
Abstract
Activation of protein kinase B (PKB, also known as Akt) by phosphorylation at serine-473 and threonine-308 promotes cell survival in multiple in vitro and in vivo models where neuronal death is seen, including traumatic brain injury (TBI); however, whether PKB is activated in humans after TBI was heretofore unknown. Activated PKB inhibits apoptogenic factors and is involved in the regulation of several transcription factors. Accordingly, we examined phosphorylation of the PKB signaling pathway in humans as well as rats after TBI using phosphospecific antibodies. Increased phosphorylation of PKB and PKB substrates was detected in injured brain from both humans and rats. In humans, increased phosphorylation of the PKB signaling pathway-related proteins Bad and forkhead transcription factor (FKHR) was detected in patients with TBI versus controls. In rats, increased phosphorylation of FKHR, inhibitor of κBα, and cyclic adenosine monophosphate responsive element binding protein (CREB) was detected after TBI versus controls. The deoxyribonucleic acid-binding activity of CREB was also enhanced after TBI in rats. Increased phosphorylation of PKB and PKB substrates was identified in neurons and other cell types by immunohistochemistry in both humans and rats. These data show increased phosphorylation of PKB, PKB substrates, and related proteins after both experimental and clinical TBI, suggesting either activation of the PKB signaling pathway or reduced phosphatase activity in both species.
Introduction
Programmed cell death, or apoptosis, is a major mechanism of neuronal cell loss after traumatic brain injury (TBI). Typical deoxyribonucleic acid (DNA) fragmentation and morphological changes of apoptotic neurons are detectable in experimental TBI (Clark et al, 1997a; Colicos and Dash, 1996; Fox et al, 1998; Rink et al, 1995) as well as in human brain after severe head injury (Clark et al, 1999, 2000a; Smith et al, 2000). Studies have shown that apoptosis after TBI is controlled both at the transcriptional and posttranscriptional levels. Changes in the expression profile of bcl-2 family genes and the activation of caspases contribute to the regulation of neuronal apoptosis after TBI (Clark et al, 1997a, 2000; O'Dell et al, 2000; Wennersten et al, 2003; Yakovlev et al, 1997). Additional regulation, both procell survival and procell death, is provided at the posttranslational level, particularly via phosphorylation.
Protein kinase B (PKB; also known as Akt), a serine/threonine kinase involved in diverse signal-transduction pathways, is constitutively expressed in the brain. Activation of PKB via phosphorylation may regulate apoptosis initiated by the intrinsic cell death pathway via downstream phosphorylation and inhibition of the prodeath Bcl-2-related protein Bad (Datta et al, 1997; Henshall et al, 2002) and caspase-9 (Zhou et al, 2000). Other proteins involved in the PKB signaling pathway include forkhead family transcription factors (Linseman et al, 2002; Zheng et al, 2000, 2002) and cyclic adenosine monophosphate (AMP) responsive element binding protein (CREB) (Brami-Cherrier et al, 2002; Du and Montminy, 1998), indicating that the PKB signaling pathway is also capable of regulating cell death and survival at the gene transcription level. Activation of CREB, by phosphorylation at Ser133, may contribute to neuronal survival in vitro (Walton and Dragunow, 2000), and in vivo studies suggest that CREB might be important for hippocampal plasticity and cognitive function (Dash et al, 2002; Wu et al, 2003). Activation of PKB is shown to protect neurons against ischemic injury and is associated with increased neuronal survival after experimental TBI in mice (Noshita et al, 2002; Yano et al, 2001), although the mechanisms are not fully defined. The aim of the current study was to investigate the changes in phosphorylation of PKB, PKB substrates, and related phosphorylated proteins as a surrogate marker of activation of the PKB signaling pathway after both clinical and experimental TBI.
Materials and methods
Human Brain Tissue Samples
This protocol was approved by the Institutional Review Board at the University of Pittsburgh. Informed consent was obtained from the legal next of kin for each patient, or approval was granted for existing tissue specimens. Patients in the study were admitted to the University of Pittsburgh Medical Center between August 1995 and January 2000. Brain tissue samples were obtained from patients (n = 15) who underwent a decompressive craniotomy as part of the management of life-threatening intracranial hypertension after TBI, clinical or radiographic evidence of cerebral herniation or impending cerebral herniation, or significant mass effect. The samples were collected after surgical resection of the injured area of brain. Tissue was divided into two portions, for biochemical assays and immunohistochemistry. For biochemical assays, the tissue was immediately frozen and stored at −70°C until analysis. For immunohistochemistry, the tissue was immersion fixed in 4% paraformaldehyde for 48 h, then cryoprotected in graded sucrose before optimal cutting temperature (OCT) embedding. Control samples for biochemical assays were frozen postmortem brain-bank specimens obtained from patients who died of causes not related to TBI (n = 6). Samples were from the temporal lobe cortex and were originally collected between April and October 1996. For immunohistochemical controls, the temporal cortex was obtained postmortem from subjects without brain injury or neurological disorders (n = 5, age 50 to 75 years) or patients with a clinical and neuropathological diagnosis of Alzheimer's disease (AD, n = 5, age range 76 to 87 years), who were enrolled in the University of Pittsburgh Alzheimer's Disease Research Center brain tissue bank. The latter were used as positive controls, given previous reports showing activation of PKB in patients with AD (Pei et al, 2003; Rickle et al, 2004).
Experimental Model of Traumatic Brain Injury
A previously described controlled cortical impact (CCI) model with secondary hypoxemic insult (Clark et al, 1997b) was used. Adult male Sprague-Dawley rats were anesthetized with 4% isoflurane, intubated, and mechanically ventilated with 2.0% isoflurane/66% N2O/balance O2. A femoral arterial catheter was inserted for monitoring of blood pressure and blood sampling. A craniotomy was made over the left parietal cortex. A temperature probe (Physiotemp, Clifton, NJ, USA) was inserted through a burr hole into the left parietal cortex. Rats were maintained at a brain temperature of 37°C ± 0.5°C and allowed to equilibrate under anesthesia (1.1% isoflurane/66% N2O/balance O2) for 30 mins. After removal of the bone flap, injury was produced using the CCI device (Dixon et al, 1991). For all studies a 5.0-mm rounded impact tip, depth of penetration of 2.5 mm, velocity of 4.0 ± 0.2 m/sec, and duration of deformation of 50 ms, was used. To produce hypoxemia, air and oxygen were blended to achieve an FiO2 of 0.11 beginning 1 min after CCI and maintained for 30 mins during which time the bone flap was replaced. After 30 mins, catheters and probes were removed, anesthesia was discontinued, and rats were allowed to awaken. Rats were extubated, placed in supplemental O2 for 30 mins, then returned to their cages. Naive rats were used as controls.
Western Blot Analysis
Western blotting was performed as described previously (Zhang et al, 2003). Proteins were denatured, separated through sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto polyvinyl difluoride (PVDF) membranes (BioRad, Hercules, CA, USA). After blocking with 5% skimmed milk in 1 × Tris buffered saline (TBS), membranes were incubated in primary antibodies against PKB, phosphorylated PKB (p-PKB) at serine 473 (Ser473) or threonine 308 (Thr308), phosphorylated BAD (p-BAD) at Ser136, phosphorylated CREB (p-CREB) at Ser133, phosphorylated inhibitor of κBα (p-IκBα) at Ser32/36, forkhead transcription factor (FKHR), or phosphorylated FKHR (p-FKHR) at Ser256, all from Cell Signaling (Beverly, MA, USA) overnight at 4°C. Washed membranes were incubated in horseradish peroxidase-conjugated secondary anti-rabbit or anti-mouse antibodies (Santa Cruz Biotechnologies, Santa Cruz, CA, USA) and immunoblotted proteins were detected using chemiluminescent reagents (NEN, Boston, MA, USA). Relative optical densities (ROD) for protein bands of interest were determined using an image analysis system and software (Kodak Image Station, Rochester, NY, USA).
Immunohistochemistry in Human Brain Tissue
Immunohistochemistry for p-PKB and p-PKB substrates was conducted using a free-floating technique as previously described in tissues from eight TBI patients, five control patients, and five AD patients (Ikonomovic et al, 2004). Briefly, fixed brain tissue samples were cut on a freezing sliding microtome. Sections were preblocked with 10% normal serum and 0.1% Triton X-100 in 0.1 mol/L PBS. Sections were incubated overnight at 4°C with antibodies against p-PKB (Ser473) and p-PKB substrates (Cell Signaling, Beverly, MA, USA). Sections were then incubated in the affinity-purified biotinylated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) secondary antibody diluted in 5% normal serum and 0.1% Triton X-100 in 0.1 mol/L PBS at 4°C for 1 h. The peroxidase reaction was developed using a Diaminobenzimide Substrate Kit with nickel enhancement (Vector, Burlingame, CA, USA). Sections from TBI, control, and AD temporal cortex were processed simultaneously. As a negative control, omission of primary antibodies resulted in complete absence of immunostaining.
Immunohistochemistry in Rat Brain Tissue
Immunohistochemistry was performed as described previously (Clark et al, 2000b). Briefly, naive rats and rats at 2, 6, 24, 72 h and 7 days after TBI (n = 3/group) were anesthetized as described above and perfused with 200 mL ice-cold heparinized saline, followed by 500 mL 2% paraformaldehyde. Brains were removed, immersion-fixed for 30 mins, cryoprotected in 30% sucrose, then frozen in precooled isopentane suspended in liquid nitrogen. Coronal brain sections (10 μm) were incubated at 4°C overnight in a 1:50 dilution of primary antibodies against PKB, p-PKB (Ser473), or p-PKB substrates (Cell signaling, Beverly, MA, USA), then incubated in a 1:200 dilution of anti-rabbit secondary antibody conjugated with Alexa488 (Molecular Probes, Eugene, OR, USA) for 1 h. For dual labeling of neurons, anti-NeuN (Chemicon, Temecula, CA, USA) at a dilution of 1:600 was mixed and incubated with each primary antibody, followed by a 1:50 dilution of rhodamine-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA). In sections from each specimen, the primary antibody was omitted to assess for nonspecific binding of the secondary antibody.
Electrophoretic Mobility Shift Assay (EMSA)
An EMSA was used to determine the DNA-binding activity of CREB in hippocampal tissue samples from rats after TBI. Naïve rats (n = 5) and rats of 2, 6, and 24 h after TBI (n = 3/group) were anesthetized as described above, then killed. Ipisilateral hippocampi were dissected on ice and proteins were extracted immediately. Nuclear proteins were extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce, Rockford, IL, USA). In all, 8 μg of nuclear proteins from each sample was incubated with radioactive 3′-end-labeled consensus oligonucleotides for CREB (5′-AGA GAT TGC CTG ACG TCA GAG AGC TAG-3′) using a gel shift assay system (Promega, Madison, WI, USA). Nonlabeled oligonucleotides were used as cold competitors to verify the specificity of binding. The reactions were separated on nondenatured 5% TBE gels. Protein-DNA complexes were detected by autoradiography.
Data Analysis
For human brain samples, comparisons regarding relative protein abundance between control and TBI groups determined by Western blot were made using a t-test or Mann-Whitney rank sum test if data were not normally distributed or failed tests of equal variance, and immunohistochemical data were descriptively analyzed. For rat brain samples, both Western blot and immunohistochemical data were descriptively analyzed, since Western blot data represent pooled samples. Comparisons of the ROD of CREB binding via EMSA between groups were made using one-way ANOVA. A P < 0.05 was considered significant.
Results
Patient Demographics
Samples used in this study were from the University of Pittsburgh Brain Trauma Research Center tissue bank. The majority of the samples (6/6 control patients and 15/17 TBI patients) were from patients where separate tissue was used to report the presence of cell death markers after TBI (Zhang et al, 2003). For the control patients, age = 52.2 ± 22.1 years, there were 4 women and 2 men, and postmortem time = 10.5 ± 5.7 h. For the TBI patients, age = 42.5 ± 16.5, there were 3 women and 12 men, median initial Glasgow Coma Scale score (CCS) = 6 (3 to 15), and time from injury to resection = 17.1 ± 28.4 h. There was no difference in the ages of the control group compared with the TBI group (P = 0.269). Mechanism of injury included nine motor vehicle-related accidents, four falls, and two assaults. Detailed demographic data can be found in Zhang et al (2003).
Effect of Postmortem Time on Protein Kinase B-Phosphorylated Proteins
Protein phosphorylation is a covalent modification and an energy-dependent process. Since the control brain tissues were removed postmortem (2.5 to 20 h), our concern was that detectable phosphorylated protein levels could decrease during postmortem time due to energy depletion and/or protein phosphatase and proteome degradation. To address this issue, a brain sample obtained via biopsy from a TBI patient was cut into four pieces that were stored at 4°C to mimic morgue conditions. Proteins were then extracted at 0, 2, 6, and 24 h from these pieces to mimic different postmortem times. Western blot analysis of p-PKB substrates was then performed to determine whether the level of PKB-phosphorylated proteins decayed over time when stored at 4°C. As shown in Figure 1, protein phosphorylation did not decrease over time, as detected using the antibody against p-PKB substrates.
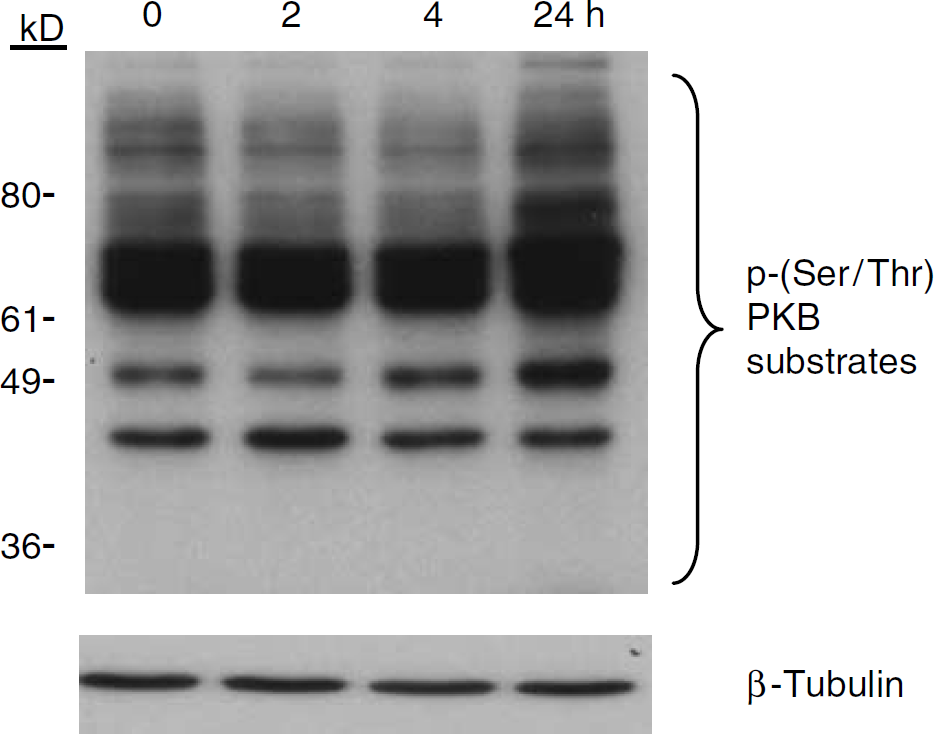
Effect of time on protein phosphorylation as determined using Western blot analysis and an antibody against PKB substrates. Human brain tissue was stored at 4°C to mimic postmortem conditions. Proteins were extracted at 0, 2, 6 and 24 h. Relative phosphorylation of PKB substrates did not appear to diminish with increasing refrigeration time.
Increased Phosphorylation of Protein Kinase B and its Substrates after Traumatic Brain Injury in Humans
Western blotting of human TBI brain samples revealed that phosphorylation of PKB at Thr308 was increased after TBI compared with controls (49.2 ± 32. 4 versus 4.4 ± 8.9 ROD, respectively; P = 0.002; Figure 2). Unphosphorylated PKB was also increased in TBI patients compared with control patients using the anti-PKB antibody (81.7 ± 33.5 versus 36.9 ± 39.7 ROD, respectively; P = 0.02; Figure 2). Western blotting using an antibody against PKB phosphorylated at Ser473 was unsuccessful in our hands. Activation of PKB by phosphorylation was supported by the increase in phosphorylation of its downstream substrates in injured brain. Multiple phosphorylated proteins were detected in many of the TBI patients ranging from 36 to 80 kDa, but were not detected in controls (124.1 ± 82.3 versus 1.6 ± 1.3 ROD of the ~40, 50, 60, and 70 kDa bands, respectively; P < 0.001; Figure 2). Activation of PKB and/or other kinase signaling pathways was also supported by increased phosphorylation of the transcription factor FKHR (phosphorylated at Ser256; 31.0 ± 49.8 versus 0.0 ± 0.0 ROD, TBI versus control patients, respectively; P = 0.01; Figure 2) and the Bcl-2-related protein BAD (phosphorylated at Ser136; 14.8 ± 17.7 versus 0.0 ± 0.0 ROD, TBI versus control patients, respectively; P= 0.006; Figure 2).
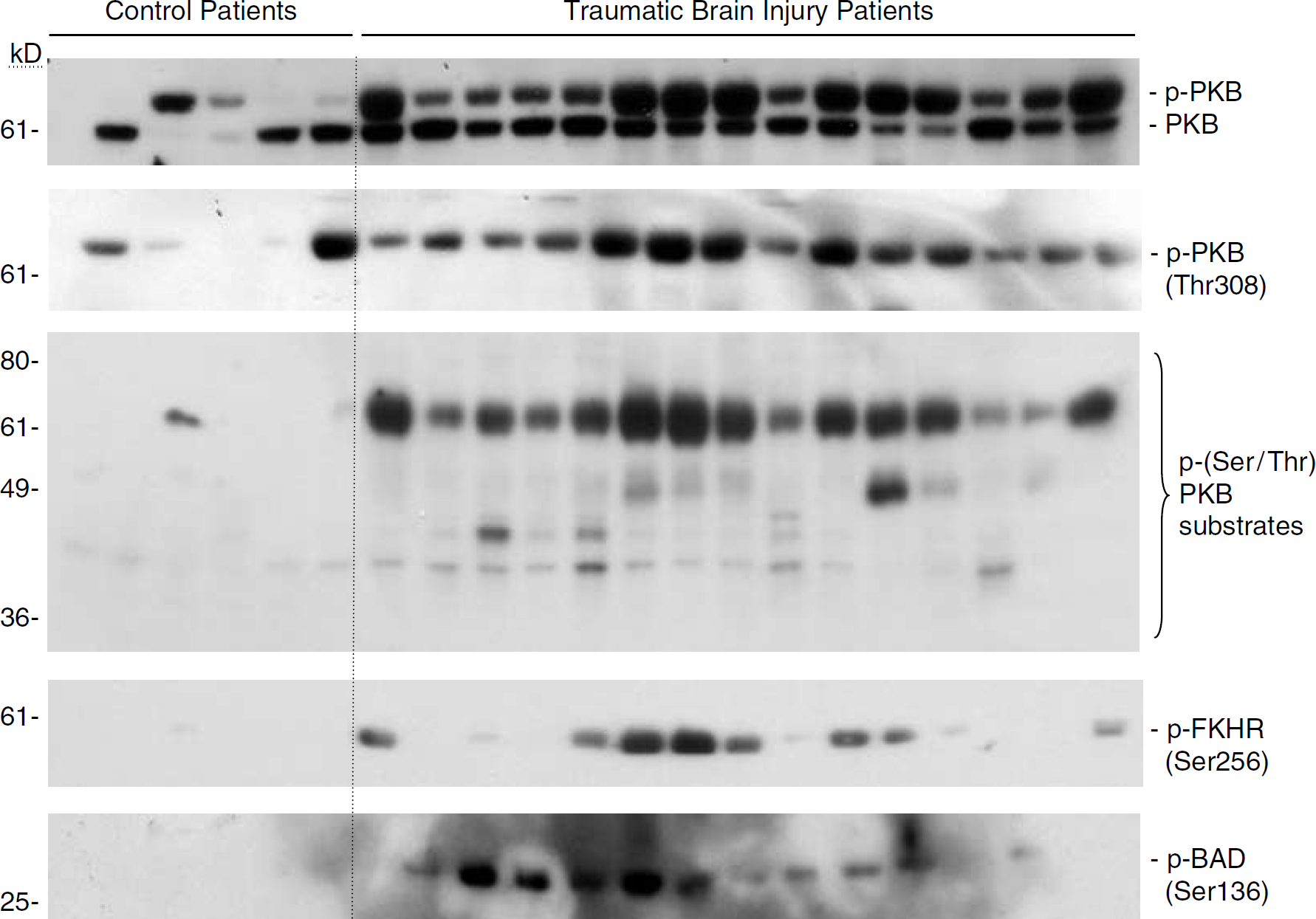
Relative levels of PKB, phosphorylated (p) PKB, p-PKB substrates, and the related proteins p-FKHR and p-BAD in brain tissues from TBI patients undergoing decompressive craniotomy (n = 15) and brain bank control patients (n = 5), as determined using Western blot analysis and phosphospecific antibodies. Relative levels of p-PKB, PKB substrates, p-FKHR, and p-BAD appeared to be increased in TBI versus control patients.
To examine cellular localization of p-PKB and p-PKB substrates after TBI in humans, immunohistochemical analysis was performed using samples from temporal cortex from a subset of the TBI patients. Minimal p-PKB (Ser473) and p-PKB substrate immunoreactivity was detected in controls (Figures 3A and 3I, respectively). In contrast, brain sections from TBI patients showed marked increases in both p-PKB and p-PKB substrate immunoreactivity, which were localized to the soma and processes of numerous cell elements with morphological characteristics of either neurons (Figures 3C to 3D and 3K to 3L, respectively) or glia (Figures 3E to 3H and 3M to 3P, respectively). Increased p-PKB and p-PKB substrate immunoreactivity was also found in the temporal cortex from AD patients, where labeled cell bodies and processes with neuronal morphology were seen (Figures 3B and 3J, respectively). This is consistent with previous reports showing activation of the PKB signaling pathway in patients with AD (Pei et al, 2003; Rickle et al, 2004).
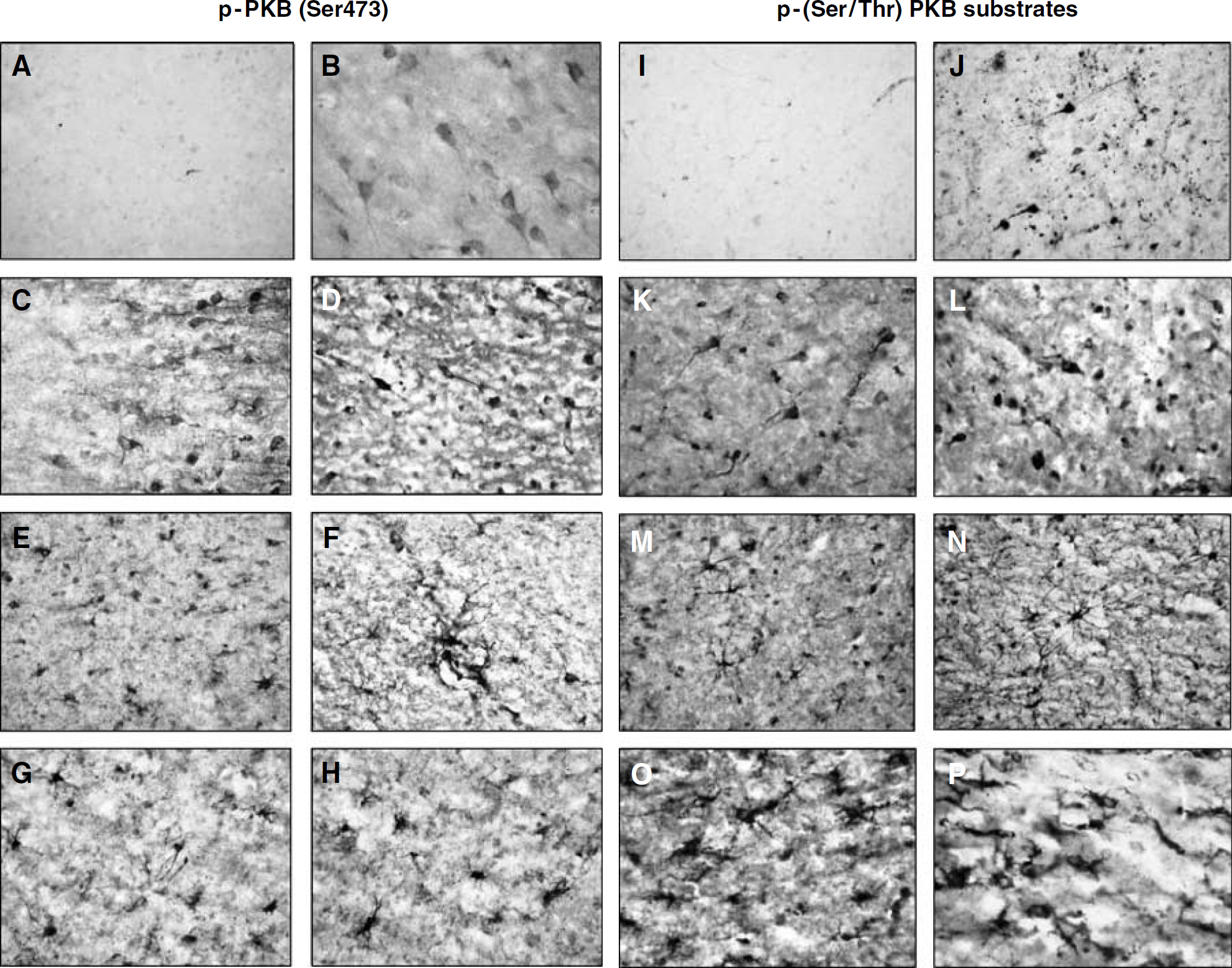
Cellular localization of p-PKB (
Increased Phosphorylation of Protein Kinase B and its Substrates after Traumatic Brain Injury in Rats
Limitations of our human studies are the substantial variability of clinical presentation seen in TBI patients requiring surgical intervention, and the difficulty in obtaining age-matched, disease-free biopsy controls. To corroborate the findings using injured brain from humans, an established rat brain trauma model (Clark et al, 1997b) was used to examine the time course and cellular expression of the PKB signaling pathway. Western blot analysis using pooled samples (n = 3 to 5/group) showed relative levels of total PKB in naïve rat cortex and hippocampus that did not appear to change at 2 to 72 h after TBI (Figure 4A). Baseline phosphorylation of PKB at Ser473 was detected, and relative levels appeared to increase in both ipsilateral cortex and hippocampus at 6 h after TBI (Figure 4A). In contrast to p-PKB (Ser473), minimal phosphorylation of PKB at Thr308 was detected at baseline, whereas relative levels appeared to be increased in both ipsilateral cortex and hippocampus at 72 h after TBI (Figure 4A). Relative protein levels of FKHR appeared to remain constant before and after TBI in both cortex and hippocampus; however, a modest increase in phosphorylation of FKHR at Ser256 was apparent in ipsilateral cortex at 72 h and in ipsilateral hippocampus at 6 and 24 h after TBI versus naïve controls (Figure 4A). In our hands, it did not appear that the antibody used to detect p-BAD in human tissues reacted with p-BAD in rat tissues (data not shown). Western blotting also showed increased phosphorylation of IκBα and CREB, primarily in the ipsilateral hippocampus after TBI versus control (Figure 4A). As determined by Western blotting using the antibody against p-PKB substrates, PKB activation appeared to increase at 6 h in both ipsilateral cortex and hippocampus after TBI versus naïve control (Figure 4B). For all Western blots, equal protein loading was examined by stripping the membrane and reprobing with an antibody against β-tubulin (Figure 4B).
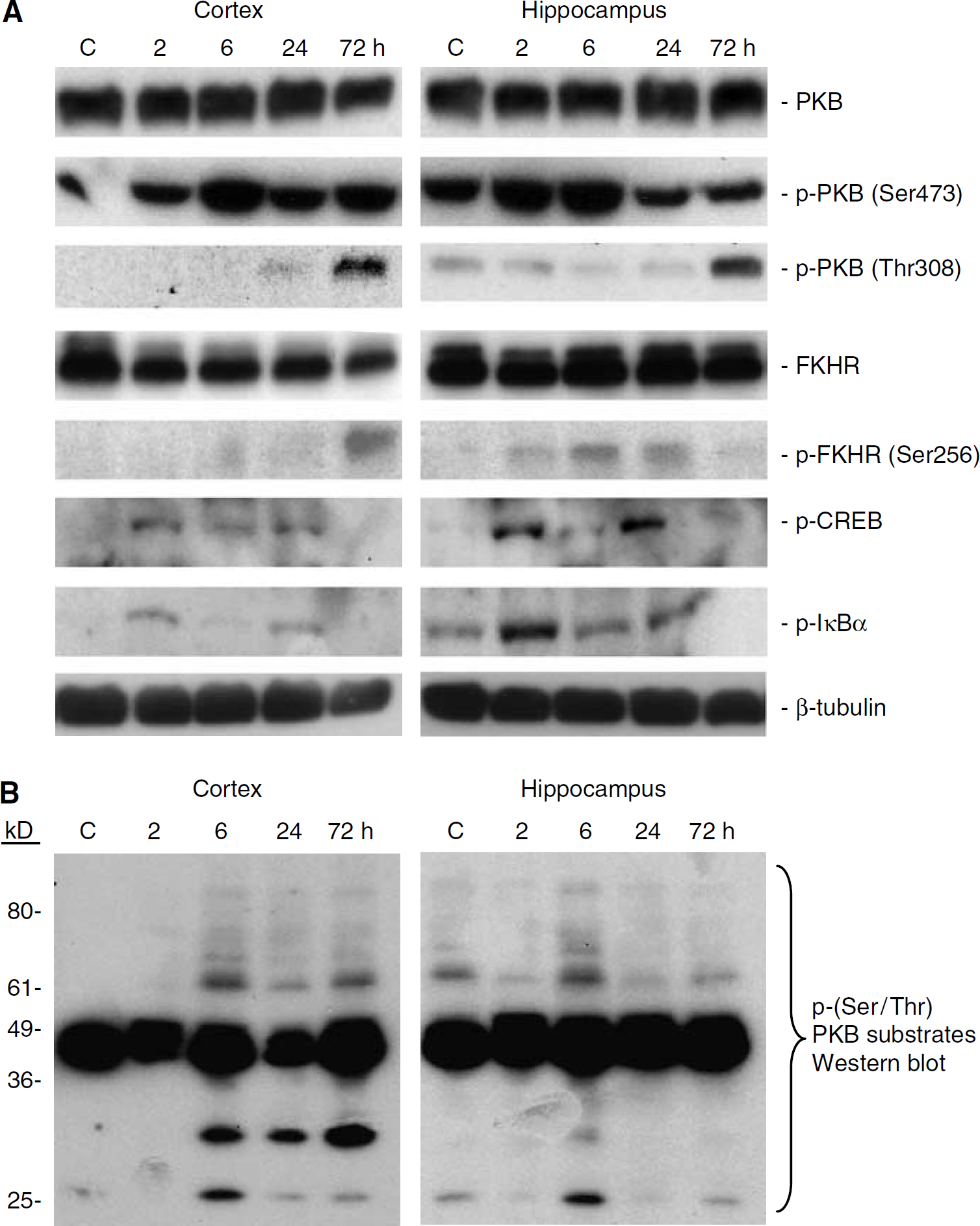
Relative levels of PKB, p-PKB (Ser473 and Thr308), p-PKB substrates, and the related proteins FKHR, p-FKHR, p-CREB, and p-IκBα in pooled ipsilateral cortical and hippocampal tissue samples (n = 3/group) from naïve rats and rats at 2, 6, 24, and 72 h after CCI, as determined using Western blot analysis and phosphospecific antibodies. (
Increased Phosphorylation of Protein Kinase B and its Substrates in Neurons after Traumatic Brain Injury in Rats
Dual-label immunohistochemistry was used to determine whether increased p-PKB and p-PKB substrate expression was occurring in neurons. Consistent with Western blot data, increased p-PKB (Ser473) and p-PKB substrate immunoreactivity was seen in the cortex adjacent to the contusion and ipsilateral hippocampus at 6 h after TBI versus control (Figures 5 and 6). In the hippocampus, increased p-PKB immunoreactivity appeared to occur in all hippocampal regions (CA1 to 4, subiculum, and dentate gyrus) compared with naïve control (Figures 5F versus 5A), whereas increased p-PKB substrate immunoreactivity appeared to be confined to the CA1 hippocampus and subiculum at 6 h after TBI compared with naïve control (Figures 6F versus 6A). Dual labeling with the neuronal marker NeuN showed that the majority of cells with increased p-PKB (Figures 5G to 5J) and p-PKB substrate (Figures 6G to 6J) immunoreactivity within the pericontusional cortex and ipsilateral hippocampus were neurons. Immunostaining in sections incubated without primary antibodies showed no immunoreactivity (data not shown).
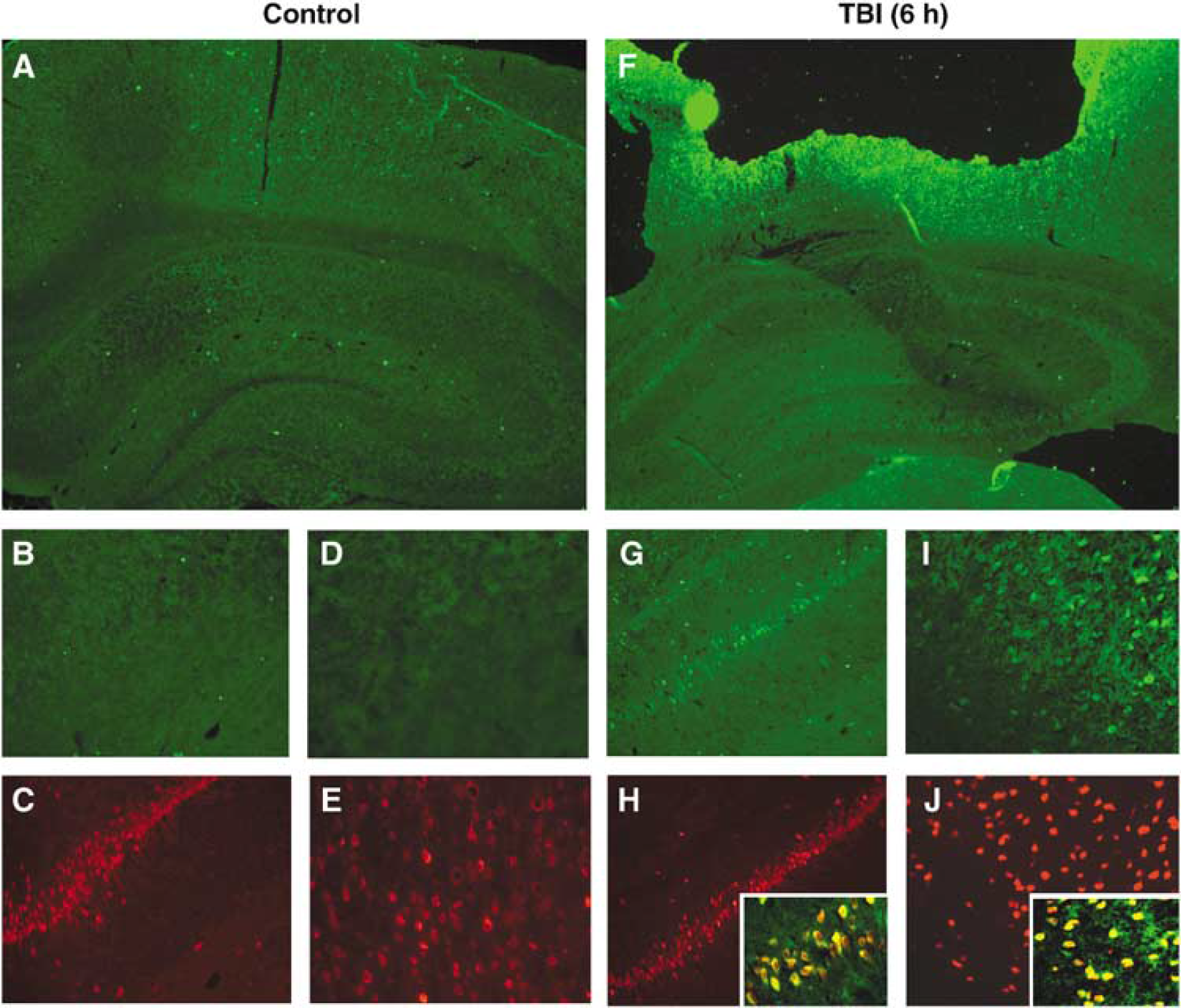
Immunofluorescent staining of p-PKB (Ser473) in naïve rats and rats 6 h after CCI (n = 3/group). Increased p-PKB immunoreactivity (green) is seen in the ipsilateral cortex and hippocampus 6 h after TBI (
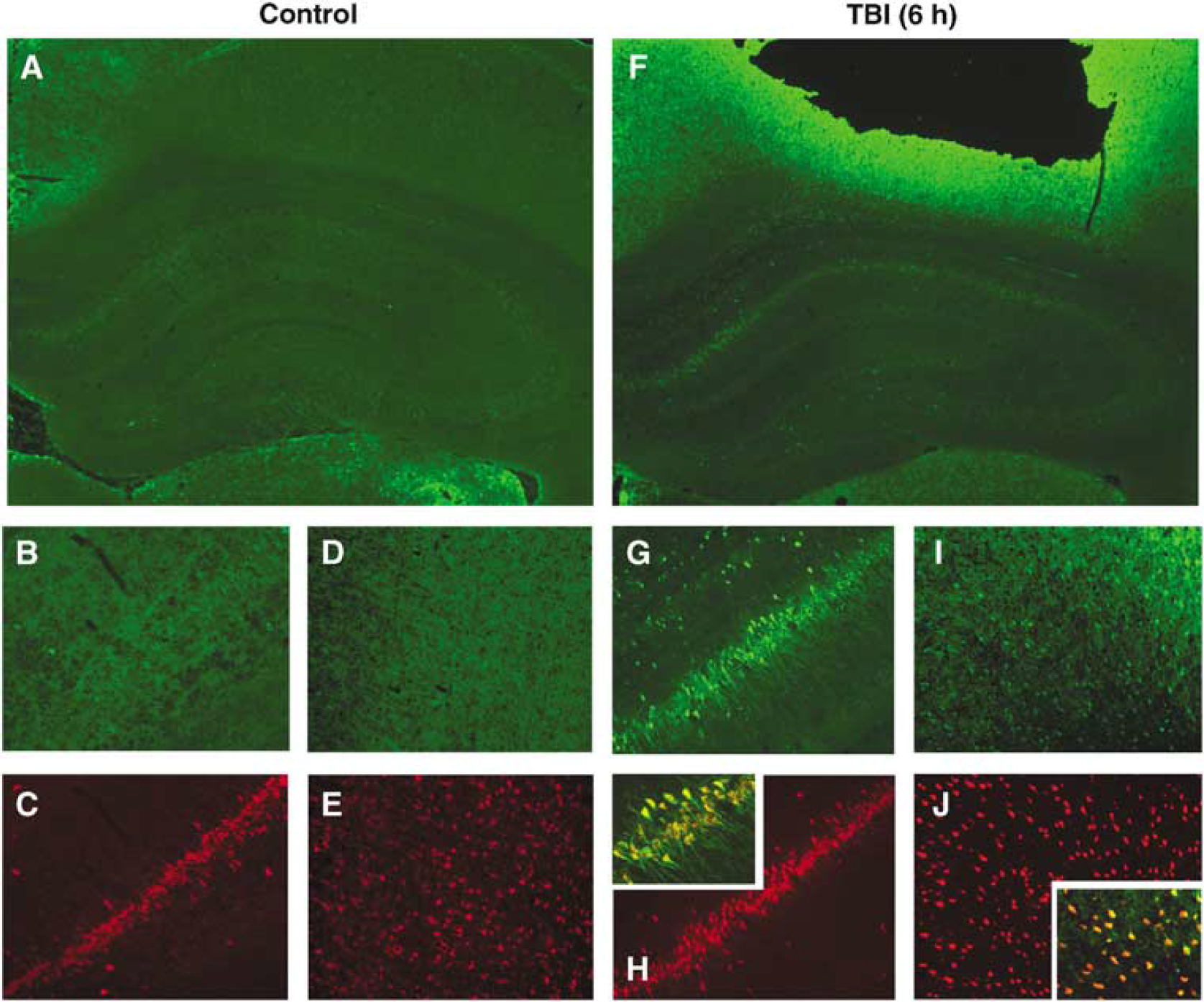
Immunofluorescent staining of p-PKB substrates in naïve rats and rats 6 h after CCI (n = 3/group). Increased p-PKB substrate immunoreactivity (green) is seen in the ipsilateral cortex and hippocampus 6 h after TBI (
Increased Cyclic Adenosine Monophosphate Responsive Element Binding Protein-Deoxyribonucleic Acid-Binding Activity in Hippocampus after Traumatic Brain Injury in Rats
The DNA-binding activity of CREB was determined in the ipsilateral hippocampus by EMSA, since CREB activity is thought to be either regulated or coregulated by the PKB signaling pathway (Du and Montminy, 1998). Cyclic adenosine monophosphate responsive element binding protein-DNA binding increased at 2, 6, and 24 h after TBI compared with naïve controls (all P < 0.05 versus control; Figure 7). The specificity of the CREB-DNA binding was verified using cold competitive and noncompetitive probes.
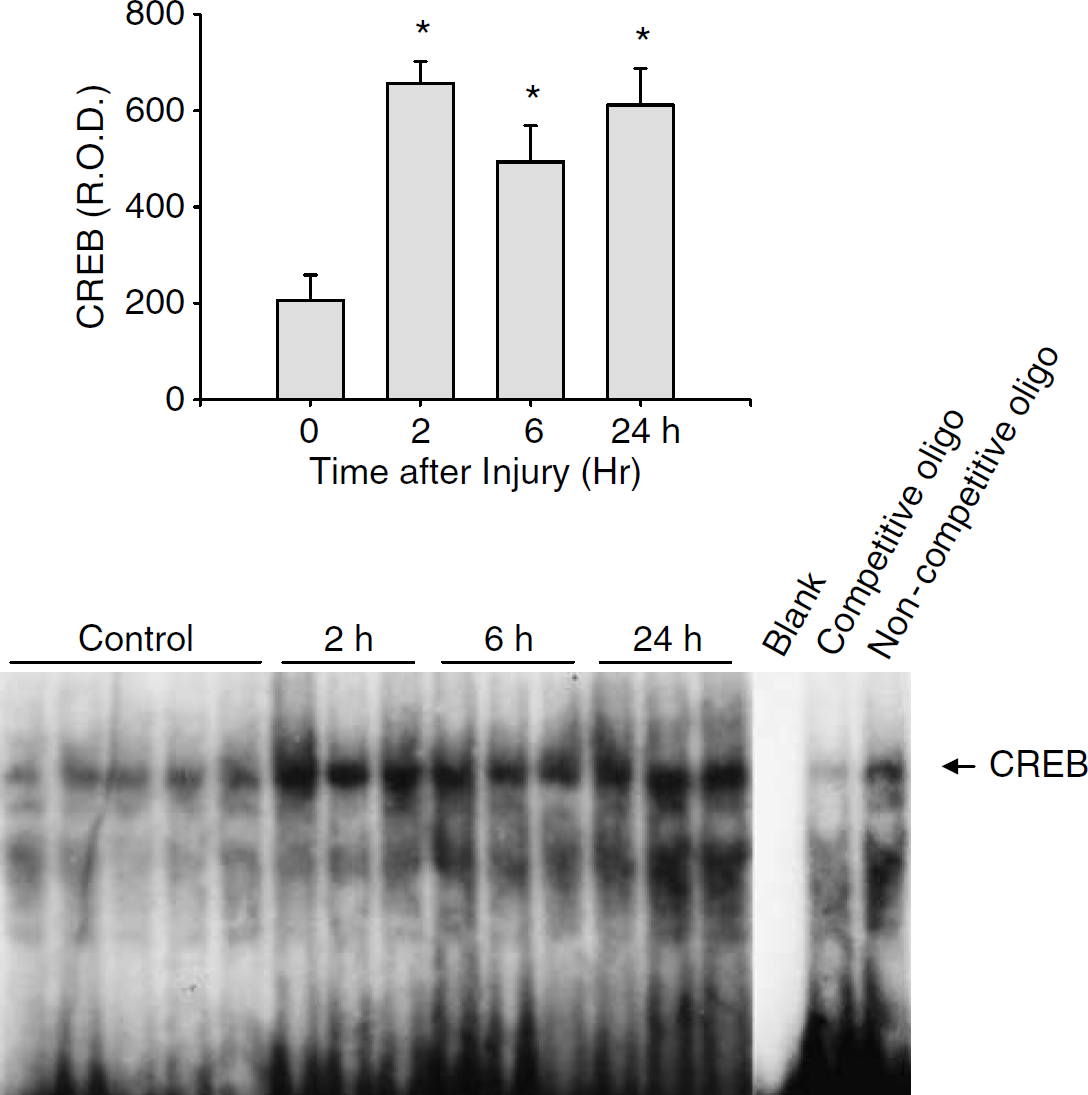
Increased CREB-DNA binding in nuclear homogenates from ipsilateral hippocampus from naïve rats (n = 5) and rats at 2, 6, and 24 h after CCI (n = 3/group) as determined using an EMSA. Relative optical densities were determined for each group. Increased CREB-DNA binding was observed at 2, 6 and 24 h after CCI versus control (*P < 0.05 versus control).
Discussion
The major finding of this study is that in humans, PKB, PKB substrates, and many PKB signaling pathway-related proteins show increased phosphorylation in injured brain after trauma compared with controls, suggesting stimulated activation of the PKB signaling pathway. Alternatively, TBI could result in decreased counterregulatory phosphatase activity. Increased phosphorylation of PKB and its substrates appears to occur in neurons. Results using human tissues and phosphospecific antibodies were corroborated using an established and well-controlled model of TBI in rats. Taken together, these data support activation of the PKB signaling pathway in mammalian brain after TBI. To our knowledge, this is the first description of activation of the PKB signaling pathway in human brain after acute injury.
Evidence of disturbed phosphoinositide-3-kinase (PI3-K)/PKB signaling after TBI has been discussed in a limited number of reports using experimental models of TBI. After TBI in mice, increased phosphorylation of PKB at Ser473 in the ipsilateral CA1 hippocampus and cortex was observed, along with increased phosphorylation of BAD and glucose synthetase kinase-3 (Noshita et al, 2002). After mild TBI in immature rats, alteration of phosphorylated PKB enzyme substrates was also reported in the ipsilateral hippocampus (Jenkins et al, 2002). The present study shows increased phosphorylation of PKB at Thr308 in patients at various time points after TBI compared with controls. In rats after TBI, phosphorylation of PKB at Thr308 was a relatively late finding (72 h), in contrast to phosphorylation at Ser473 which appeared to occur much earlier (6 h) after TBI. Alteration in phosphorylation of substrates either directly phosphorylated by PKB or those downstream within the PKB signaling pathway (FKHR, BAD, IκBα and CREB) supports activation of PKB after TBI in mammalian brain.
The viral oncogene Akt, encoding for PKB/AKT, was first identified in the genome of the retrovirus AKT8, which induces a murine thymic lymphoma (Staal and Hartley, 1988; Staal et al, 1977). Its human homologue AKT1 was subsequently identified in a human gastric adenocarcinoma (Staal, 1987). The amino-acid sequence of human PKB is similar to protein kinase A (PKA) and protein kinase C (PKC) (Coffer and Woodgett, 1991). At least three PKB isoforms have been identified, termed AKT1/PKBα, AKT2/PKBβ, and AKT3/PKBγ, as well as C-terminal splice variants of PKBβ and PKBγ. Protein kinase B contains an N-terminal pleckstrin (PH) domain, which is responsible for binding to phosphoinositol 3,4,5-triphosphate (PIP3), a catalytic domain that is nearly identical to those of PKA and PKC, and a C-terminal regulatory domain (Coffer et al, 1998). The binding of PIP3 to the PH domain of PKBα facilitates the translocation of PKBα to the cell membrane and its phosphorylation by 3-phosphoinositide-dependent kinase 1 (PDK1) at Thr308 of the catalytic loop. The other phosphorylation site is Ser473 within the C-terminal regulatory domain. It is believed that phosphorylation of both sites is required for insulin and IGF-1 to fully activate PKBa (Alessi et al, 1996).
Protein kinase B phosphorylates its protein targets at specific serine and/or threonine residues in the peptide sequence. Its signaling pathway is important in cell survival, protein synthesis, glucose metabolism, nutrition and growth, neuronal synaptic plasticity, and cognitive function (Chan, 2004; Dash et al, 2002; Noshita et al, 2002; Perazzona et al, 2004). Thus, altered PKB signaling could have many functional consequences after mild and moderate brain injury, in addition to severe TBI. Multiple PKB signaling mechanisms important in cell survival have been proposed. Phosphorylation of the Bcl-2 family member BAD (Datta et al, 1997) and caspase-9 (Cardone et al, 1998) modifies intrinsic apoptotic machinery at both pre- and postmitochondrial levels, promoting cell survival over cell death. Activated PKB also inhibits p53 by phosphorylating mouse double minute-2 (Ogawara et al, 2002) and human double minute-2 (Ashcroft et al, 2002). Moreover, the regulation of protein synthesis may also play a role in the cell survival function of PKB signaling (Inoki et al, 2002). In brain, PKB plays an essential role in nerve growth factor-dependent neuronal survival after ischemic and excitotoxic insults (Jin et al, 2003; Lenhard et al, 2002; Siren et al, 2001). Several studies reported increases in phosphorylation of PKB at Ser473 across models of experimental brain ischemia (Jin et al, 2000; Noshita et al, 2001; Ouyang et al, 1999; Shibata et al, 2002). Neurons with increased PKB phosphorylation were TUNEL negative (Noshita et al, 2001), suggesting a prosurvival role for PKB. Phosphorylation of PTEN, a negative modulator of PKB, is increased after ischemia, providing additional evidence for PKB activation after ischemia (Omori et al, 2002).
While the direct functional consequences of increased PKB signaling and neuronal survival after TBI were not assessed in the present study, some cautious inferences can be made. Related to the human studies, cells with increased phosphorylation of PKB and PKB substrates within tissue that was not clearly necrotic appeared morphologically intact (Figure 3). Related to rat studies, cells with increased phosphorylation of PKB and PKB substrates were seen in the contusion penumbra and regions of hippocampus, where cell survival predominates over cell death in this model (Clark et al, 1997b). This is more apparent in the studies related to PKB substrates, where hippocampal regions with increased immunoreactivity included the subiculum and CA1 hippocampus, areas with limited neuronal death compared with dentate gyrus and CA3 hippocampus, where increased PKB substrate immunreactivity was sparse (Figure 6). These data, together with the study by Noshita et al (2001) showing that neurons with increased PKB activity likely survive after cerebral ischemia, support a prosurvival role for PKB signaling after TBI in humans and rats. Caution remains in order, however, and further studies showing cause and effect are necessary.
Multiple cell signaling pathways are altered after TBI, in addition to PKB, and significant crosstalk occurs between these pathways. While reports dealing directly with TBI are somewhat limited in number, several important studies exist, including demonstrated roles for the mitogen-activated protein kinase (Dash et al, 2002; Mori et al, 2002) and PKC (Armstead, 2003) pathways. As an example of potential crosstalk, CREB, in addition to being modulated by PI3-K/PKB, can also be phosphorylated by PKA (Zanassi et al, 2001), extracellular signal-regulated kinase (Cancedda et al, 2003), calcium/calmodulin-dependent protein kinase (Kasahara et al, 2001), and dephosphorylated by protein phosphatases (Bito et al, 1996). In the present study, the sustained increase in CREB-DNA binding in hippocampus is particularly intriguing, given the fact that Bcl-2 is a downstream target of CREB (Riccio et al, 1999), and we have previously shown that Bcl-2 is upregulated in both humans and rats after TBI (Clark et al, 1997a, 1999, 2000a).
In conclusion, the PKB signaling pathway is activated in both humans and rats after TBI. The precise roles of increased PKB activation in terms of cell survival and function, the regulation of PKB signaling by kinases and phosphatases, and crosstalk between cell signaling pathways after both experimental and clinical TBI remain to be determined. Based on evidence of a robust response of the PKB signaling pathway after TBI, further study appears warranted.
Footnotes
Acknowledgements
We thank Barbara A Isanski for technical assistance and Ava Puccio for management of the tissue bank.