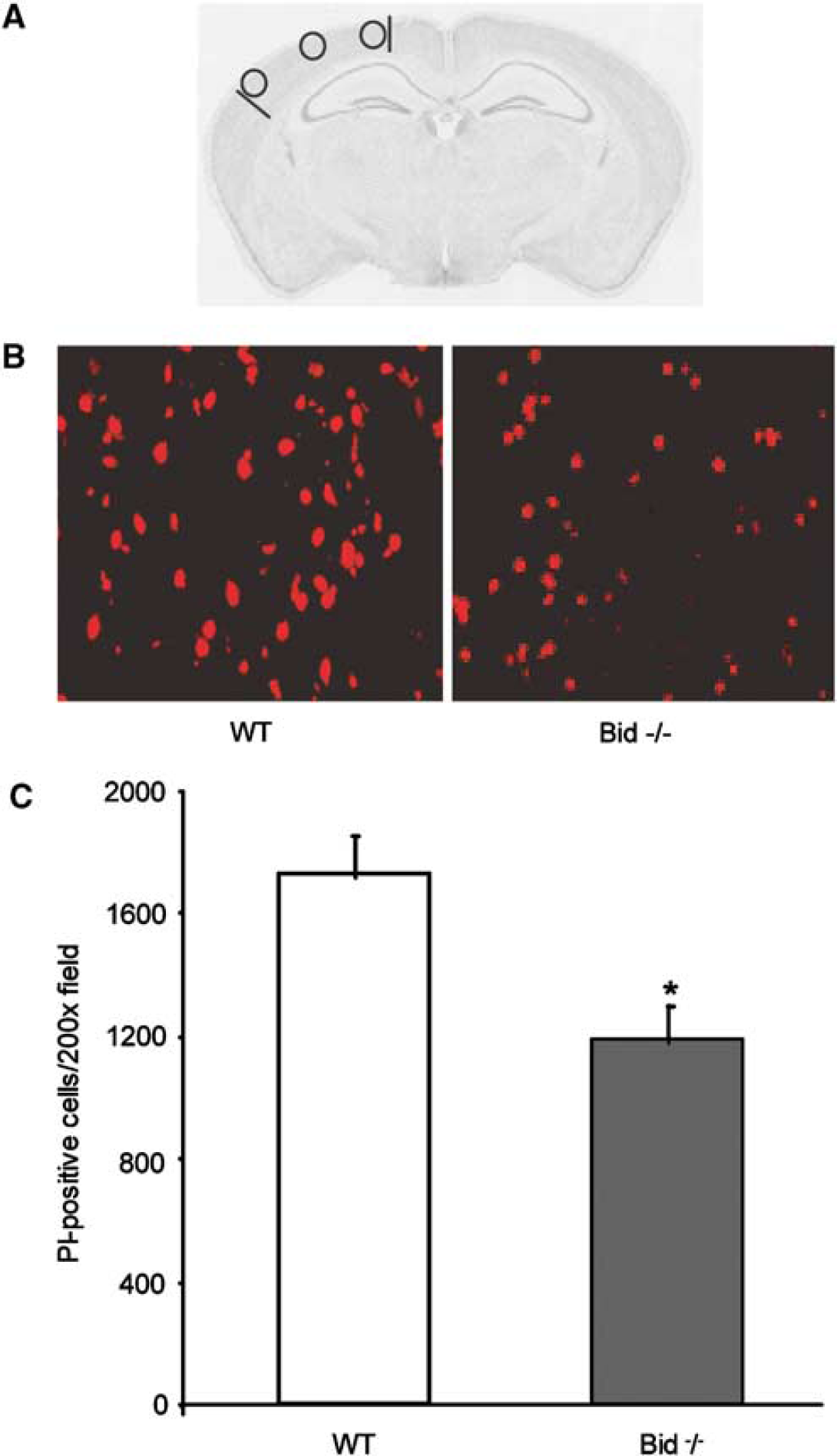
Quantification of Pl-positive cells at 6 h after CCI. (
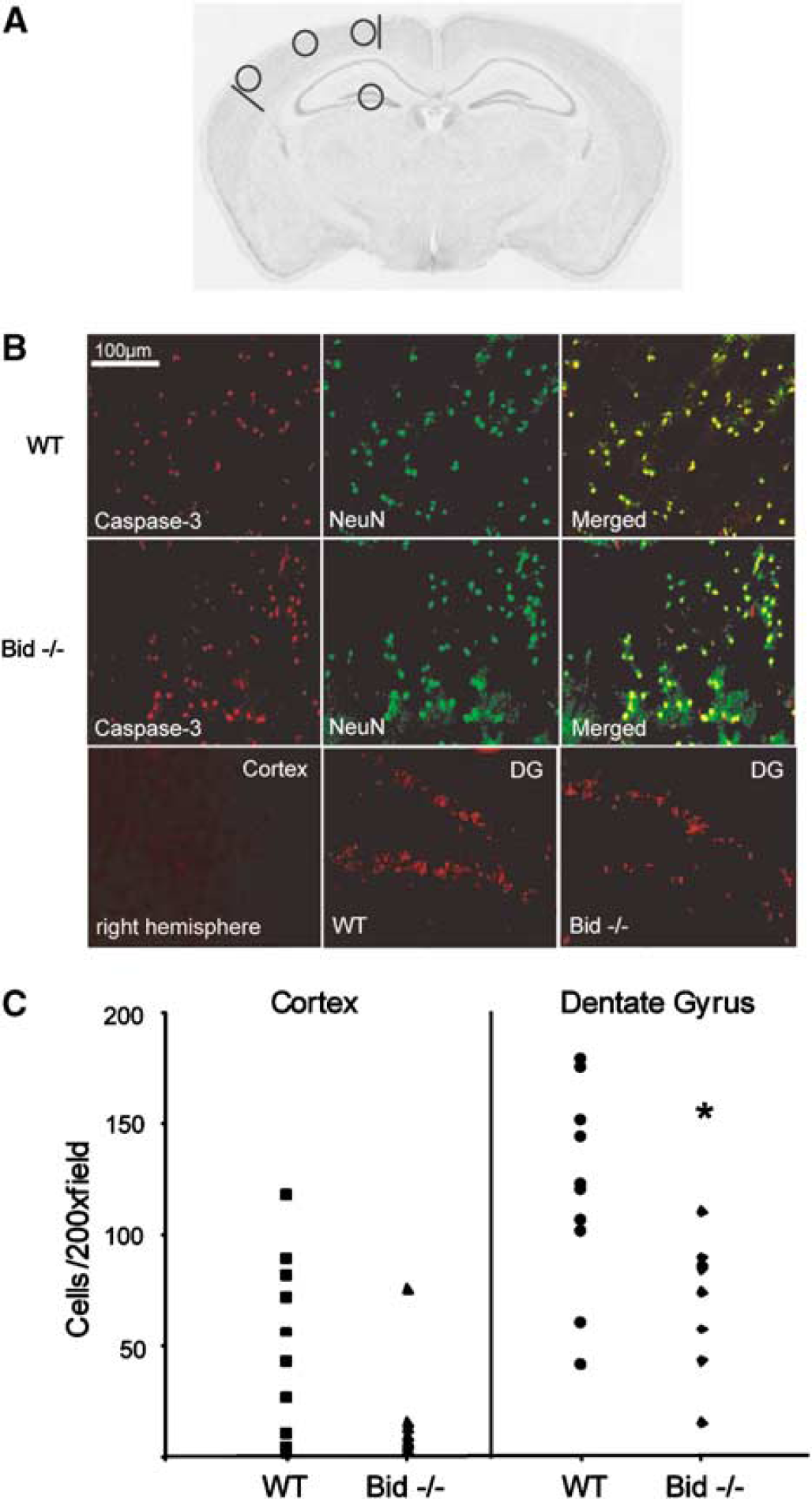
Cleaved caspase-3 expression in the cortex and dentate gyrus 48 h after CCI in Bid–/– and WT mice (magnification × 200). (
As Bid has been reported to play a role in caspase-3 activation after ischemic brain injury, we assessed the effect of Bid knockout on cleavage of caspase-3 to its active form (p18) in our CCI model. Cleaved caspase-3 immunostaining was not detected in injured or uninjured brain regions at 6 or 24 h in wild-type or Bid–/– mice (data not shown). At 48 h after CCI, p18 immunostaining was detected in the core and particularly the margins of the contusion in injured parietal cortex, as well as in dentate gyrus, in wild-type and Bid–/– mice (Figure 3). Immunostaining was not observed in contralateral (uninjured) hemispheres. Double labelling with the neuronal marker anti-NeuN revealed that many of the caspase-3-positive cells were neurons (Figure 3B). No difference in the number of cortical caspase-3-positive cells was observed between groups; however, in dentate gyrus, caspase-3-positive cells were decreased in Bid–/– versus WT mice at 48 h after CCI (P < 0.05; Figure 3C). The reduction in caspase-3-positive cells in Bid–/– mice was not explained by between-group differences in cell loss in regions of dentate gyrus with overt cell death, as Bid–/– and WT mice had similar numbers of cells remaining in regions of dentate gyrus where caspase-3-positive cells were assessed (Bid–/–, 149.7 ± 17.8 cells/ × 400 field; WT, 126 ± 10.3 cells/ × 400 field, P = 0.31).
We next assessed the effect of Bid knockout on acute and long-term lesion volume and total brain tissue loss. Lesion volume at 12 days was significantly reduced in Bid–/– (5.9 ± 0.4 mm3) versus WT (8.4 ± 0.4 mm3) mice (P < 0.001; Figure 4). However, no differences in lesion volume at 30 days or in total brain tissue loss at 40 days after CCI were observed between Bid–/– and WT mice (Figures 4B and 4C).
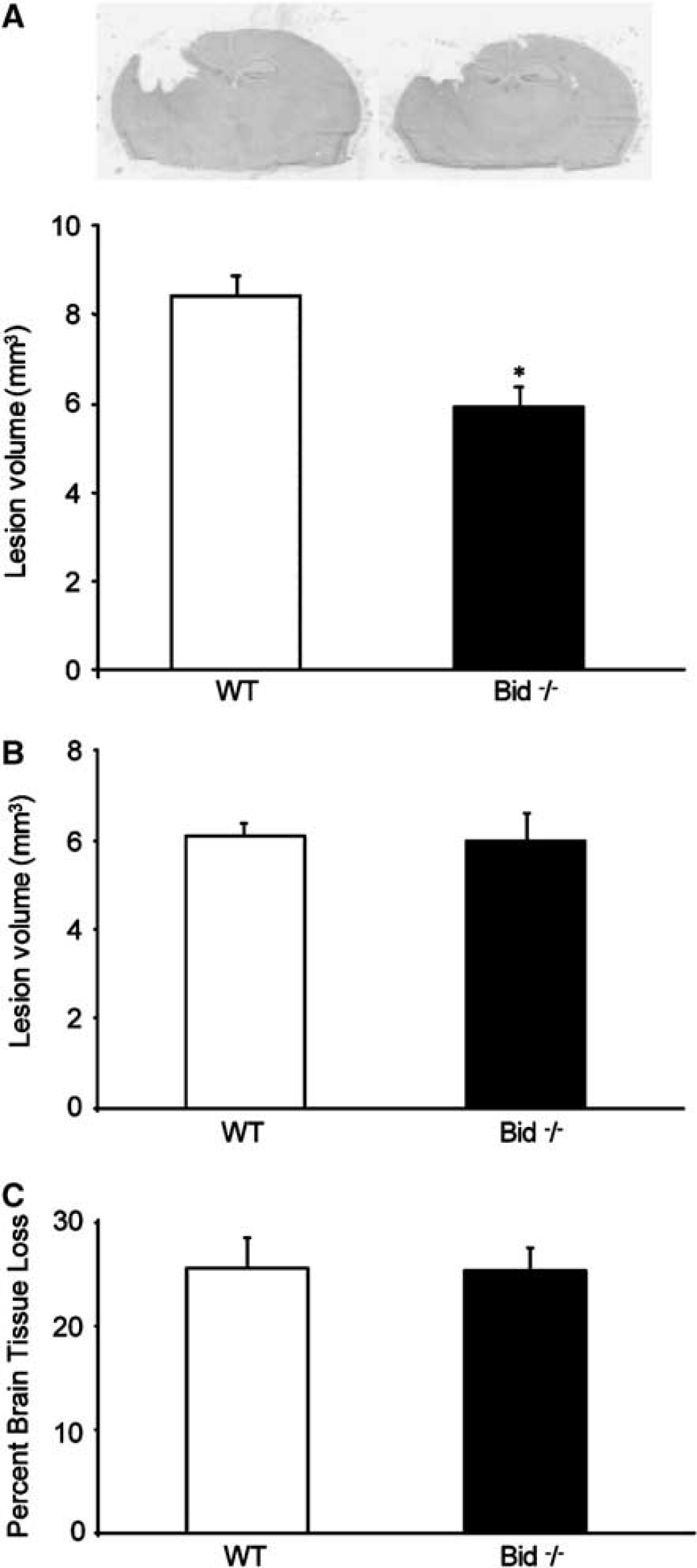
Lesion volume and overall brain tissue loss in Bid–/– and WT mice at 12, 30, or 40 days after CCI. (
To assess the effect of Bid knockout on functional neurologic outcome after CCI, we used the wire grip test and a MWM paradigm. Naïve Bid–/– and WT mice did not differ in wire grip test performance (Figure 5A). Compared with naive mice, motor performance was impaired after CCI in both Bid–/– and WT mice (P < 0.05; Figure 5A); however, no difference between groups was observed in motor score or latency (Figure 5B) over the experimental period. Latency to the hidden and visible platform in the MWM paradigm likewise was similar in naive Bid–/– and WT mice (Figure 5C). Both groups performed worse in the MWM after CCI compared with naïve (P < 0.05), but no significant between-group differences were observed in Bid–/– and WT mice in hidden or visible platform trials after CCI (Figure 5C).
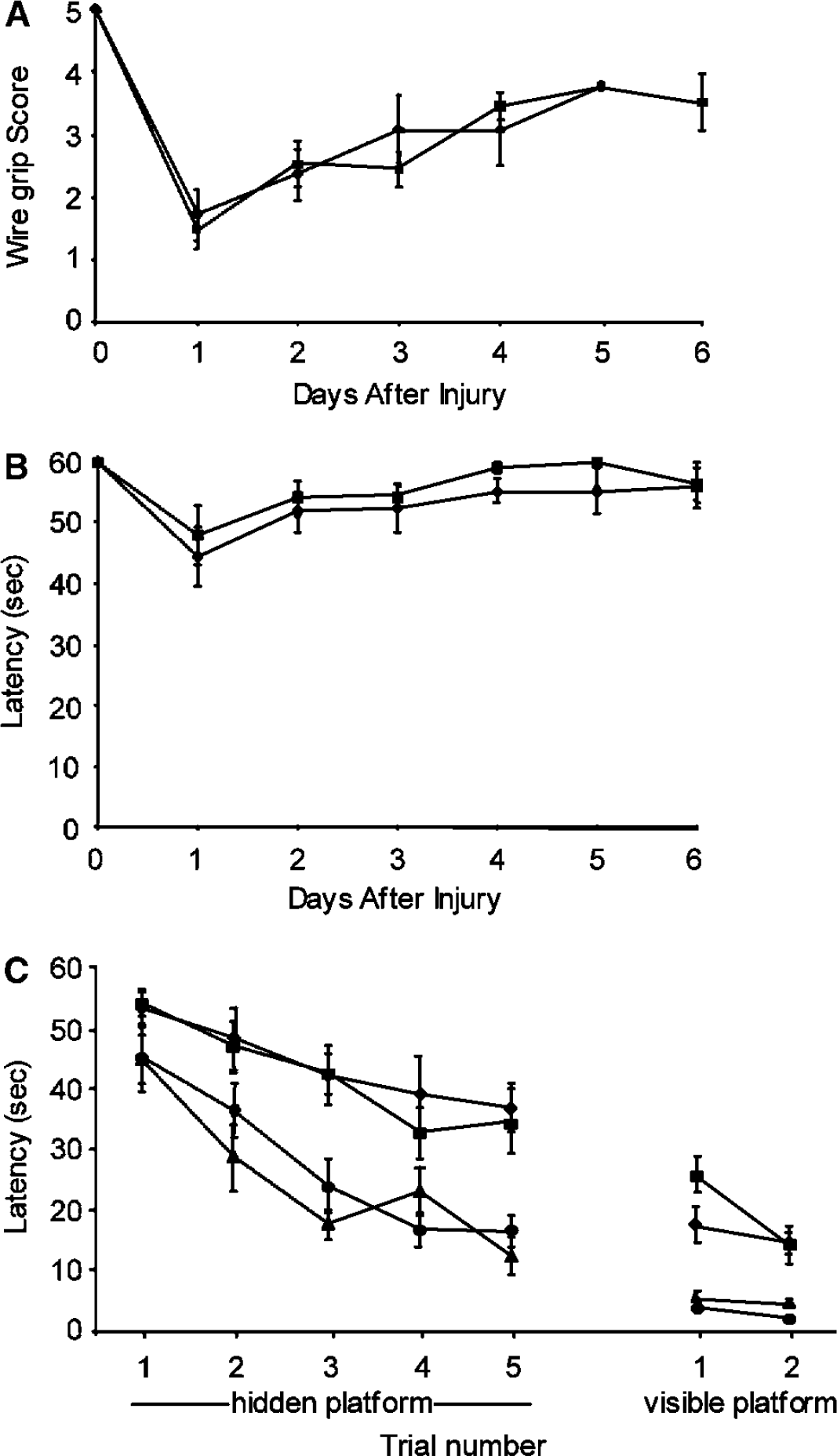
Behavioral testing in mice with Bid gene deletion and wild-type controls. Vestibulo-motor function was assessed by wire grip test at 1 to 6 days after CCI in Bid–/– (-▪-) and WT (-⧫-) mice. Compared with the pretrauma baseline, both groups had impaired wire grip scores and latencies after CCI (P < 0.05), but no differences were observed between groups in wire grip score (
Discussion
Based on previous studies showing that Bid is cleaved in ischemic and traumatic mammalian cortex (Franz et al, 2002; Krajewska et al, 2004), and a known role for Bid in ischemia-induced brain cell death (Plesnila et al, 2001; Yin et al, 2002), we tested the hypothesis that Bid mediates traumatic brain cell death after CCI. We found that genetic deletion of Bid reduced acute cellular injury and traumatic lesion volume, but that the magnitude of protection was less than that reported in experimental stroke, in which Bid–/– mice had 67% less infarction volume compared with WT (Plesnila et al, 2001; Yin et al, 2002). Cultured Bid–/– neurons were previously reported to be resistant to death induced by oxygen/glucose deprivation, suggesting that at least part of the observed reduction in postischemic and posttraumatic cell death in vivo reflects intrinsic differences in sensitivity of Bid–/– neurons to metabolic insults (Plesnila et al, 2001). The present study extends the observations that Bid is cleaved early in neurons after CCI in rats (Franz et al, 2002), and shows that Bid mediates acute posttraumatic cell death and tissue damage, but not longer-term lesion size (30 days) or ultimate brain tissue loss (40 days) in the CCI model.
To assess for a possible association between acute cellular injury and Bid deficiency, PI was used in vivo to label injured brain cells with altered plasmalemma permeability after CCI. Propidium iodide is normally excluded from uninjured cells by intact plasmalemma function, but acutely injured cells may become permeable to PI, which produces bright red fluorescence when bound to DNA or RNA (Unal-Cevik and Dalkara, 2003). As plasmalemma damage is a hallmark of necrosis, PI-positive cells may represent necrotic cell death. However, cerebral ischemic injury in mice produces PI-positive cells that are necrotic, late apoptotic (due to late onset plasmalemma failure), or a hybrid cell death phenotype with features of necrosis and apoptosis (Unal-Cevik and Dalkara, 2003; Unal-Cevik et al, 2004). In addition to a well-known role in apoptotic cell death, recent studies have suggested that full-length Bid can mediate necrosis outside the central nervous system by noncaspase mechanisms (Wang et al, 2003). Our finding that Bid–/– mice had significantly fewer PI-positive cells at 6 h after CCI compared with WT suggests that Bid might also be involved in both apoptotic and necrotic cell death after TBI. This idea is supported by a recent study showing that Bid is cleaved within 10 mins of global cerebral ischemia in dogs by apoptotic as well as necrotic cell death mechanisms (Krajewska et al, 2004). However, PI labelling alone is insufficient to identify mode of cell death, and further studies are needed to distinguish whether Bid plays a role in apoptotic versus nonapoptotic cell death in the CCI model.
We hypothesized that Bid knockout would reduce caspase-3 cleavage after CCI, particularly if mitochondrial-dependent cell death predominates as in experimental stroke (Yin et al, 2002). We found that Bid is not absolutely required for cleavage and activation of caspase-3 in traumatic mouse brain, but that Bid knockout reduced the number of cells expressing cleaved caspase-3 in dentate gyrus at 48 h after injury, a time when expression of cleaved caspase-3 is maximal in our CCI model. These data are not explained by differences in numbers of cells remaining in the dentate gyrus of Bid–/– and WT mice; indeed, Bid–/– mice did not differ from WT in the number of cells remaining in the injured dentate gyrus compared with wild-type animals.
In contrast to the findings in dentate gyrus, we did not observe an effect of Bid knockout on cortical p18 immunostaining. These results might be due to sampling error, as caspase-3 expression is extremely variable in the cortex of wild-type mice at 48 h after CCI (Figure 3). Alternatively, involvement of other proapoptotic bcl-2 family members, such as bax, bcl-xs, and bad, could compensate for lack of Bid and promote cortical cell death in Bid knockout mice. Another possibility is that caspase-3 might be upstream of caspase-8 in traumatic cerebral cortex, as has been reported in non-neuronal cells (Aouad et al, 2004). Robust Fas-Fas-associated death domain protein (FADD)-caspase-8 DISC assembly occurs early after cortical contusion in mice and can be detected in brain samples removed from patients with severe TBI (Qiu et al, 2002). Moreover, caspase-8 and caspase-3 cleavage is detectable in TdT-mediated dNTP nick end labelling (TUNEL)-positive neurons in mice at 48 h after CCI (Qiu et al, 2002). Adult mice deficient in TNF-alpha and Fas receptor lack immunohistochemically detectable cleaved caspase-3 in injured cortex at 48 h after CCI, suggesting that death receptor activation and the extrinsic death pathway play a role in caspase-3 activation after TBI (Bermpohl et al, 2003). Our findings do not, however, rule out other mechanisms for caspase-3 cleavage in the CCI model.
The finding that caspase-3 cleavage, but not overall cell loss, in the dentate gyrus at 48 h was different between Bid–/– and WT mice raises the possibility that caspase-3 might not be essential for posttraumatic cell death in this brain region. Although Bid deficiency influenced early markers of cellular injury in cortex (PI labelling) and dentate gyrus (caspase-3 cleavage), our study does not directly assess the role of plasmalemma damage or caspase-3 activation in the pathogenesis of posttraumatic cell death. In a model of fluid percussion TBI, plasmalemma damage occurred in some brain cells with no detectable ultrastructural damage (Singleton and Povlishock, 2004), and a role for caspase-3 in the pathogenesis of posttraumatic cell death remains unproven. It is likely that Bid influences multiple mechanisms of cell death in both cortical and subcortical brain regions, and thereby reduces the overall lesion size (measured at 12 days); however, the exact mechanisms by which Bid influences cell death in the CCI model remain to be determined.
The acute reduction in tissue loss observed in Bid–/– mice at 12 days after CCI did not translate into reduce tissue loss at later time points (30 or 40 days), or to differences in functional outcome, as assessed by motor and MWM tests within the same time period. It is likely that the larger lesion size observed in mice at 12 days versus 30 days results from intra-laboratory variability in lesion volume produced by CCI, and not by time-dependent differences in tissue loss; even at 1 year after CCI, lesion size increases only by 50% at the level of injury used in our laboratory, and it is known that intralaboratory variability can be 30% or more with respect to lesion size in the CCI model (Clark et al, 2000). Dissociation between histopathology and functional recovery has been described in a number of TBI models, including fluid percussion injury and CCI (Lyeth et al, 1990; Whalen et al, 1999). Caspase inhibitors and overexpression of bcl-2 in particular have been associated with a magnitude of postinjury tissue sparing similar to that seen in Bid–/– mice, without significant effects on posttraumatic motor or MWM performance after CCI (Clark et al, 2000; Raghupathi et al, 1998). In contrast, adult mice genetically deficient in TNF-alpha and Fas receptor had decreased posttraumatic lesion size similar to Bid–/–, but also had reduced motor dysfunction and improved memory acquisition compared with wild-type mice after CCI (Bermpohl et al, 2003). The results of the present study suggest that Bid modulates acute cell death but does not take part in secondary injury processes that mediate functional outcome, or progression of brain tissue loss, in the mouse CCI model.
We conclude that Bid contributes to acute cell death and early lesion size, but not to lesion size and eventual brain tissue loss measured at later time points, or to functional outcome, in the mouse CCI model. Thus, targeting Bid alone might not be a clinically useful therapeutic strategy to improve tissue damage or mitigate functional outcome after TBI.
Footnotes
Acknowledgements
The authors thank Dr John Reed and Dr Stan Krajewski of the Burnham Institute, La Jolla, California, for providing the AR53 and AR54 reagents used in this study.