
Abstract
Heat shock protein (Hsp)70 can suppress both necrosis and apoptosis induced by various injuries in vivo and in vitro. However, the relative importance of different functions and binding partners of Hsp70 in ischemic protection is unknown. To explore this question, we tested the ability of Hsp70-K71E, an adenosine triphosphate (ATP)ase-deficient point mutant, and Hsp70-381-640, a deletion mutant lacking the ATPase domain and encoding the carboxyl-terminal portion, to protect against ischemia-like injury in vivo and in vitro. Heat shock protein 70-wild type (-WT), -K71E, −381-640, and control vector plasmid LXSN were expressed in primary murine astrocyte cultures. Astrocytes overexpressing Hsp70-WT, -K71E, or −381-640 were all significantly protected from 4 h combined oxygen-glucose deprivation and 24 h reperfusion when assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay or propidium iodide staining and cell counting (P < 0.05). Brains of rats were transfected with plasmids encoding Hsp70-WT, -K71E, −381-640, or LXSN 24 h before 2 h middle cerebral artery occlusion followed by 24 h reperfusion. Animals that overexpressed either of the mutant proteins or Hsp70-WT had significantly better neurological scores and smaller infarcts than control animals. Protection by both mutants was associated with reduced protein aggregation, as assessed by ubiquitin immunohistochemistry and reduced nuclear translocation of apoptosisinducing factor. The results show that the carboxyl-terminal portion of Hsp70 is sufficient for neuroprotection. This indicates that neither the ability to fold denatured proteins nor interactions with cochaperones or other proteins that bind the amino-terminal half of Hsp70 are essential to ischemic protection.
Introduction
Heat shock proteins (Hsps) play important roles in normal cellular function and survival. They are molecular chaperones expressed constitutively and induced in response to various types of stress, including heat shock, ischemia, oxidative stress, glucose deprivation, and exposure to toxins and heavy metals (Kiang and Tsokos, 1998). Heat shock protein 70 has a 44 kDa amino-terminal adenosine triphosphate (ATP)ase domain, and a carboxyl-terminal domain that contains the 18 kDa peptide or substrate-binding domain, followed by a 10 kDa stretch terminating in the highly conserved EEVD sequence (O'Brien et al, 1996; Ohno et al, 2004; Rajapandi et al, 1998; Wang et al, 1993). We took advantage of two mutants of Hsp70 to test the importance of two aspects of Hsp70 function in protection from ischemic injury, interaction sites within the amino-terminal domain, and the importance of facilitating protein folding.
Previous studies have shown that Hsp70 overexpression protects cells from death induced by various insults that cause either necrosis or apoptosis, including hypoxia and ischemia/reperfusion, by inhibiting multiple cell death pathways (Giffard and Yenari, 2004; Steel et al, 2004). Enforced overexpression with viral vectors, as a transgene, or pharmacological induction of Hsp70, all decrease injury after cerebral ischemia and protect both neurons and glia (Giffard et al, 2004; Hoehn et al, 2001; Lu et al, 2002; Rajdev et al, 2000). In addition, there is strong evidence that Hsp70 can protect cells from toxicity due to misfolded, aggregated proteins associated with neurodegenerative diseases (Dong et al, 2005; Muchowski and Wacker, 2005; Tidwell et al, 2004). For example, overexpression of Hsp70 suppresses degeneration and improves motor function in a transgenic mouse model of SCA1 (Cummings et al, 2001). Similarly, the overexpression of Hsp70 reduces the toxicity of mutant α-synuclein in Parkinson's disease (Auluck et al, 2002). However, it is not clear whether the protective effect of Hsp70 is caused by direct interaction with misfolded protein or, alternatively, as a result of antiapoptotic and antinecrotic effects of Hsp70.
Possible protective actions of Hsp70 include prevention of protein aggregation, refolding partially denatured proteins, anti-inflammatory effects, and inhibition of cell death pathways (for recent reviews, see Garrido et al, 2001; Giffard and Yenari, 2004; Mosser and Morimoto, 2004). In response to multiple intrinsic cell death signal transducing pathways, cytochrome c, which is released to the cytosol from mitochondria, interacts with Apaf-1, leading to apoptosome formation and downstream caspase-3 activation (Adams and Cory, 2002; Jaattela, 2004). Heat shock protein 70 has been reported to act at several stages of the apoptotic pathway; our data indicate that Hsp70 inhibits release of cytochrome c from mitochondria and hence acts upstream of the formation of the apoptosome (Steel et al, 2004). Another mitochondrial protein involved in apoptosis is apoptosisinducing factor (AIF). Apoptosis-inducing factor is synthesized as a 67 kDa precursor and converted to mature AIF (57 kDa) on mitochondrial import by removal of the N-terminal mitochondria localization signal (Susin et al, 1999). Apoptosis-inducing factor is a key to the conserved caspase-independent pathway of cell death. Heat shock protein 70 overexpression can sequester AIF (Gurbuxani et al, 2003; Ravagnan et al, 2001) and reduce neonatal ischemic brain injury (Matsumori et al, 2005). Heat shock protein 70 has been shown to act on both caspase-dependent and -independent cell death pathways. Since both apoptotic and necrotic cell death are involved in ischemic brain injury, the ability of Hsp70 to reduce multiple types of cell death makes it an appealing candidate for brain protection.
Heat shock protein 70 can assist assembly and disassembly of protein complexes, and can facilitate presentation of substrates for degradation by the proteosome. A large number of proteins are known to interact with different domains within Hsp70. Both nascent polypeptide chains and hydrophobic peptides and regions of proteins are substrates for the peptide-binding domain of Hsp70 (Erbse et al, 2004; Wang et al, 1993). A previous report showed that Hsp70 lacking the ATPase domain was still capable of protecting cells from heat (Li et al, 1992). We have shown that an ATPase-deficient mutant of Hsp70 (K71E), in which the lysine at position 71 is replaced by glutamic acid (Rajapandi et al, 1998), is also capable of protecting cells from heat stress (unpublished data). The 18 kDa peptide-binding domain of Hsp70 alone is sufficient for high-affinity binding (Wang et al, 1993). However, deletion of the C-terminal EEVD residues can result in loss of substrate-binding activity, suggesting that correct folding of the protein is important for stable substrate association (Freeman et al, 1995). An Hsp70 deletion mutant missing the ATPase domain was shown to interact physically with AIF and protect cells from death induced by serum withdrawal or staurosporine, whereas the peptide-binding domain deletion mutant of Hsp70 was not protective, suggesting that the carboxyl-terminal portion of Hsp70 is necessary and sufficient to neutralize AIF (Ravagnan et al, 2001). The relative importance of the ATP-binding domain compared with the peptide-binding domain of Hsp70 in ischemic protection is currently unknown. To begin to address this question, we have begun structure-function studies of Hsp70 to identify which activities and which protein-protein interactions are most relevant to protection from specific insults.
We previously reported that astrocytes expressing elevated levels of inducible Hsp70 are protected from oxygen-glucose deprivation (OGD), hydrogen peroxide (H2O2) exposure, or hyperthermic insult (Papadopoulos et al, 1996; Xu and Giffard, 1997). We believe that the important role played by astrocytes in the brain and in sustaining neuronal function warranted study of mechanisms to protect this critical cell type. We also wished to test the ability of nonviral DNA delivery into brain as a potential protective strategy in vivo. In the present study, we used a folding-deficient point mutant, Hsp70-K71E, which retains the potential to associate with some amino-terminal protein interaction partners. Binding sites for some antiapoptotic cochaperones including BAG1 have been shown to be in the amino-terminal domain and the amino-terminal domain of Hsp70 is required for protection by BAG1 (Townsend et al, 2004). To test the importance of the amino-terminal domain, we used a deletion mutant lacking the first 380 amino acids of Hsp70-wild type (WT), which therefore lacks all interaction sites in the amino-terminal domain, but which still retains the substrate-binding domain and the carboxyl-terminal EEVD sequence. The two mutants let us test (1) the importance of protein folding to ischemic protection and (2) the importance of protein-protein associations requiring the amino-terminal domain to ischemic protection both in primary culture and in vivo against middle cerebral artery occlusion (MCAO).
Materials and methods
All experimental protocols performed on animals were performed according to protocols approved by the Stanford University Animal Care and Use Committee and in accordance with the NIH guide for the care and use of laboratory animals.
In Vitro Experiments
Primary cortical astrocyte cultures: Astrocytes were isolated from postnatal 1- to 3-day Swiss Webster mice (Simonsen, Gilroy, CA, USA), as described previously (Xu and Giffard, 1997). Neonatal mice were anesthetized, brains removed, and the cortices dissected free of meninges and hippocampi. After trituration, the cells were collected by centrifugation at 700 g for 5 mins and dissociated cells were plated in 15 mm Falcon Primaria 24-well plates (Becton Dickinson, Lincoln, IL, USA) at a density of 2 hemispheres per 24 multiwell, in Eagle's minimal essential medium (Gibco, Grand Island, NY, USA) supplemented with 10% equine serum (Hyclone, Logan, UT, USA), 10% fetal bovine serum (Hyclone), 21 mmol/L glucose (Sigma, St Louis, MO, USA), and 10 ng/mL epidermal growth factor (Sigma). The cultures were maintained in a 37°C humidified incubator with 5% C02 in room air atmosphere. Cultures were used after 2 weeks for experiments.
Overexpression of heat shock protein 70-wild type and mutants: Human inducible Hsp70-WT, -K71E or −381-640 was cloned downstream of the 5′ LTR promoter in the LXSN backbone. Retrovirus was produced using ²−2 or phoenix packaging cells (Xu and Giffard, 1997). Astrocyte cultures from Swiss Webster mice were infected 36 to 48 h after plating by exposure to medium from the packaging cells (after 0.2 μm filtration) and containing 8 μg/mL polybrene (Sigma, St Louis, MO, USA) for 24 h. Infections were repeated twice. Astrocytes expressing human Hsp70-WT, -K71E, −381-640, or control vector LXSN were selected in 400 μg/mL G418 (Sigma) for 5 days. The infected astrocytes were grown for 2 weeks, during which time they were fed every 5 days with plating medium containing 100 μg/mL G418. Experiments were performed at least three times on cultures from different dissections.
Immunocytochemistry: Expression of Hsp70-WT, -K71E, or −381-640 was confirmed by fluorescence immunocytochemistry. Cells were fixed with 4% formaldehyde for 30 mins. After three washings in phosphate-buffered saline (PBS), cells were blocked with 3% bovine serum albumin (BSA) in PBS/0.2% Triton X100 (TX100) for 60 mins, then incubated with mouse monoclonal anti-Hsp70 monoclonal antibody (Stressgen, Victoria, BC Canada, 1:200) overnight at 4°C. Cells were then washed with PBS and incubated with a rhodamine-conjugated goat anti-mouse IgG for 1 h at room temperature. Fluorescence was examined using an Axiovert 200 mol/L fluorescence microscope (Carl Zeiss, Germany).
Immunoblot: Equal 50-μg protein samples were electrophoresed on 12% polyacrylamide gels and electrotransferred onto Immobilon PVDF membrane (Millipore, Bedford, MA, USA) using a mini Trans-Blot cell (BioRad). After transfer, the membranes were blocked with 5% nonfat dry milk and incubated overnight at 4°C with monoclonal antibody to Hsp40, Hsp60, Hsp70, or Hsp90 (1:1,000, Stressgen Biotechnology, San Diego, CA, USA) and reprobed with monoclonal antibody to β-actin (1:2000, Chemicon). A horseradish peroxidase-conjugated anti-mouse secondary antibody (1:2000, Cell Signaling, Beverly, MA, USA) and a chemiluminescence substrate system (Amersham, Beverly, MA, USA) were used to visualize the immunolabeled bands. Gels were also stained with Coomassie brilliant blue after transfer to verify equal loading and transfer.
Oxygen-glucose deprivation: Cortical astrocytes were subjected to OGD as described previously (Dugan et al, 1995). Cultures were placed in an anaerobic chamber (Coy Laboratory Products Inc., Grass Lake, MI, USA) with an atmosphere of 5% CO2, 5% H2, and 90% N2. The culture medium was replaced three times with deoxygenated, glucose-free balanced salt solution (BSS0), pH 7.4, containing Phenol Red (10 mg/L) and the following (in mmol/L): NaCl 116, CaCl2 1.8, MgSO4 0.8, KCl 5.4, NaH2PO4 1, NaHCO3 14.7, and HEPES 10. Oxygenated BSS5.5 containing 5.5 mmol/L glucose in BSS was used for uninjured controls incubated in the normal incubator. Cultures were kept at 37°C in the anaerobic chamber for 3 to 4 h. Oxygen tension in the chamber was monitored with an oxygen electrode and was kept under 0.1%. Oxygen-glucose deprivation was ended by adding glucose to the culture medium to a final concentration of 5.5 mmol/L and returning the cultures to normoxia for 24 h.
Cell viability assays: Cell viability was assessed with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) absorbance as described previously (Sun et al, 2001). Astrocytes were incubated for 2 h at 37°C with MTT at 0.5 mg/mL. After incubation, the medium was discarded and MTT formazan crystals dissolved by addition of 500 μL DMSO and absorbance at 570 nm determined in a spectrophotometer. Results were expressed as percentage of uninjured control, which was considered 100%. Cell viability was also examined with propidium iodide (PI) (Sigma Chemicals, St Louis, MO, USA) staining. Cells were stained with PI for 5 mins at room temperature and then fixed with 4% paraformaldehyde (PFA) after washing with PBS. 4',6'-Diamidino-2-phenylindole (DAPI) was used for counterstaining. Both PI-positive and -negative cells were counted in five × 20 microscopic fields per well and the percent of PI-positive cells expressed as a percent of the total number of cells.
In Vivo Experiments
Overexpression of heat shock protein 70-wild type, -K71E, and −381-640 in brain: DNA/lipid complex was prepared as described previously (Hecker et al, 2001). Adult male Sprague-Dawley rats (280 to 310 g) were anesthetized by face mask with isoflurane in 70% N2O and 30% O2 and placed in a stereotaxic frame with a rat head holder. Heat shock protein 70-WT, -K71E, −381-640 or vector with the cationic lipid (l:3 μg/μL), or lipid alone was infused into the right lateral cerebral ventricle (0.48 mm posterior to bregma, 1.5 mm lateral to midline, 3.7 mm deep from dura) at a speed of 2 μL/mins via a burr hole. Total volume infused was 30 μL. The needle was left in place for 5 mins, then the bone wound was closed with bone wax after the injections. Anesthesia was discontinued, and rats were returned to their cages. The day after, 24 h after the injection, rats were either killed for immunohistochemical analysis or subjected to focal ischemia.
Focal cerebral ischemia: Ischemia was induced using the suture occlusion technique, as previously described (Sun et al, 2003). Male, 280 to 310 g Sprague-Dawley rats (n = 13 per group; Simonsen Laboratories, Gilroy, CA, USA) were anesthetized with isoflurane in 70% N2O/30% O2 using a mask. The neck was incised in the midline, the left external carotid artery (ECA) was carefully exposed and dissected, and 19 to 20 mm of a 3-0 monofilament nylon suture with the tip rounded by a flame was inserted from the ECA into the right internal carotid artery to occlude right middle cerebral artery (MCA) at its origin. After 120 mins, the suture was removed to allow reperfusion, the ECA was ligated, and the wound was closed. Sham-operated rats underwent an identical procedure, except that the suture was not inserted. Rectal temperature was maintained at 37.0°C ± 0.5°C using a servo-controlled heating pad. Arterial blood gases, blood hemoglobin, glucose concentrations, and blood pressure were monitored. After surgery, rats were allowed to regain consciousness and were monitored throughout the 24 h recovery period. In all, 13 animals were studied per group, all had neurological assessment at 24 h, 10 were used to assess infarct volume, and 3 per group were used for immunohistochemical staining.
Neurological assessment: The neurological status of each rat was evaluated at 24 h after 2 h MCAO according to a neurological grading score as described previously (Sun et al, 2003). Grades were based on the degree of contralateral hemiparesis: (0) no observable deficit, (1) any amount of consistent contralateral forelimb flexion, (2) reduced resistance to lateral push toward the paretic side, or (3) circling behavior toward the paretic side. Neurological assessment was performed immediately before the killing to assess infarct volume.
Quantification of infarct volume: At 24 h after MCA occlusion and assessment of neurological score, rats (n = 10 per group) were anesthetized with isoflurane and decapitated. Brains were removed and sectioned coronally at 2-mm intervals in a cryostat. Sections were incubated in 2% 2,3,5-triphenyltetrazolium chloride (TTC) in saline for 20 mins at 37°C and transferred to 4% paraformaldehyde overnight. To determine infarct volume at 24 h by TTC staining, six slices per rat were analyzed by a masked observer using the NIH Image program. To correct for the effects of cerebral edema and of differential shrinkage resulting from tissue processing, the infarction area in each section was calculated by subtracting the area of the healthy, uninfarcted (TTC-stained) tissue in the ipsilateral hemisphere from the area of the contralateral hemisphere as described previously (Sun et al, 2003; Swanson et al, 1990). Infarction volume was determined by summing the infarction areas of all sections and multiplying by the slice thicknesses.
Immunohistochemistry: Sham-operated or ischemic rats after 24 h reperfusion were anesthetized, then perfused with 0.9% saline, followed by 4% paraformaldehyde in PBS (pH 7.4), brains were then quickly removed, postfixed and embedded in paraffin. The paraffin-embedded sections (6 μm thick) were deparaffinized with xylene and rehydrated with ethanol. Fluorescence staining was performed as described previously (Sun et al, 2003). Briefly, deparaffinized sections were incubated with blocking buffer, 3% BSA in PBS/0.2% TX100 for 1 h at room temperature and subsequently with the following primary antibodies: rabbit polyclonal anti-AIF (1:200, Chemicon, Temecula, CA, USA), mouse monoclonal anti-NeuN (Neuronal Nuclei) (1:500, Chemicon), mouse monoclonal anti-ubiquitin (1:500, Chemicon), mouse monoclonal and rabbit polyclonal anti-Hsp70 (1:200, Stressgen), both specific for stress-inducible Hsp70, and rabbit polyclonal anti-GFAP (1:4, Immunostar, Inc.). The secondary antibodies were rhodamine-conjugated rat-absorbed donkey-anti-mouse IgG and fluorescein isothiocyanate (FITC)-conjugated anti-rabbit and anti-mouse IgG (Jackson ImmunoResearch; 1:200). Controls included omitting the primary antibody. Fluorescence images were recorded with an Axiovert 200 mol/L fluorescence microscope (Carl Zeiss, Germany) or Zeiss LSM510 confocal microscope (Carl Zeiss, Germany).
Assessment of redistribution of ubiquitin and apoptosis-inducing factor: Immunoreactivity for ubiquitin and AIF was visualized on sections from ischemic rats and sham rats, and the number of cells per × 20 field showing either the normal pattern or altered staining pattern were counted by a masked observer in three fields per slide in the penumbra, and one slide was counted for each of three animals per group. In the case of ubiquitin, normal staining was both nuclear and cytoplasmic and diffuse, redistribution of ubiquitin staining after ischemia was present if there was reduced nuclear labeling and irregular patchy staining in the cytoplasm. For AIF normal staining was perinuclear, while after ischemia some cells showed AIF immunoreactivity colocalized with PI nuclear staining.
Klenow and cresyl violet staining in ischemic brain: Cell death was assessed by cresyl violet staining (Sun et al, 2003) and Klenow staining (Roche Molecular Biochemicals, Indianapolis, IN, USA). Single- and double-strand DNA breaks were detected in paraffin-embedded 6 μm sections using the Klenow fragment of DNA polymerase I, as described previously (Jin et al, 1999). To determine nonspecific background labeling, some sections were incubated without the Klenow fragment, and normal brain sections were also analyzed. The number of Klenow-positive cells was counted in at least three random microscopic fields (× 20) for each section and three animals per group in the cortical region of the penumbra based on cresyl violet staining. Mean values were expressed as the total number of stained cells in each microscopic field.
Statistical Analysis
Data were expressed as mean ± s.e.m. values. Differences between groups were analyzed by analysis of variance (ANOVA) followed by Newman-Keuls or Bonferroni test for multiple comparisons. Significance was established at P < 0.05. Differences in neuroscores were determined by ANOVA for nonparametric variables.
Results
Heat Shock Protein 70-Wild Type, -K71E, and −381-640 Reduce Cell Death Induced by Oxygen-Glucose Deprivation
Heat shock protein 70-wild type, -K71E, −381-640, or control vector LXSN without Hsp70 was expressed in primary astrocyte cultures using retroviral-mediated infection. Immunostaining with antibody against Hsp70 amino acid residues 556 to 568 showed that there was robust expression (> 95% cells) after G418 selection after retroviral infection (Figures 1B to 1D). We performed westerns to assess whether overexpression of these proteins might be associated with induction of a stress response by determining whether there were changes in levels of Hsp40, 60, or 90. We observed no changes in the levels of these other proteins when Hsp70 was overexpressed compared with uninfected cells (sham; Figure 1E). After 4 h of OGD followed by 24 h of recovery in the presence of oxygen and glucose, most of the cells showed shrunken, condensed, and sometimes fragmented nuclei when examined with PI staining (Figure 2B, arrows). There were fewer cells with PI stained, condensed, fragmented nuclei in Hsp70-K71E-, and −381-640-overexpressing cultures than in vector control cultures (Figures 2B to 2D) indicating reduced apoptosis. Quantitative analysis showed that both Hsp70-K71E and −381-640 significantly reduced cell death and increased viability when assessed by either PI staining (Figure 2E) or by MTT absorbance (Figure 2F). The extent of protection by the mutants was comparable to that seen with Hsp70-WT.
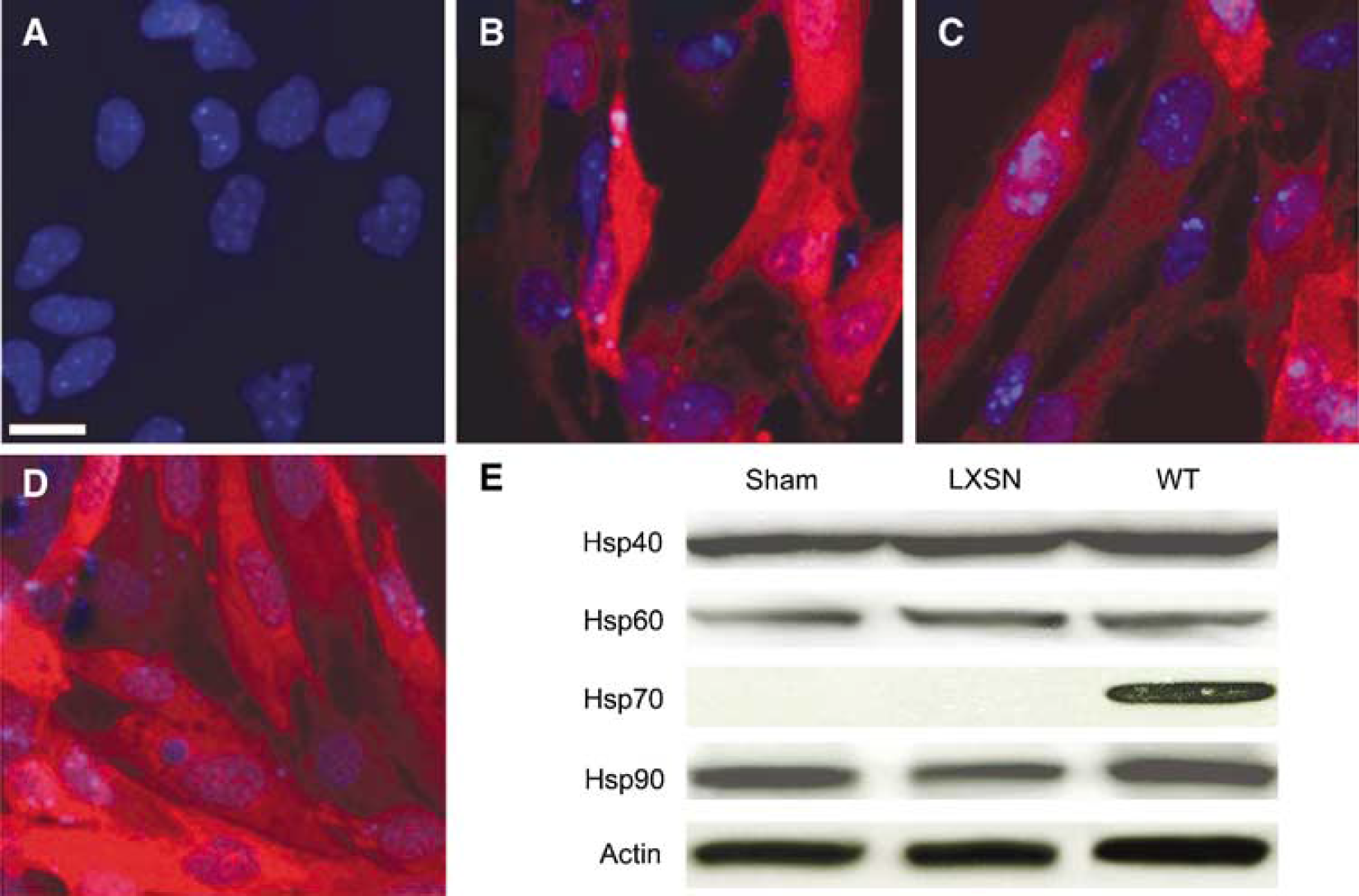
Expression of Hsp70-WT, K71E, or −381-640 in cultured astrocytes. Primary cortical astrocytes were infected with retrovirus encoding Hsp70-WT, -K71E, −381-640, or vector LXSN. Heat shock protein 70 immunostaining showed that Hsp70 proteins were expressed in > 95% of the astrocytes after selection (
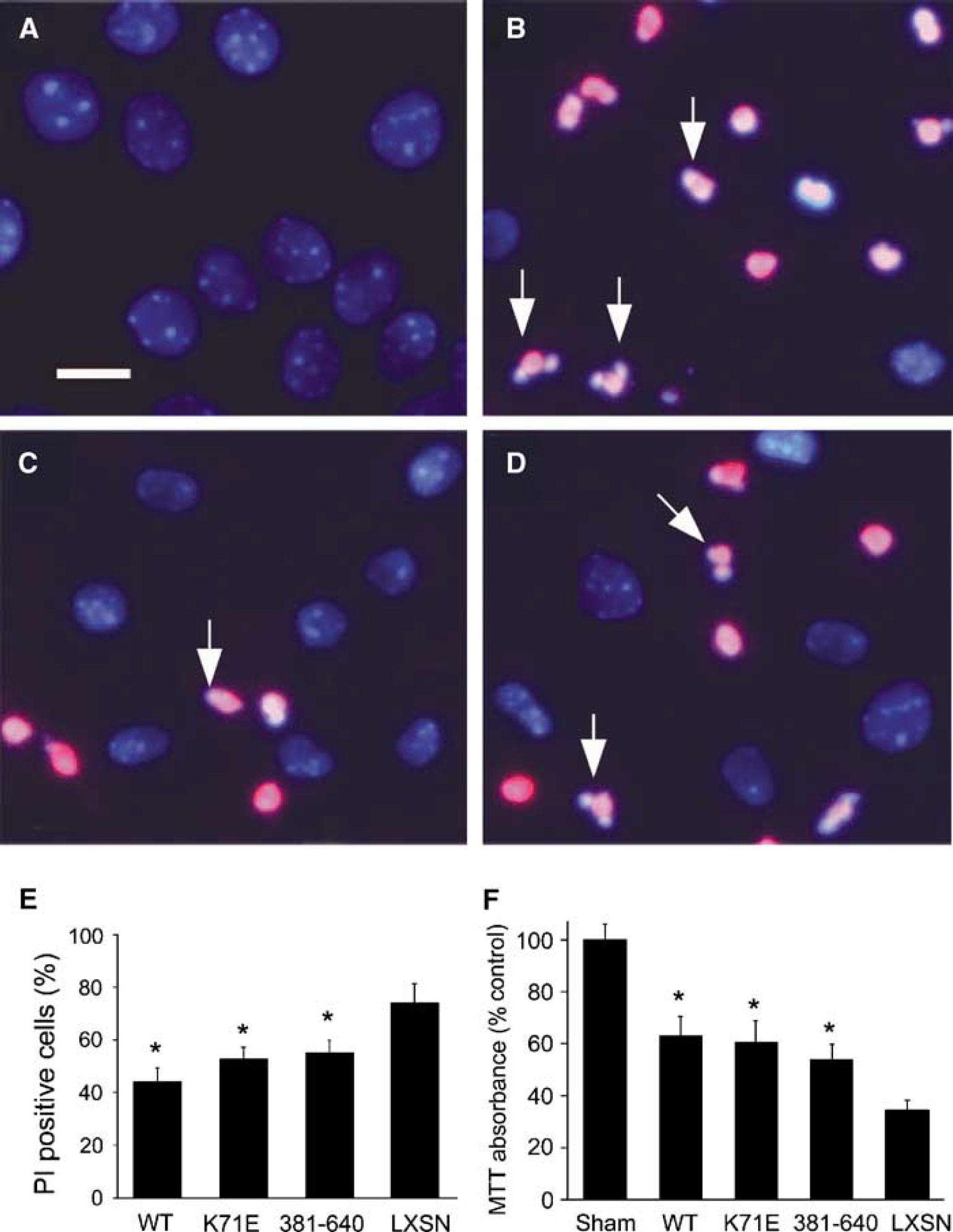
Expression of Hsp70-WT, K71E, or-381-640 protects astrocytes from OGD injury. Heat shock protein 70-overexpressing astrocytes were subjected to OGD for 4 h followed by 24 h recovery. Cell viability was significantly increased in the Hsp70-WT, K71E, and −381-640 groups compared with vector LXSN control, as assessed by PI staining and cell counting (
Heat Shock Protein 70-K71E and −381-640 Inhibit Protein Aggregation in Astrocytes after Oxygen-Glucose Deprivation
There is increased protein aggregation with ischemia and ATP depletion inhibits function of the proteosome and ATP-dependent chaperones, and so ubiquitinated aggregates accumulate in the cell. Ubiquitin immunoreactive aggregates were previously observed in ischemic brain (Hu et al, 2001). To test whether protection by Hsp70-K71E or −381-640 expression correlates with inhibition of protein aggregation, redistribution of ubiquitin immunostaining was used to assess the percentage of cells with evidence of protein aggregation. In uninjured control cells, ubiquitin immunoreactivity was present in both the cytoplasm and nucleus (Figure 3A). After 3 h OGD followed by 24 h recovery, the immunolabelling pattern changed from a relatively even distribution to a heterogeneous pattern with coarse clumps and small patches scattered throughout the cytoplasm, and with marked reduction of nuclear staining in most cells (Figure 3B), whereas the number of cells with evidence of protein aggregation was significantly reduced in Hsp70-WT-, -K71E-, or −381-640-expressing cultures compared with LXSN vector-expressing cultures (Figure 3D).
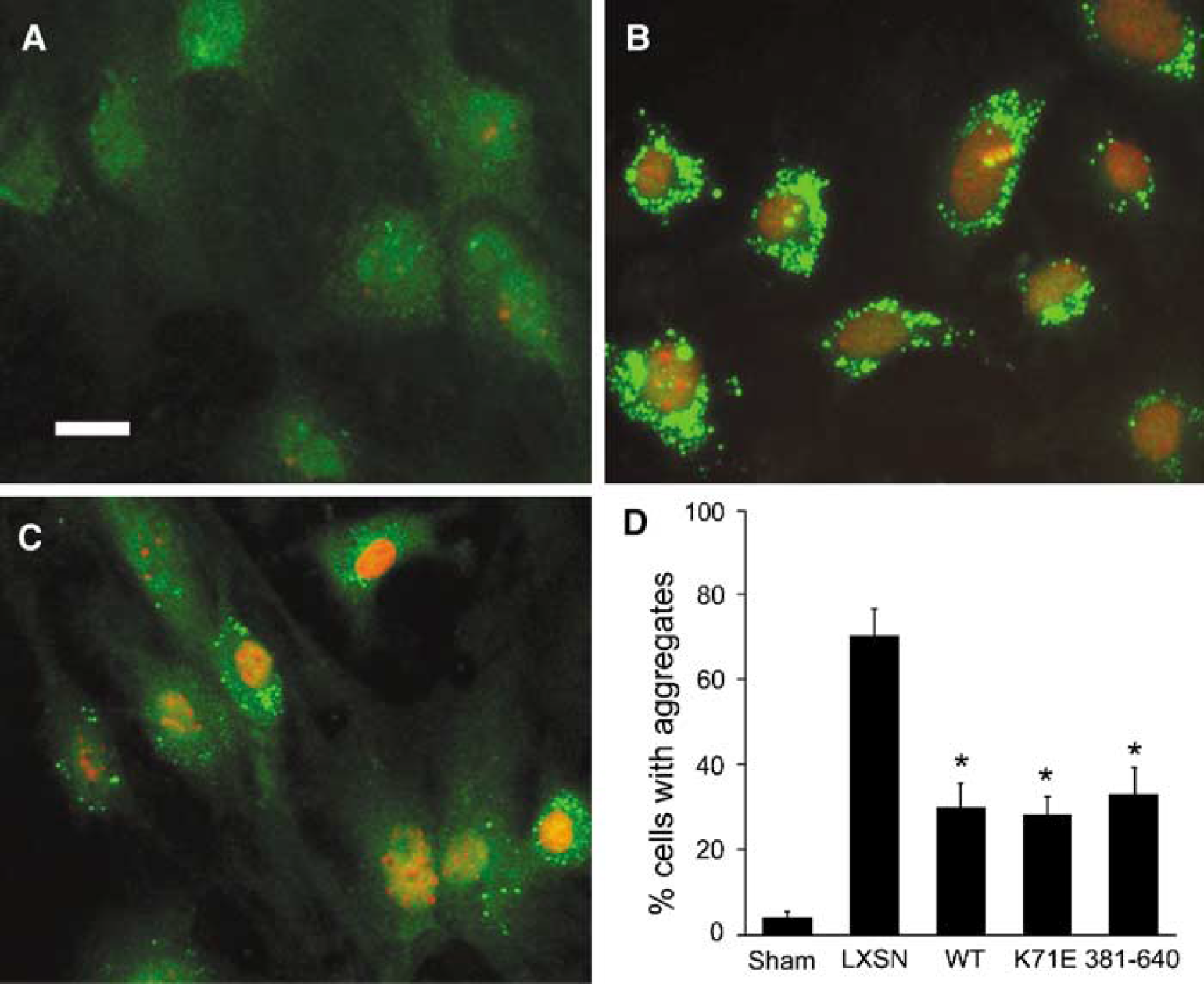
Heat shock protein 70-WT, -K71E, and −381-640 reduce OGD-induced protein aggregation. Primary cortical astrocytes were infected with retrovirus encoding Hsp70-WT, K71E, −381-640, or vector LXSN, and ischemic injury was induced with 3 h OGD, followed by 24 h recovery. Photomicrographs of astrocytes labeled with antiubiquitin show diffuse ubiquitin immunoreactivity in both the nucleus and cytoplasm in uninjured control cells, sham (
Expression of Heat Shock Protein 70-Wild Type and Mutants in Neurons and Astrocytes in Brain
Since both Hsp70-K71E and −381-640 were protective in primary astrocyte cultures, we then tested whether these two mutants could protect the brain from focal cerebral ischemia in vivo. Plasmid DNA encoding the two Hsp70 mutants, Hsp70-WT, or empty vector LXSN was stereotactically injected into the right lateral cerebral ventricle with nonviral cationic lipid as described previously (Hecker et al, 2001). The rats were killed 24 h after microinjection, perfused, and the brains were removed. The identity of Hsp70-K71E-expressing cells was determined by double immunostaining with antibodies against cell type-specific markers. Heat shock protein 70-K71E expression was observed in the cytoplasm and the processes of both neurons (Figures 4A to 4C) and astrocytes (Figures 4D to 4F). In addition to immunohistochemistry of sections, we performed Western blots on brain extracts from both cortex and striatum to show the increase in expression of Hsp70 in these regions at 24 h. In addition, we probed for Hsp40, 60, and 90 to rule out a general heat shock response after injection of a plasmid, leading to forced overexpression of a protein. We were unable to detect changes in any of these three proteins at 24 h after injection of plasmid encoding Hsp 70-WT (Figure 4G).
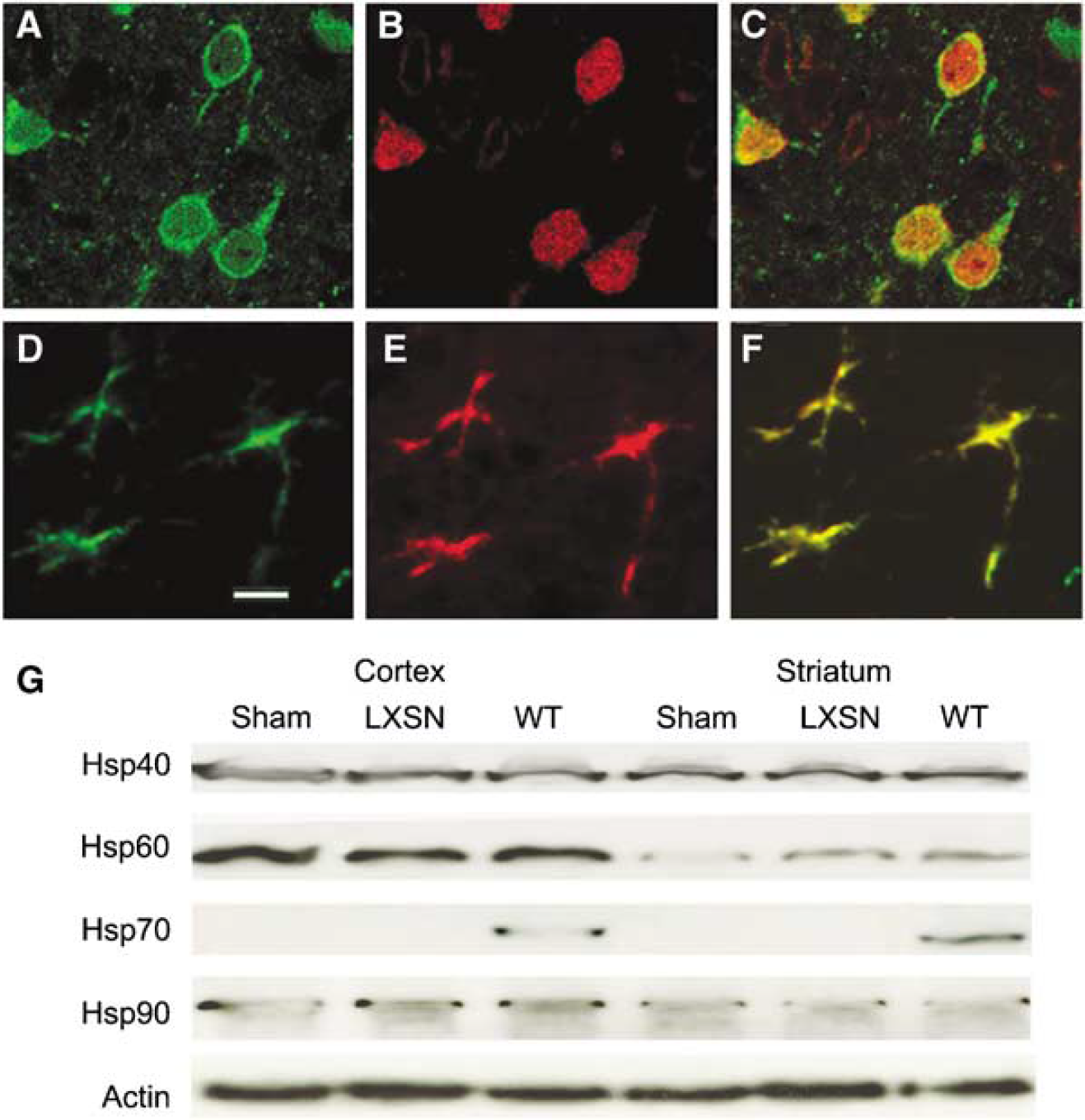
Heat shock protein 70 K71E was expressed in neurons and astrocytes after nonviral DNA transfection using cationic lipid in rat brain. A mixture of DNA and cationic lipid was stereotactically infused into the right lateral ventricle. Rats were killed 24 h later and paraffin sections were stained with Hsp70 antibody and visualized with fluorescein or rhodamine-labeled secondary antibody. Double staining showed colocalization of Hsp70-K71E (
Heat Shock Protein 70 Mutants Decrease Infarct Size and Improve Neurological Outcome
The plasmids encoding Hsp70-WT, -K71E, −381-640, or vector LXSN alone were mixed with lipid, then stereotactically microinjected into the right lateral ventricle. Focal cerebral ischemia was induced 24 h after injection, since Hsp70 mutants were expressed at that time (Figure 4), as determined by immunocytochemistry and Western blotting. There was no difference with respect to mean arterial blood pressure, arterial partial pressure of O2 or CO2, arterial blood pH, or blood glucose concentration between groups (data not shown). Triphenyltetrazolium chloride (TTC) staining of the four groups showed that animals that received Hsp 70-WT, -K71E, or −381-640 had significantly smaller infarct volumes compared with vector LXSN control, as shown in Figures 5A and 5B. There were no statistically significant differences in infarct volume between the vector LXSN group and a lipid-only group (data not shown). Rats that overexpressed Hsp70-WT or mutants displayed better neurological function and neurological scores were significantly decreased compared with vector-control rats (Figure 5C).
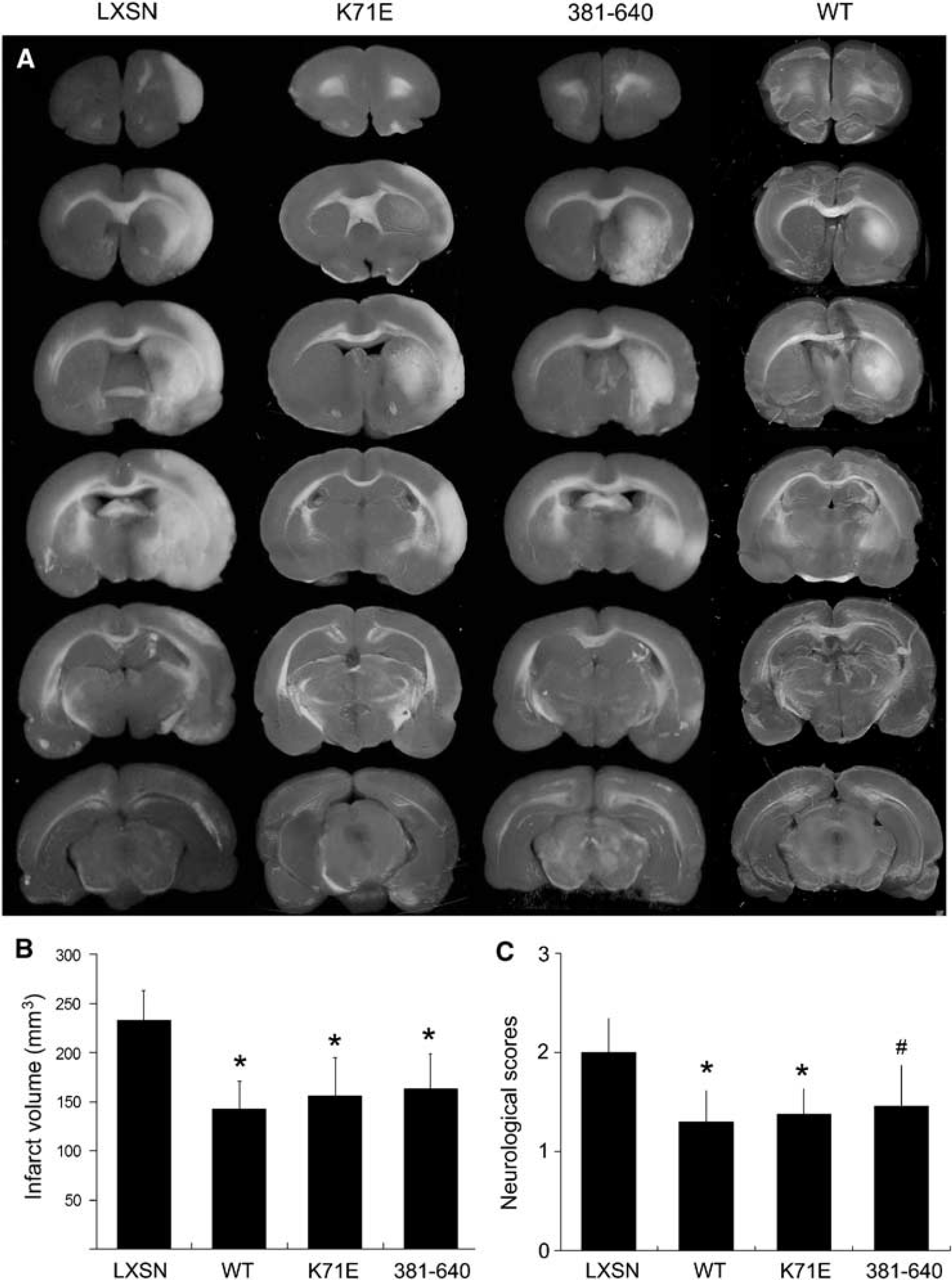
Heat shock protein 70-wild type, -K71E, and −381-640 reduce infarct volume and improve neurological outcome after transient MCAO. Heat shock protein 70-wild type, -K71E, −381-640, or vector LXSN was infused into the right lateral cerebral ventricle followed 24 h later by MCAO. Infarct volume and neurological scores were assessed at 24 h reperfusion after 2 h MCAO. (
Heat Shock Protein 70-Wild Type and Mutants Decrease Cell Injury after Middle Cerebral Artery Occlusion
The effect of these two mutants as well as of Hsp70-WT on cell death after ischemic injury was then evaluated morphologically with cresyl violet staining and Klenow staining of paraffin sections from brains of the five groups of rats. Groups of three rats for each condition were studied. Ischemic cell death involves both necrosis and apoptosis (Giffard and Yenari, 2004). When we evaluate morphology in the penumbral region, normal cells are pyramidal or round in shape (Figure 6A) and do not stain with Klenow labeling (Figure 6F), whereas ischemic dead cells have acidophilic cytoplasm, as well as dark, shrunken, and polygonal nuclei (Figure 6B, arrowheads) and show positive Klenow staining (Figure 6G). Overexpression of Hsp70-WT, -K71E, or −381-640 was associated with a reduction in cellular injury (Figures 6C-E), as shown by morphological criteria—cell shrinkage and nuclear pyknosis, and an increased number of cells with normal morphology, as well as by reduction of the number of cells showing DNA strand breaks by Klenow staining (Figures 6H to 6J). Quantitative analysis showed that the number of cells with evidence of DNA strand breaks was decreased in the Hsp70-WT, -K71E, or −381-640 treated groups compared with vector LXSN (Figure 6K).
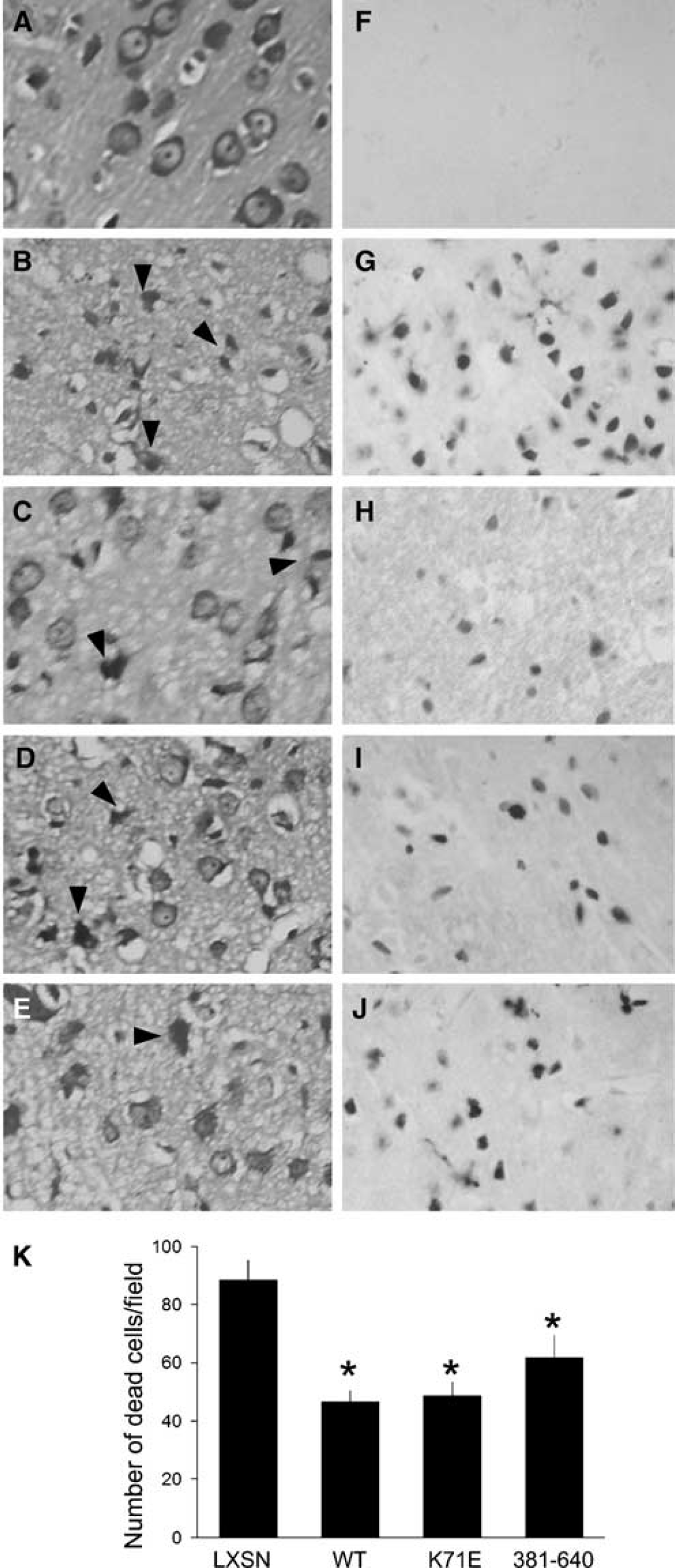
Heat shock protein 70-wild type, -K71E, and −381-640 reduce histological cell damage after transient MCAO in rats. Cell injury was evaluated with cresyl violet (
Heat Shock Protein 70-K71E and −381-640 Inhibit Apoptosis-Inducing Factor Nuclear Translocation after Middle Cerebral Artery Occlusion
Since AIF translocation has been noted early after MCAO in mice (Plesnila et al, 2004), and Hsp70 can inhibit this translocation, we tested whether the carboxyl-terminal domain of Hsp70 could reduce AIF translocation. Apoptosis-inducing factor was localized to mitochondria in normal control brain sections (Figure 7A), as observed by fluorescence immunostaining. After 2 h MCAO followed by 24 h reperfusion, AIF was often seen localized to condensed and fragmented nuclei (Figure 7B, arrows), suggesting that AIF was released from mitochondria and translocated to nuclei after ischemic injury in the rat. Fewer cells showing AIF nuclear translocation were observed in sections from Hsp70-381-640-overexpressing brains (Figure 7C, arrows) compared with sections from vector LXSN brains (Figure 7B). More cells retained a perinuclear, mitochondrial distribution. Cell counting showed that both Hsp70-K71E and −381-640 significantly decreased AIF nuclear translocation (Figure 7G). These results show that AIF nuclear translocation occurs in the penumbra after transient focal ischemia, and that the carboxyl-terminal portion of Hsp70 is sufficient to reduce this.
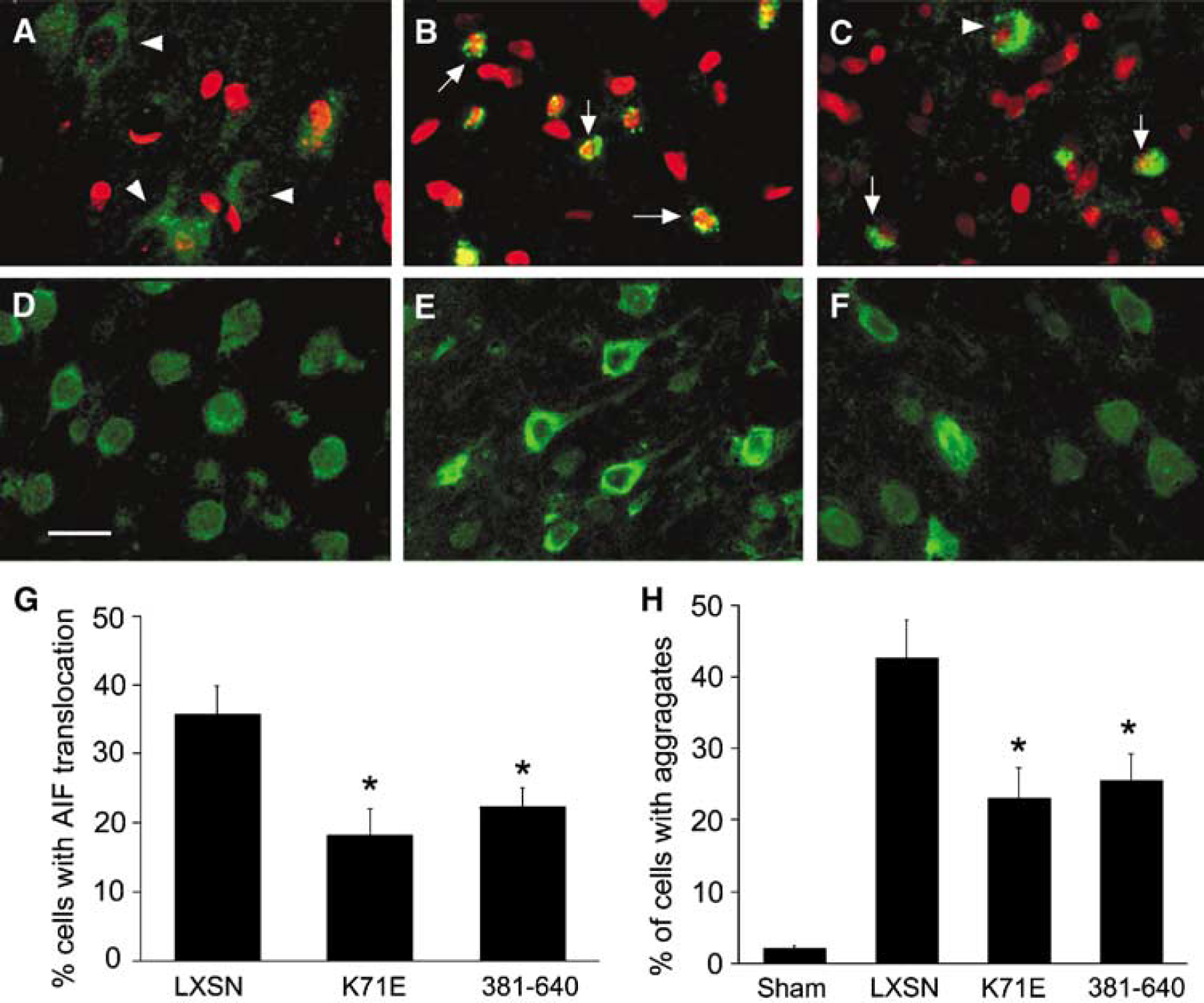
Heat shock protein 70 K71E and −381-640 reduce protein aggregation and AIF translocation in ischemic brain. Paraffin-embedded sections (6 μm) were prepared from animals killed at 24 h reperfusion after MCAO. Representative fluorescence staining with antibody against AIF (
Heat Shock Protein 70-K71E and −381-640 Inhibit Protein Aggregation after Focal Ischemia
Ubiquitin immunostaining was performed in brain sections from the animals subjected to 2 h MCAO followed by 24 h reperfusion. Ubiquitin immunoreactivity is observed in both the nucleus and cytoplasm of cells in sham control brain sections (Figure 7D). After ischemia, the injured cells showed redistribution of ubiquitin staining with increased cytoplasmic staining and decreased nuclear staining (Figure 7E). These results are consistent with the observations in primary cultures shown in Figure 3 above and prior results (Giffard et al, 2004; Hu et al, 2001; Ouyang et al, 2005). Heat shock protein 70-381-640 markedly reduced the number of cells with evidence of protein aggregation (Figure 7F). Both Hsp70-K71E and −381-640 significantly decreased ubiquitin redistribution (Figure 7H). The results found here after both in vivo and in vitro injury indicate that overexpression of the Hsp70 carboxyl-terminal domain is sufficient to reduce protein aggregation after ischemic injury.
Discussion
In this report, we studied two Hsp70 mutants: (1) Hsp70-K71E, a point mutation that abrogates ATP binding and renders the protein deficient in folding ability, and (2) Hsp70-381-640, a deletion mutant lacking the first 380 amino acids of Hsp70, which includes the entire ATP-binding domain and binding sites for several interacting proteins. Both mutants can bind denatured proteins and maintain their solubility, but are unable to fold them. We find that overexpression of Hsp70-K71E or −381-640 protects against ischemia-like injury of primary cultured murine astrocytes and reduces focal ischemic injury induced by transient MCAO in vivo. These results are consistent with prior reports showing that a deletion mutant, containing the peptide-binding domain of Hsp70 but lacking the ATPase domain, is still capable of protecting cells from heat (Li et al, 1992), serum withdrawal (Ravagnan et al, 2001), heat-stress-induced apoptosis (Volloch et al, 1999), and tumor necrosis factor (TNF)α (unpublished results). We did not observe a general stress response after forced overexpression of these proteins either in cultured astrocytes or in brain. Despite this, it is still possible that other changes in gene expression or signaling secondarily induced by overexpression of Hsp70-WT or mutants, but not by the backbone LXSN, resulted in a protective effect.
The molecular and biochemical basis of the protective effect of Hsp70 against ischemia is not well understood, although several hypotheses have been advanced (Giffard and Yenari, 2004). It is widely accepted that Hsp70 can inhibit both necrosis and apoptosis, while its downregulation is sufficient to facilitate the induction of cell death (Lee et al, 2004; Nylandsted et al, 2000). Stressinduced apoptosis can proceed through a mitochondria-dependent signaling cascade that involves release of cytochrome c, Apaf-1, and activation of caspase proteases (Cain et al, 2002; Zou et al, 1997). Apoptosis can also be triggered by release of AIF from mitochondria, which then goes to the nucleus to trigger caspase-independent cell death. Apoptosis-inducing factor translocation has been shown to occur early after focal ischemia (Plesnila et al, 2004; Zhao et al, 2004) and is implicated in poly(ADP ribose) polymerase-mediated cell death (Wang et al, 2004; Yu et al, 2002) such as occurs with excitotoxicity, an important component of ischemic neuronal death. A recent report found that Hsp70 could sequester AIF in neonatal hypoxic/ischemic injury (Beere et al, 2000; Li et al, 2000; Matsumori et al, 2005; Ravagnan et al, 2001; Saleh et al, 2000).
Heat shock protein 70 can physically interact with AIF, resulting in suppression of caspase-independent cell death (Matsumori et al, 2005; Ran et al, 2004; Ravagnan et al, 2001). The point mutant Hsp70-K71E may still interact with most of the Hsp70-specific cochaperones and interacting proteins and retain anti-cell death activity based on several mechanisms. Heat shock protein 70-381-640 is missing all binding sites in the ATPase domain, but retains those interaction sites present in the carboxyl-terminal 259 amino acids, including the binding site for AIF (Schmitt et al, 2003). ATP is not required to interact with AIF and an Hsp70 deletion mutant lacking the carboxyl-terminal peptide-binding domain failed to interact with AIF and failed to confer protection from induced apoptosis (Ravagnan et al, 2001). Apoptosis-inducing factor nuclear translocation was previously observed in focal ischemia in mouse (Plesnila et al, 2004). We found nuclear translocation of AIF in the penumbra after transient focal ischemia, and this was reduced by expression of both Hsp70 mutants. The results described above from Ravagnan are consistent with our finding that both -K71E and −381-640 inhibit AIF translocation to nuclei, which in this case reduced ischemic cell death. Interestingly, the extent of protection by the deletion mutant was generally similar to that of Hsp70-WT and -K71E.
The interaction of Apaf-1 with cytochrome c and ATP, leading to activation of caspase 9, has been shown to be inhibited by Hsp70. However, despite some reports of direct interaction of Hsp70 with the apoptosome (Beere et al, 2000; Saleh et al 2000), it is our view that Hsp70 acts primarily by preventing cytochrome c release from the mitochondria, and that the reported ability of Hsp70 to block caspase-9 activation in cytosols was due to the high salt concentration in the Hsp70 preparation (Steel et al, 2004). In other studies, we have shown that both the Hsp70-K71E and −381-640 mutants can protect cells from heat and TNFα-induced apoptosis (unpublished). The interaction with AIF is known to require the peptide-binding carboxyl-terminal domain (Ravagnan et al, 2001). Our results suggest that inhibition of the AIF pathway is sufficient to block ischemia-induced cell death. This is consistent with earlier data showing that deletion of the carboxyl-terminal substrate-binding domain abolished the protective effect of Hsp70 while deletion of the ATPase domain did not prevent protection from apoptosis induced by serum withdrawal or staurosporin (Ravagnan et al, 2001) or heat (Volloch et al, 1999). In contrast, the ATPase domain was required for protection from etoposide and doxorubicin, deaths presumed dependent on Apaf-1 (Ravagnan et al, 2001).
Another activity retained in the Hsp70 mutants that might be relevant to protection from ischemic injury is inhibition of protein aggregation. Heat shock protein 70 is a molecular chaperone that, without ATP, can bind denatured proteins and maintain their solubility, while ATP hydrolysis is required to facilitate folding of nascent or denatured proteins (O'Brien et al, 1996; Ohno et al, 2004). If nascent or denatured polypeptides cannot be folded successfully, they must be degraded by the ubiquitin-proteasome system. However, this process is likely impaired during ischemia due in part to ATP depletion and potentially in part due to inhibition of the proteosome by protein aggregates (Bence et al, 2001). Protein aggregation and misfolding are now recognized to play an important role in a variety of diseases affecting the central nervous system, including acute injury such as stroke, as well as in chronic neurodegeneration such as Huntington's, Alzheimer's, and Parkinson's diseases. Protein aggregates commonly contain ubiquitin immunoreactivity, suggesting that proteins targeted for degradation that fail to be degraded may end up in aggregates.
Heat shock protein 70 overexpression in Rat-1 cells can protect against nuclear protein aggregation, independent of the ATP-binding domain (Stege et al, 1994). We show here that both Hsp70-K71E and −381-640 do reduce protein aggregation, as assessed by ubiquitin immunohistochemistry in cultured astrocytes and in brain sections from rats subjected to ischemic injury. These results are consistent with the idea that ATPase activity and indeed the ATPase domain of Hsp70 are not required for ischemic protection. Furthermore, since ATP levels decrease rapidly with the onset of ischemia, the activity of Hsp70-WT will be influenced by ATP levels, while the mutants will consistently help maintain the solubility of unfolded proteins without increasing ATP demand when supply is reduced by ischemia and the amount of unfolded or aggregated protein may increase. Our results suggest that Hsp70 mimetics that contain the peptide-binding region might be used to reduce injury from cerebral ischemia.
The results presented here identify the peptide binding domain of Hsp70 as sufficient for protection from ischemia in vivo and in vitro. The main findings are that expression of exogenous Hsp70-K71E or −381-640 (a) protects primary astrocytes against ischemic insults in vitro; (b) reduces infarct size and improves neurological function in vivo; (c) decreases evidence of protein aggregation; and (d) inhibits AIF nuclear translocation. The protection from injury seen with the mutants is comparable to that seen with Hsp70-WT. Both inhibition of protein aggregation and reduction of AIF-dependent cell death likely contribute to the protection we observed. The significant protective effect of Hsp70-381-640 raises the possibility that peptides such as the carboxyl-terminal domain of Hsp70 might be a useful therapeutic strategy for the treatment of stroke and neurodegenerative diseases.
Footnotes
Acknowledgements
We thank Dr Lois Greene for providing Hsp70-K71E.