
Abstract
The amyloid-beta peptide (
Introduction
Alzheimer's disease (AD) is a neurodegenerative disease that results from the progressive deposition of fibrillar amyloid-β peptide (Aβ) in senile plaques in the brain (Wisniewski and Wegiel, 1995; Yankner, 1996). While considerable attention has focused on parenchymal Aβ accumulation and neuronal degeneration in AD brains (Yankner et al, 1989; Behl et al, 1994), Aβ also accumulates in the cerebrovascular walls of AD patients and in nondemented elderly individuals, leading to cerebral amyloid angiopathy (Perlmutter et al, 1994; Wisniewski et al, 2000). One of the most widely recognized complications of amyloid angiopathy is hemorrhagic strokes (Walker, 1997). Amyloid-β peptide is toxic to cerebral endothelial cells (CECs) (Price et al, 1997; Preston et al, 1998; Xu et al, 2001; Yin et al, 2002b), and may contribute to alterations in cerebral blood flow (Iadecola et al, 1999). Amyloid-β peptide causes CEC death with morphological and molecular features suggestive of apoptosis (Blanc et al, 1997; Hase et al, 1997; Suo et al, 1997; Xu et al, 2001; Yin et al, 2002b). In particular, mitochondrial dysfunction has been prominently shown in CECs exposed to Aβ (Xu et al, 2001; Yin et al, 2002b). However, the molecular mechanism underlying Aβ-induced mitochondrial dysfunction and apoptosis in CECs remains to be fully characterized.
Akt (a.k.a. protein kinase B), a serine/threonine kinase with oncogenic properties (Burgering and Coffer, 1995; Franke et al, 1995), serves as an antiapoptotic regulator, controlling the balance between survival and apoptosis in growth factor-mediated cytoprotection (Burgering and Coffer, 1995; Ahmed et al, 1997; Dudek et al, 1997). Akt is activated in a PI3-kinase-dependent manner by phosphorylation at two regulatory sites: threonine 308 (Thr 308) and serine 473 (Ser 473) (Franke et al, 1995, 1997; Klippel et al, 1997; Sable et al, 1998). Fully activated Akt, in turn, functions to promote cell survival by selectively phosphorylating and inactivating several downstream proapoptotic targets including the BH3-only protein, Bad (Datta et al, 1997; del Peso et al, 1997). While a role for Akt has been established in growth factor-mediated survival, little is known regarding its potential role under conditions of pathological neurodegeneration, such as that seen in cerebral amyloid angiopathy.
Mitochondrial dysfunction has been implicated as a key event in apoptosis in a number of cell death paradigms (Green and Reed, 1998). Several cellular processes characterize mitochondrial dysfunction and activation of apoptosis in Aβ-induced CEC death. These include mitochondrial release of cytochrome c (Xu et al, 2001), Smac (Yin et al, 2002b), and endonuclease G (Endo G) (as shown in the present study). Cytochrome c triggers caspase activation through interaction with Apaf-1 (Liu et al, 1996; Li et al, 2000). Second mitochondria-derived activator of caspase (Smac, a.k.a. DIABLO) relieves Inhibitor of Apoptosis Proteins (IAPs) from inhibiting caspases (Du et al, 2000; Verhagen et al, 2000). However, mitochondrial release of Endo G causes nuclear chromatin condensation and fragmentation in a caspase-independent manner (Li et al, 2001; Cregan et al, 2002). The preferential release of select proapoptotic proteins from mitochondria might be subject to differential upstream regulation (Kandasamy et al, 2003). The Akt cascade, via its control of Bad activity, has emerged as an important regulatory mechanism upstream of mitochondria in the maintenance of cell viability. In the present study, we explore the roles of Akt and Bad and related cellular events in Aβ-induced mitochondrial dysfunction and cell death in CECs.
Materials and methods
Murine Cerebrovascular Endothelial Cell Primary Cultures
Murine CECs were prepared as described previously (Xu et al, 1992). Murine CECs migrating from the vessels were pooled to form a proliferating cell culture that was maintained in DMEM, with high glucose and
Treatment with Aβ and Other Chemicals
Aβ1–40 and Aβ1–42 are the major components of Aβ deposits in the AD brain; however, in most experimental systems, the biological effects of Aβ25–35 are comparable (Loo et al, 1993; Behl et al, 1994). We have previously shown that Aβ25–35 has approximately the same potency as Aβ1–40 in inducing cell death in CECs (Xu et al, 2001; Yin et al, 2002b). Based on these data, we employed Aβ25–35 in this study. Cerebrovascular endothelial cells were treated with 25μmol/L Aβ25–35 (Sigma, St Louis, MO, USA) in serum-free growth medium for varying times. In some experiments, the murine CECs were co-treated with wortmannin at concentrations of 0.1, 1, and 10μmol/L.
Western Blot Analysis
Subfractionization of cellular components in CECs was performed as previously reported, with little cross-contamination of fractionated proteins (Xu et al, 2001; Yin et al, 2002b). Protein samples (20 to 40μg) were electrophoresed on a 10% to 15% SDS-PAGE gel, transferred to PVDF membranes and probed with primary antibodies including rabbit anti-Endo G antibody (1:2,500, a generous gift from Dr XD Wang), goat anti-smac antibody, rabbit anti-Akt antibody, rabbit anti-phospho-Akt (Thr 308) antibody, rabbit anti-phospho-Akt (Ser 473) antibody, rabbit anti-Bad antibody, rabbit anti-phospho-Bad (Ser 136) antibody (1:1,000; Cell Signaling, Beverly, MA, USA), and mouse anti-actin antiserum (1:200; Santa Cruz, CA, USA) for 1–2 h at room temperature. The membranes were then incubated with the second antibody (1:5,000; anti-rabbit, anti-mouse or anti-goat IgG conjugated with alkaline phosphatase Promega, Madison, WI, USA) at room temperature for 1 h. The color reaction was developed using the Blot AP System (Promega, Madison, WI, USA) according to the manufacturer's instructions.
Immunofluorescent Staining
Murine CECs grown on coverslips were fixed with 4% paraformaldehyde for 30mins and washed 3 times with 0.1 mol/L PBS (pH 7.4). The cells were then incubated with a primary rabbit anti-Endo G antibody (1:500, overnight at 4°C). The next day, the cells were incubated with fluorescein-conjugated anti-rabbit IgG (1:100; Vector Labs, Burlingame, CA, USA) for 1 h. Cerebrovascular endothelial cells were counterstained with 1μg/mL of PI (Molecular Probes, Eugene, OR, USA) to visualize nuclear morphology. Slides were washed, wet mounted, and examined with an Olympus fluorescence microscope.
Generation and Purification of Tat—Akt Fusion Protein
The Tat—Akt fusion protein was prepared as described previously (Schwarze et al, 1999).
Construct design: Two mutant mouse akt plasmid cDNAs were kindly provided by Dr Tsichlis: pCMV-HA-K179m-AKT, a kinase-inactive mutant (Aktm), and pCMV-HA-myr-AKT, a constitutively active myristolated mutant (Aktca) (Franke et al, 1995). The coding region of each plasmid was amplified by PCR with the following primers: 5'-ATGAACGACGTAGCCATTG-3', and 5'-TCAGGCTGTG CCACTG-3', adding KpnI and EcoRI restriction sites at the N and C termini, respectively. Amplimer identity was confirmed by restriction digestion, then cloned into pTAT-HA using the same restriction sites. The DNA sequence was verified by sequencing analysis.
Protein production and isolation: Recombinant plasmids were transformed into Escherichia coli BL21 cells and protein expression was induced using 0.1 mmol/L isopropylthiogalactoside at 37°C for 5 to 6 h. The fusion proteins were purified using a Ni-NTA superflow agarose column (Qiagen, Valencia, CA, USA), followed by a PD-10 column, pre-equilibrated with serum-free culture medium containing 10% glycerol, and then stored in 10% glycerol/PBS at −80°C until use.
Detection of protein transduction: Cerebrovascular endothelial cells were seeded on coverslips before the Tat—Akt fusion protein was added at a final concentration of 100nmol/L, incubated for 10 to 30mins at 37°C, and washed in PBS. Cells fixed in 4% paraformaldehyde were incubated with mouse anti-HA monoclonal antibody (1:200, Santa Cruz, CA, USA), then with fluorescein-conjugated anti-mouse IgG (1:100; Vector Labs, Burlingame, CA, USA). The coverslips were washed, wet mounted, and examined with an Olympus fluorescence microscope.
Construction of Retrovirus-Mediated Overexpression of Akt in Cerebrovascular Endothelial Cells
The mutant akt cDNAs (pCMV-HA-K179m-AKT, pCMV-HA-myr-AKT) (Franke et al, 1995) were also amplified by PCR to construct viral vectors for gene transfer. The primers used were as follows: 5'-ACAGCTAGCATGAACG ACGTAGCCATTGTG-3' (antisense primer), 5'-ATATCTAG ATCAGGCTGTGCCACTGGCTG-3' (sense primer). The PCR products were cloned into a pGEM-T Easy vector (Promega, Madison, WI, USA) and isolated by digestion with NheI–XbaI restriction enzymes and subcloned into the NheI–XbaI sites of pMX-internal ribosomal entry site (IRES)-EGFP (Kitamura et al, 1995). The resulting plasmid contained the akt gene, an IRES, and an EGFP gene. For production of high-titer retroviruses expressing Akt, Phoenix ecotropic packaging cells were plated at approximately 50% to 75% confluency in 15-cm-diameter tissue culture plates and pMX-AKT-IRES-EGFP vector DNAs were transfected into Phoenix retroviral producer cells by the modified calcium phosphate method. After incubation for 2 days at 32°C, viral supernatant was collected and passed through a 0.45μm filter. Cerebrovascular endothelial cells were infected with retroviral supernatant in the presence of Polybrene at 5μg/mL and incubated for 3 h. Retroviral supernatant was removed and replaced with normal growth medium. Cells grown for 48 to 72 h were sorted by flow cytometry. Infected populations exhibiting between 60% and 75% green fluorescent cells were used for further experimentation.
RT-PCR
Bad mRNA expression was detected by RT-PCR (Yin et al, 2002a). Total RNA was isolated with RNeasy Mini Kit (Qiagen, Valencia, CA, USA). In all, 600 ng of RNA was used for the synthesis of cDNA and PCR reactions. Primers were designed based on the mouse bad sequences (forward: 5'-GGAAGACGCTAGTGCTACAG-3', reverse: 5'-GAGCCTCCTTTGCCCAAGTT-3'). The relative bad mRNA level of was normalized to endogenous cyclophilin mRNA for each sample.
Co-immunoprecipitation
Co-immunoprecipitation was prepared as described previously (Xu et al, 2001; Yin et al, 2002b). Aβ25–35-treated CECs were homogenized in lysis buffer and the supernatant was incubated with rabbit anti-Bcl-xL antibody. Protein G sepharose 4 fast flow (Pharmacia Biotech, Piscataway, NJ, USA) was added to the antigen—antibody mixture and incubated with gentle agitation for 1 to 2 h. The immunoprecipitate was washed with lysis buffer, resuspended in 6 × SDS loading buffer, separated in SDS-PAGE gel, transferred to PVDF membrane, and then analyzed by Western blotting with rabbit anti-Bad antibody (1:1,000) as described above.
Bad Knockdown in Cerebrovascular Endothelial Cells with RNA Interference
SiRNAs targeting the mouse bad gene were prepared by in vitro transcription using Silencer™ siRNA Construction Kit (Ambion, Austin, TX, USA). SiRNA was synthesized to target the coding region of mouse bad mRNA as follows: 5'-UUCUGCGAUCACGAUGUCUAU-3', and scrambled: 5'-UUGUCUCAUGUCGUCUAGACA-3'. Murine CECs were transfected 24 h after plating, using 2 to 4μL siPORT Lipid (Ambion, Austin, TX, USA) according to the manufacturer's protocol, at a final siRNA concentration of 100nmol/L. Bad mRNA and Bad protein levels were examined by RT-PCR and Western blotting 48 to 72 h after transfection.
Assessment of Murine Cerebrovascular Endothelial Cell Death
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay: Cerebrovascular endothelial cells were rinsed with HBSS and replaced with DMEM containing MTT (Sigma, St Louis, MO, USA) at a final concentration of 0.5mg/mL. The cells were then incubated for 4 h at 37°C under 5% CO2/95% air, and for another 14 h in lysis solution (10% SDS in 0.01 N HCl) at 1:1 dilution. Absorbance of the mixture was read at 540 nm using a multiplate reader. Viability was expressed as OD540 of treated cells divided by OD540 of untreated cells (Yin et al, 2002a, b).
Statistical analysis: Quantitative data are expressed as mean × s.d., based on at least three separate experiments of triplicate samples. Difference among groups was statistically analyzed by one-way analysis of variance followed by Bonferroni's post hoc t-test. Comparison between two experimental groups was based on two-tailed t-test. A P-value less than 0.05 was considered significant.
Results
Aβ25–35 Inactivation of Akt in Cerebrovascular Endothelial Cells
Akt plays a central role in cell survival by preventing the activation of a variety of proapoptotic pathways. Akt is activated in a PI3-kinase-dependent manner by phosphorylation at two regulatory sites: Thr 308 and Ser 473 (Franke et al, 1995, 1997; Klippel et al, 1997; Sable et al, 1998). To determine whether Akt inactivation is involved in Aβ25–35-induced CEC death, we first measured levels of total and phosphorylated Akt (at Thr 308 and Ser 473) by Western blot analysis. Exposure to Aβ25–35 (25μmol/L) resulted in a substantial decrease in the active forms of Akt at both Thr 308 and Ser 473, as early as 1 h after treatment; but had no effect on total Akt levels until 24 h (Figures 1A and 1B). No significant changes in Akt activation were observed in control cultures between 0 and 24 h (data not shown).
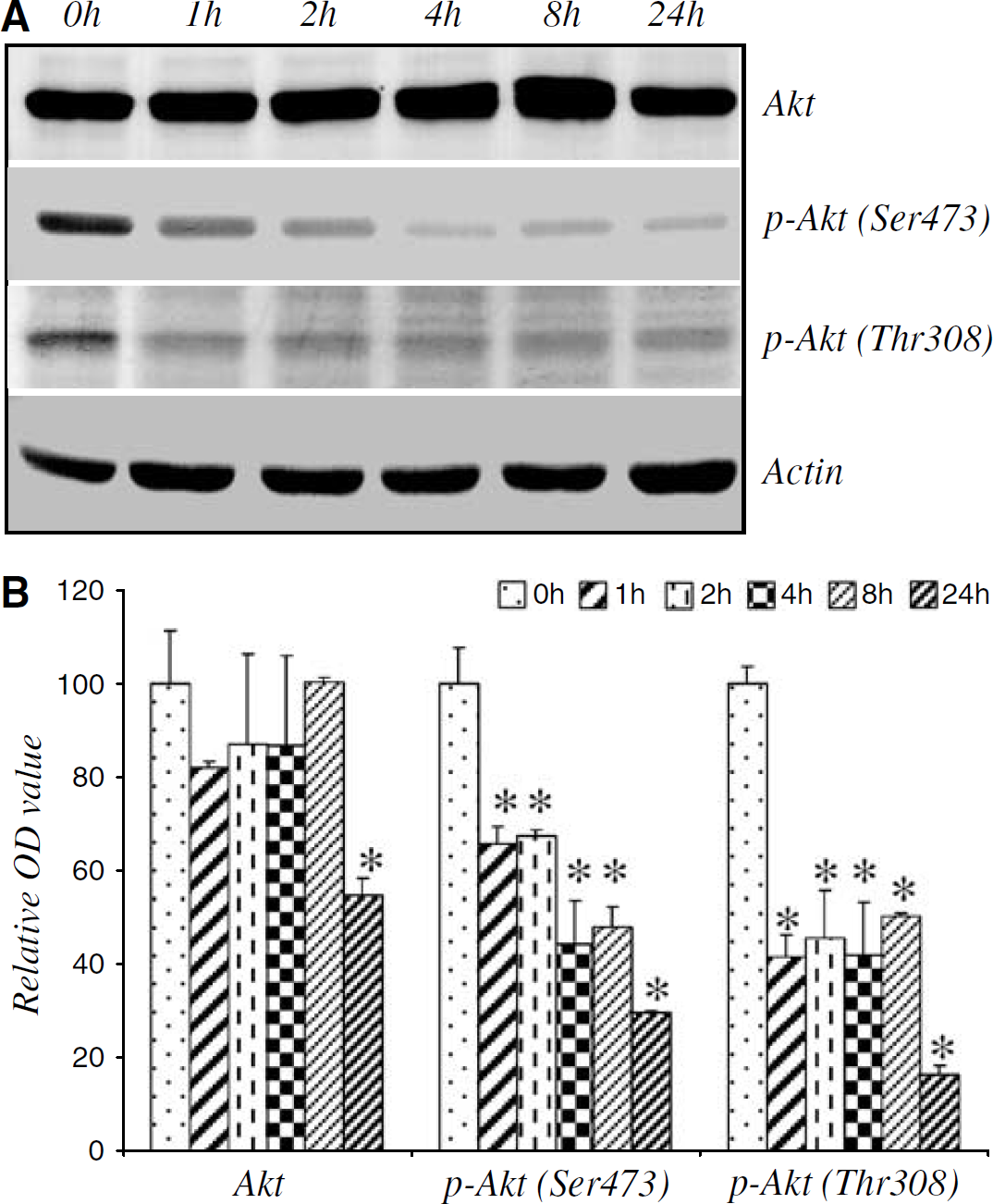
Aβ25–35 inactivation of Akt in CECs. (
Aβ25–35 Dephosphorylation of Bad in Cerebrovascular Endothelial Cells
Fully activated Akt promotes cell survival by selectively phosphorylating and inactivating several downstream proapoptotic targets including the BH3-only protein, Bad (Datta et al, 1997; del Peso et al, 1997). Under normal physiological conditions, Bad, in the phosphorylated and inactive form, is known to exist almost exclusively in the cytoplasm (Cross et al, 1995; Datta et al, 1997; del Peso et al, 1997; Hajduch et al, 1998; Brunet et al, 1999; Kaplan and Miller, 2000). Reduced Akt activity would be expected to favor the dephosphorylation of Bad (rendering the protein active). Activated Bad, recognized as an essential initiator of the apoptotic cascade (Kelekar and Thompson, 1998; Huang and Strasser, 2000), translocates to mitochondria where it binds Bcl-xL, an antiapoptotic member in the Bcl-2 family, resulting in mitochondrial membrane disruption, mitochondrial dysfunction, and subsequent cell death (Yang et al, 1995; Zha et al, 1996; Korsmeyer, 1999). In view of the inactivation of Akt by Aβ25–35, we examined if Bad was activated after exposure to Aβ25–35. Aβ25–35 decreased phospho-Bad (Ser 136) levels in the cytosolic fraction as early as 4 h after exposure, in parallel with an increase in the Bad content in the mitochondrial fraction (Figure 2A), indicating that dephosphorylated Bad translocated from the cytoplasm to mitochondria. We have previously reported that Aβ25–35 induced CEC death beginning 24 h after treatment (Xu et al, 2001), suggesting that Bad translocation preceded cell death. In addition, Bcl-xL co-immunoprecipitated with Bad in Aβ-treated CECs, suggesting a physical interaction between these two Bcl-2 family members during the apoptotic process (Figure 2B). Aβ25–35 had little effect on bad mRNA levels, suggesting that Bad activation involved primarily post-transcriptional regulation (Figure 2C). No significant change in Bad phosphorylation was observed in control cultures between 0 and 48 h (data not shown).
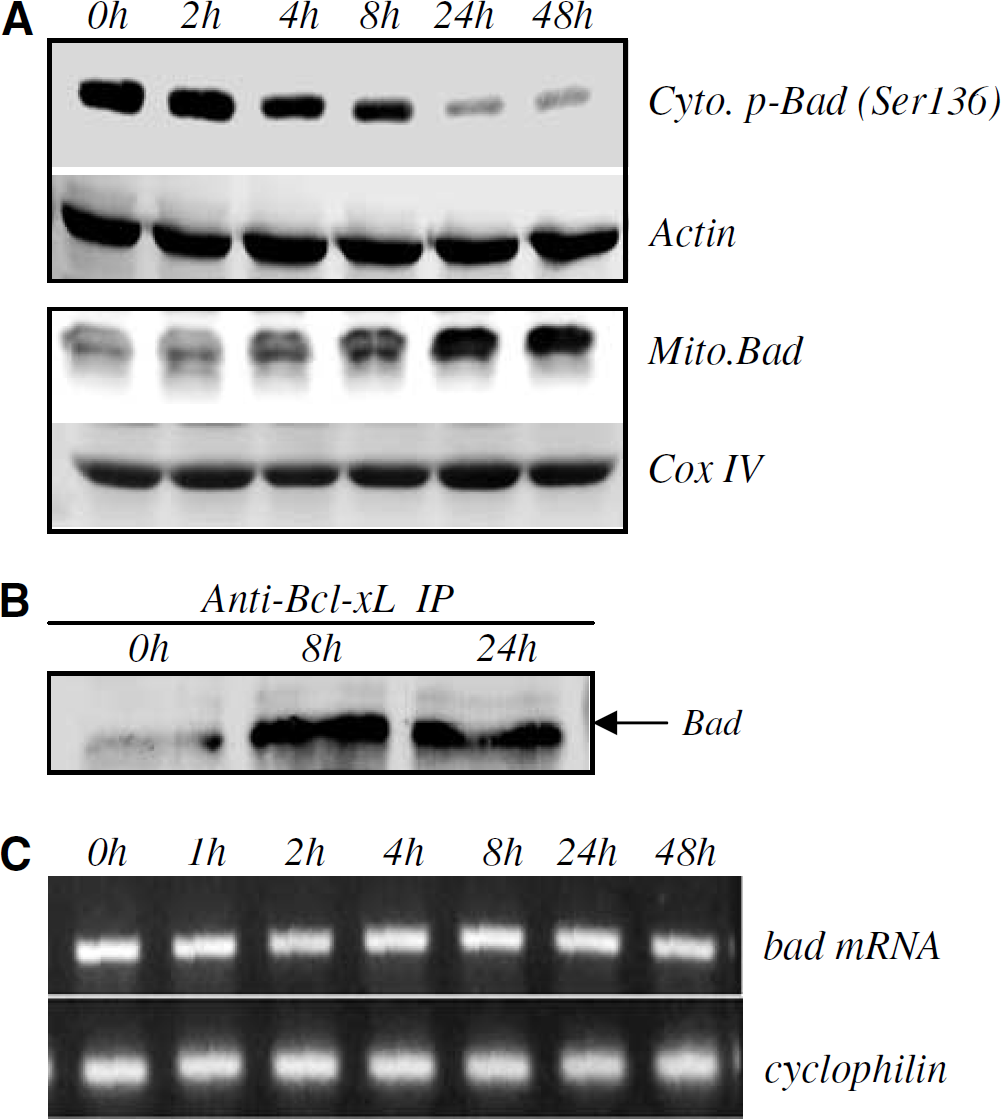
Aβ25–35 activation of Bad. Cerebrovascular endothelial cells treated with 25μmol/L Aβ25–35 at the indicated times were fractionated and examined by Western blotting with anti-Bad or anti-p-Bad (Ser 136) antibodies. Cytosolic and inactive (or phosphorylated) Bad (Cyto.p-Bad) decreased 4 h after Aβ treatment, in parallel with the appearance of Bad in the mitochondrial fraction (
Endonuclease G Translocation from Mitochondria to Nucleus After Aβ25–35 Exposure
Previous studies have shown that the release of mitochondrial intermembranous proteins after apoptotic stimuli might be under the control of some BH3-only family members, including Bad (Li et al, 2001; Wang, 2001; Madesh et al, 2002). These mitochondrial intermembranous proteins include cytochrome c, Smac and Endo G. Aβ25–35-induced Bad activation and translocation to mitochondria in CECs, as shown above, would be expected to cause mitochondrial dysfunction. We have previously shown that Aβ25–35 induced cytochrome c and Smac release, leading to CEC apoptosis (Xu et al, 2001; Yin et al, 2002b). In the present study, we found that Aβ25–35 treatment caused an increase in Endo G in the nuclear fraction and a decrease in the mitochondrial fraction, indicating translocation from mitochondria to nucleus (Figure 3A). This subcellular redistribution was confirmed by immunofluorescent staining with an anti-Endo G antibody. The punctate mitochondrial staining pattern observed in control cultures changed to a dense nuclear pattern after Aβ25-35 treatment (Figure 3B).
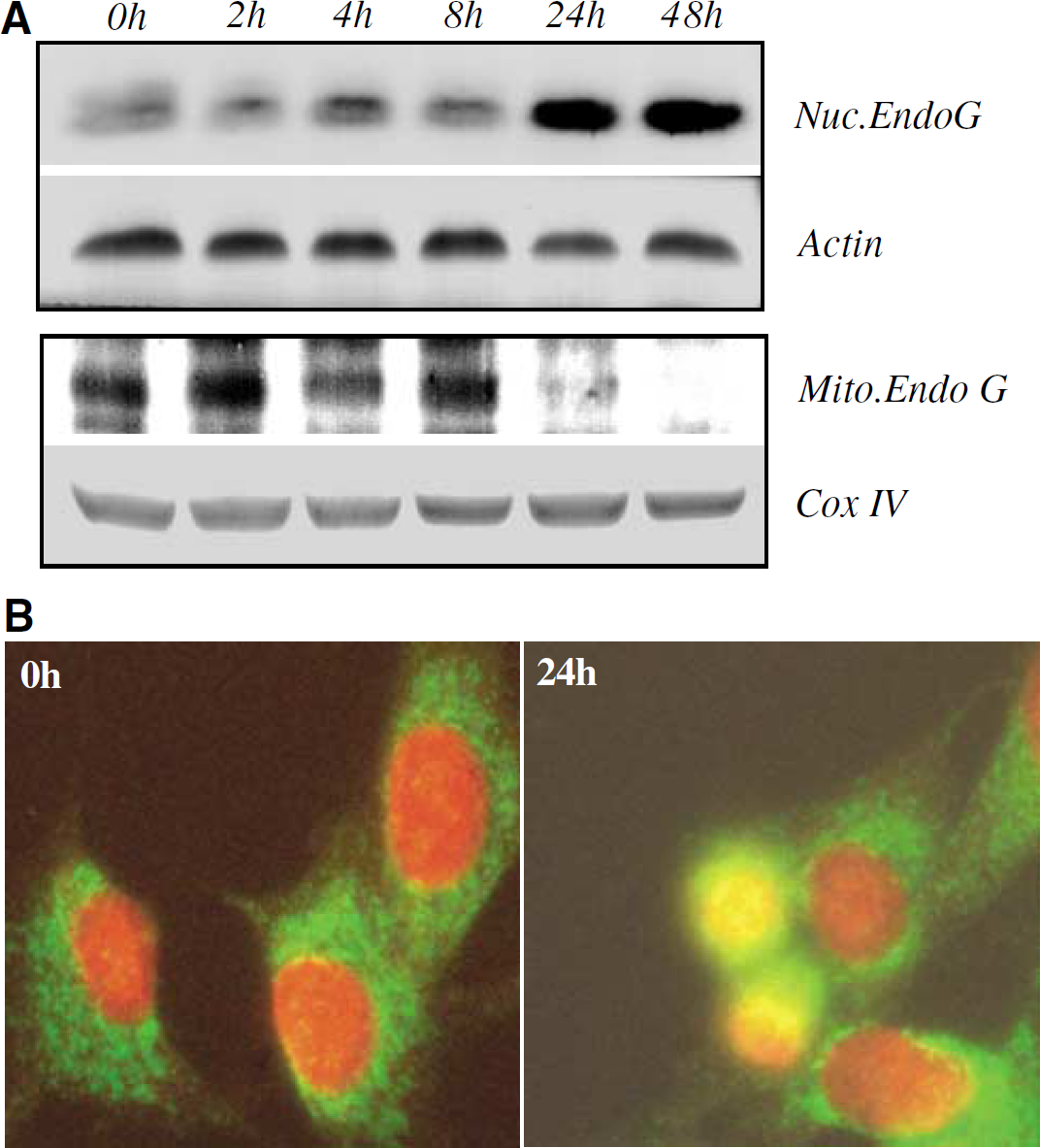
Aβ25–35-induced translocation of Endo G from mitochondria to nucleus. Cerebrovascular endothelial cells, treated with 25μmol/L Aβ25–35 and harvested at times indicated, were fractionated into mitochondrial (Mito.) and nuclear (Nuc.) extracts, separated by SDS-PAGE, and analyzed by Western blotting with anti-Endo G, anti-Actin, or anti-Cox IV antibodies (
Akt Regulation of Aβ25–35-Induced Mitochondrial Dysfunction and Cell Death
Release of mitochondrial intermembranous proteins might be regulated by multiple upstream signaling pathways (Imaizumi et al, 1999; Kaltschmidt et al, 1999; Bozyczko-Coyne et al, 2001; Martin et al, 2001). If Bad-initiated mitochondrial dysfunction (including mitochondrial release of Smac and Endo G) and CEC death is under the regulation of Akt, alteration of Akt activity would be expected to affect cellular events downstream of Bad as well. We applied three strategies to determine if Akt was involved in Aβ25-35-induced Bad activation and subsequent release of mitochondrial intermembranous proteins. The first was to determine whether inhibition of Akt affects Aβ25–35-induced mitochondrial dysfunction and cell death. Wortmannin, a PI3/ Akt inhibitor at doses between 30nmol/L and 1μmol/L (Dimmeler et al, 1998; Fujio and Walsh, 1999; Kim et al, 2000), enhanced Aβ25–35-induced Bad translocation from the cytoplasm to mitochondria in a dose-dependent manner (Figure 4A). This effect was accompanied by enhanced Endo G translocation to the nucleus, and Smac release into cytosol, in a dose-dependent manner (Figure 4A). Wortmannin also increased Aβ25–35-induced cell death (Figure 4B).
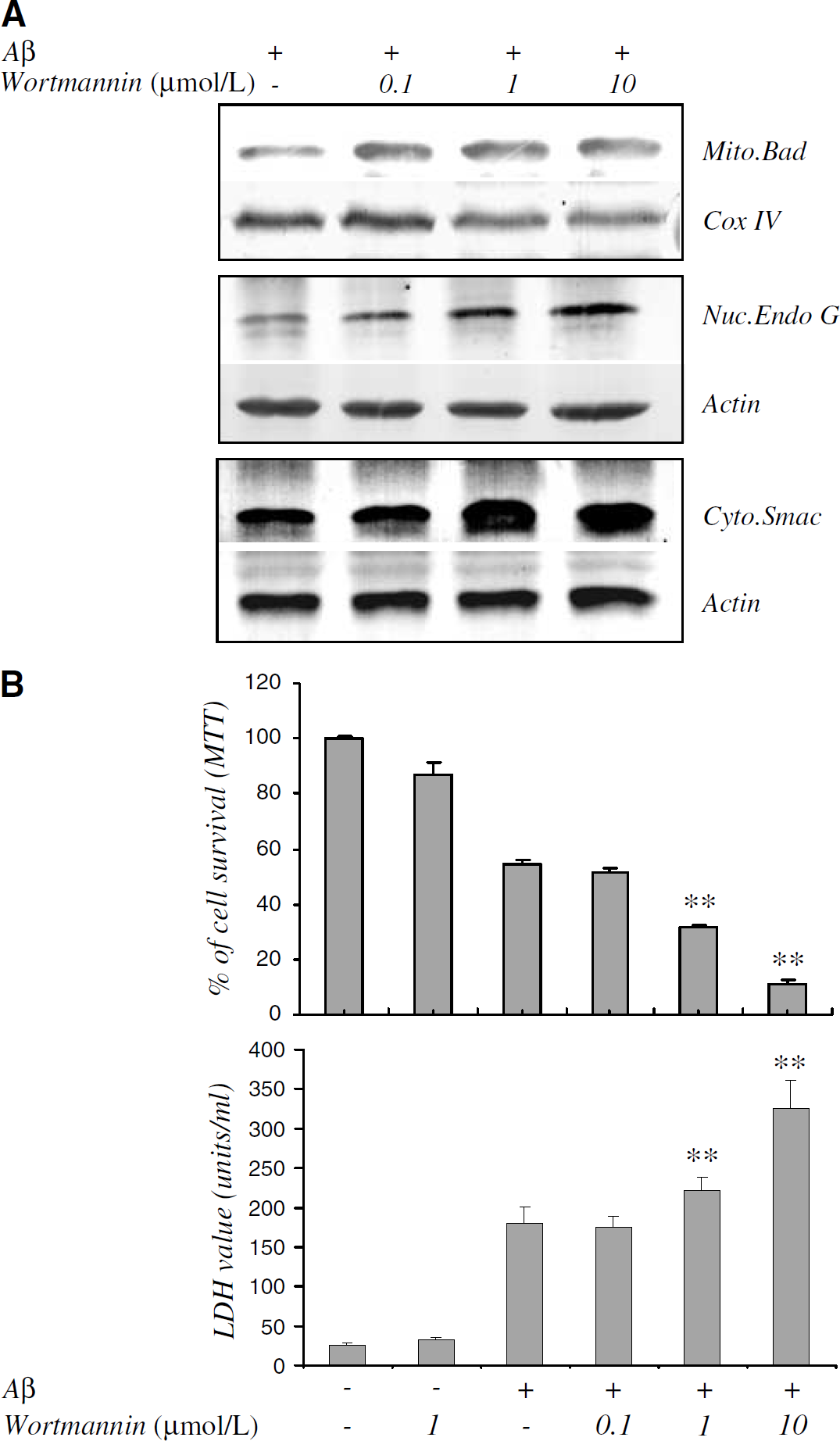
Effects of Akt inhibition on Bad translocation, mitochondrial intermembranous protein release, and CEC death induced by Aβ25–35. (
The second strategy entailed enhancement of Akt activity with cellular transfer of a constitutively active myristylated mutant Tat—Akt (Tat—Aktca) fusion protein, using a kinase-inactive mutant Tat—Akt (Tat—Aktm) as a negative control. Successful delivery of both fusion proteins (500nmol/L) into CECs was confirmed by Western blotting (Figure 5A). In addition, effective transduction of the Tat—Aktca and Tat—Aktm protein was further confirmed by immunostaining with anti-HA antibody in CECs (Figure 5B). As expected, Tat—Aktca, but not Tat—Aktm, increased phospho-Akt (Thr 308 and Ser 473) (Figure 5A). Treatment with Tat—Aktca, but not Tat—Aktm, effectively inhibited Aβ25–35-induced Bad translocation to mitochondria and subsequent mitochondrial release of Smac and Endo G to cytosol and nucleus, respectively (Figure 5C). Tat—Aktca protected CECs from Aβ25–35-induced cell death (Figure 5D), while Tat—Aktm had no effect on Aβ25–35-induced mitochondrial protein release, and subsequent cell death (Figures 5C and 5D).
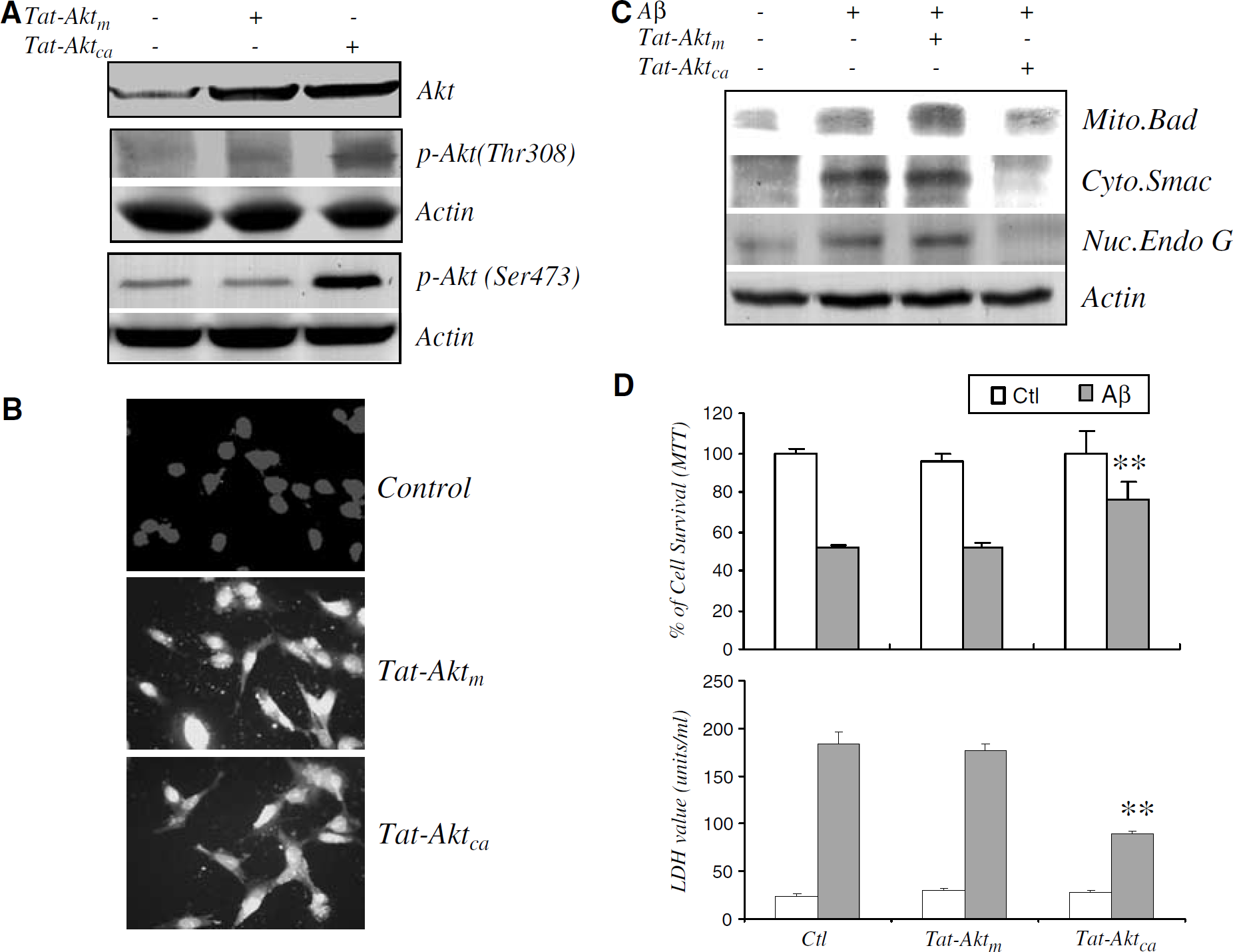
Effects of Tat—Akt fusion protein delivery on Aβ25-35-induced Bad translocation, Endo G and Smac release, and CEC death. (
The third strategy involved the upregulation of Akt activity in Aβ25–35-treated CECs using retroviral gene transfer of aktca (pMX/Aktca), or aktm (pMX/Aktm), which served as a negative control. Infection with both akt-carrying viruses increased total Akt protein levels, while only pMX/Aktca increased phospho-Akt levels in CECs (Figure 6A). Effective gene transfer was also confirmed by immunofluorescence, which revealed abundant cytoplasmic staining of the virally expressed reporter, GFP (Figure 6B). Aktca gene transfer attenuated Aβ25–35-induced Bad translocation to the mitochondria and the subsequent release of Endo G (Figure 6C) and Smac (data not shown), and cell death (Figure 6D), while gene transfer of the kinase inactive mutant aktm had none of these effects. Results of these experiments further implicate the causal role of Akt in regulating Aβ25–35-induced Bad activation and mitochondrial dysfunction, leading to CEC apoptosis.
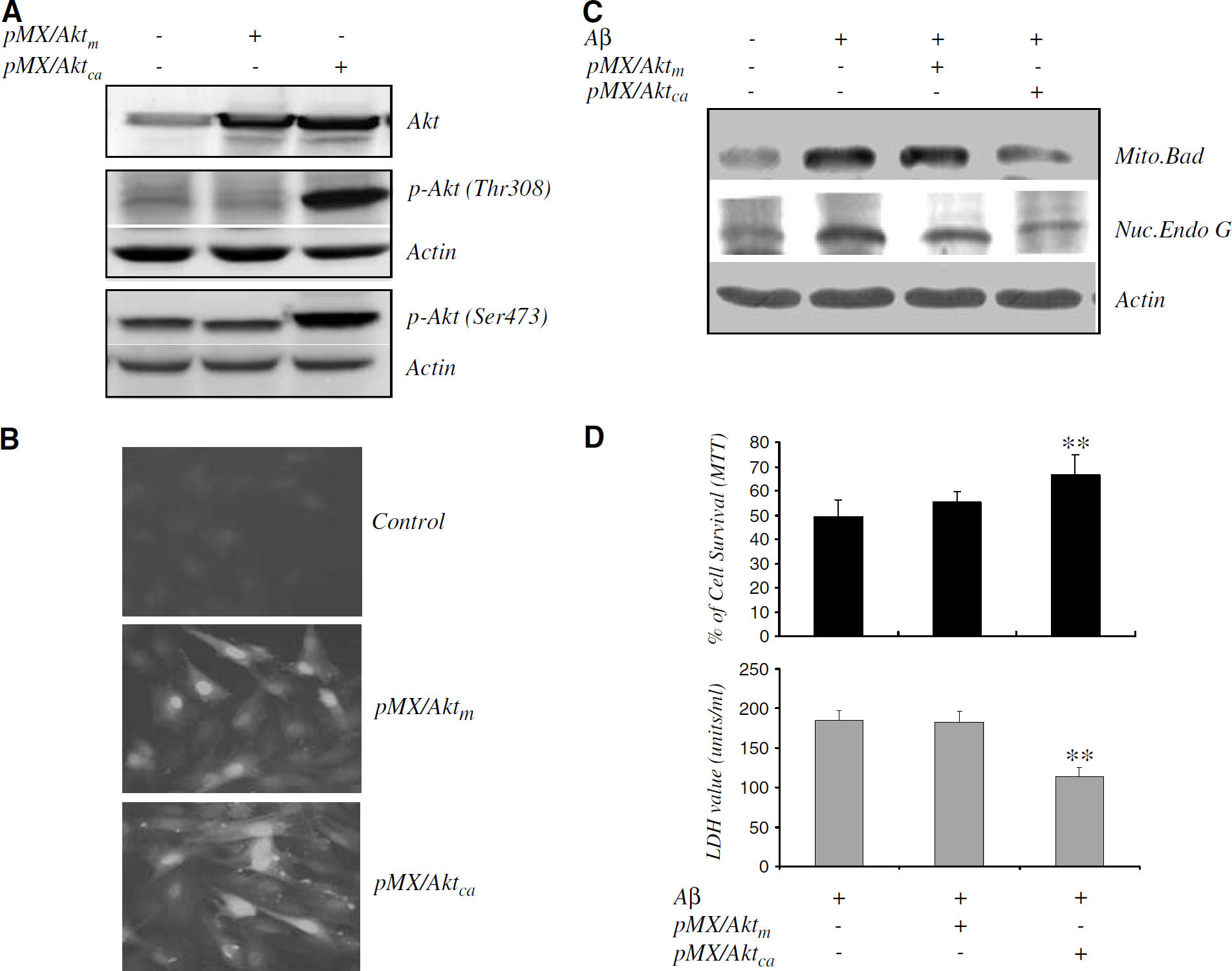
Effects of Akt upregulation by retroviral akt gene transfer on Aβ25-35-induced Bad translocation, Endo G and CEC death. (
Effects of bad Knockdown with siRNA on Mitochondrial Dysfunction and Cell Death
Since mitochondrial release of intermembranous proteins such as Smac and Endo G is under the control of BH3-only family members including Bad (Li et al, 2001; Wang, 2001; Madesh et al, 2002), we examined if Bad activation was causally related to Aβ25–35-induced mitochondrial dysfunction and CEC death. Cerebrovascular endothelial cells were treated with siRNA specific for bad without Aβ25–35 exposure. Bad mRNA levels were significantly repressed by treatment with 100nmol/L bad siRNA (Figure 7A). SiRNA treatment also decreased Aβ25–35-induced Bad translocation to mitochondria (Mito Bad, Figure 7B), and reduced Endo G translocation from mitochondria to nucleus (Nuc Endo G, Figure 7B). Smac translocation from mitochondria to cytoplasm was also decreased (Cyto Smac, Figure 7B). Finally, bad knockdown with siRNA also attenuated Aβ25–35-induced CEC death (Figure 7C). The specificity of this bad siRNA strategy was supported by the finding that scrambled bad siRNA was without effect on Aβ25–35-induced Bad activation, mitochondrial release of Smac and Endo G, and the extent of CEC death. Furthermore, bad siRNA did not alter cyclophilin mRNA levels.
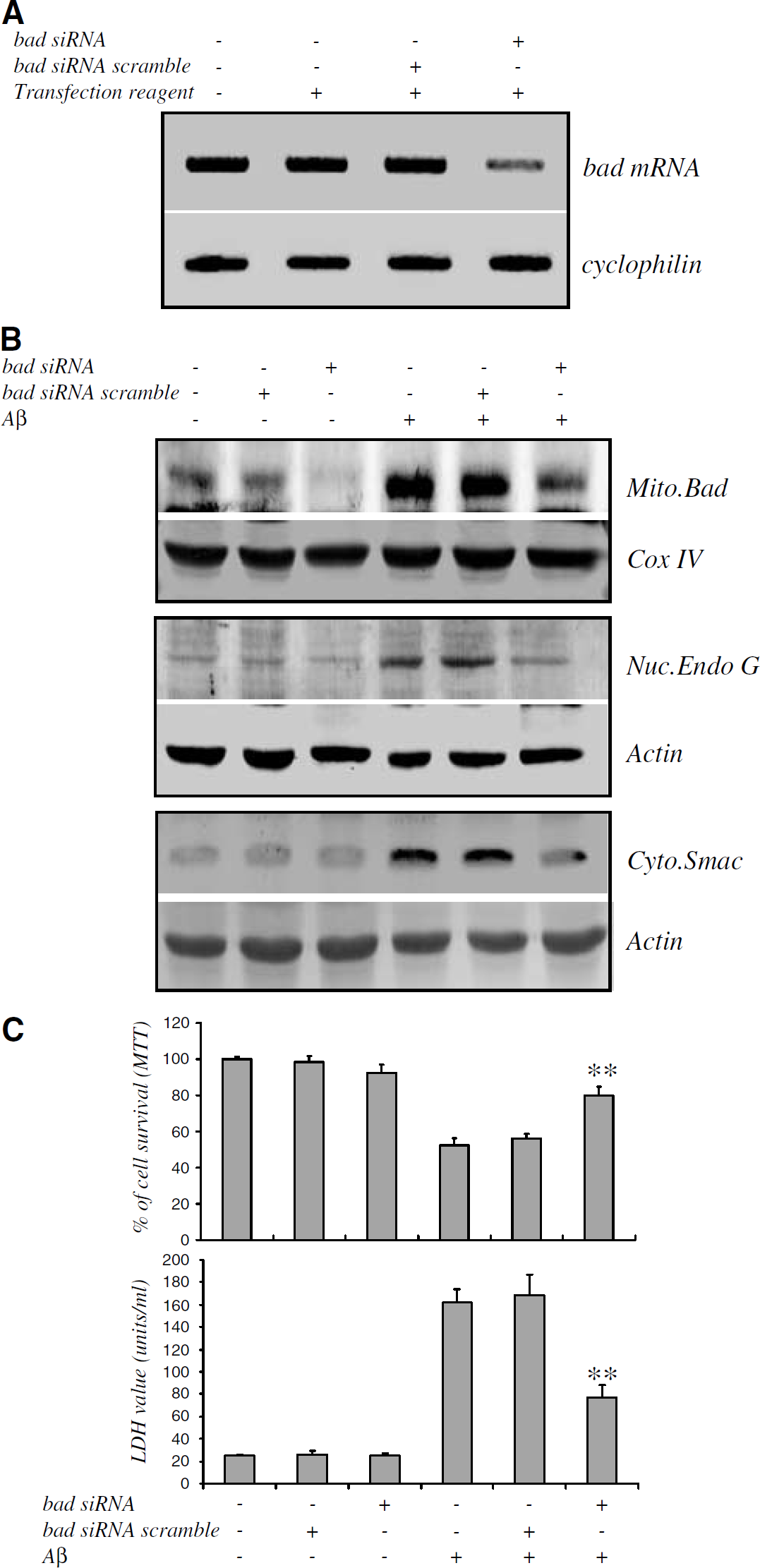
Effects of bad1 knockdown on Endo G and Smac translocation and CEC death. (
Discussion
We have shown that Aβ25–35-induced CEC death was accompanied by Akt inactivation by means of dephosphorylation, occurring within 1 h after exposure to Aβ25–35. That Akt inactivation was causally related to Aβ25–35-induced mitochondrial dysfunction and subsequent CEC apoptosis was supported by two lines of evidence. First, Akt inhibition by wortmannin exacerbated Aβ25–35-induced Bad activation, mitochondrial dysfunction, and CEC death. Second, enhancing Akt activity using a Tat—Aktca fusion protein or by viral gene transfer of a consitutively active mutant aktca attenuated Aβ25–35-induced Bad activation, mitochondrial dysfunction, and CEC death. These results suggest that Aβ25–35-induced Akt dephosphorylation activates Bad, releasing mitochondrial Endo G and Smac, and contributes to Aβ25–35-induced CEC death. The survival-promoting activity of several growth factors, including neurotrophins, is mediated by enhancement of Akt activity (Kaplan and Miller, 2000). Conversely, there is growing evidence that a number of death-promoting stimuli cause Akt inactivation. For example, N-methyl-
Although the PI3K/Akt pathway protects cells from apoptosis caused by diverse stress stimuli, its protective role in the setting of Aβ exposure is unclear, and several reports are contradictory. For example, a recent study by Wei et al (2002) shows that Aβ25-35 induced a weak activation of Akt together with JNK and ERK but not p38 kinase in SH-5Y cells. Martin et al (2001) have also shown that Akt activity was activated shortly after incubation with Aβ25–35 and Aβ1–40 with kinetics different from that of nerve growth factor in PC12 cells. However, consistent with the results of the current study, several other groups have observed down-regulation of Akt after Aβ exposure. For example, Suhara et al (2003) recently showed that virally encoded Aβ1–42 was proapoptotic and inhibitory to Akt phosphorylation in human umbilical vein endothelial cells, which was characterized by mitochondrial dysfunction, DNA condensation, and activation of caspase-3. Kubo et al (2002) also found that Aβ1–40 suppressed the PI-3K-dependent Akt phosphorylation but not mitogen-activated protein kinase phosphorylation in HeLa cells. These conflicting data might be due to differences in cell types and experimental conditions, and underscore the complexity of intrinsic apoptosis-regulatory genes in cell systems.
Akt phosphorylates a number of proteins which are known to regulate apoptosis (Cross et al, 1995; Datta et al, 1997; del Peso et al, 1997; Hajduch et al, 1998; Brunet et al, 1999; Kaplan and Miller, 2000); prominent among these proteins is the BH3-only protein, Bad. In this study, subsequent to Aβ25–35-induced Akt inactivation, we observed Bad translocation from cytoplasm to mitochondria. On translocation, we found that Bad physically interacted with Bcl-xL; Bcl-xL, in the absence of Bad, interacts with Bax to neutralize its proapoptotic activity (Zamzami et al, 1998). That Bad activation was indeed causally related to Aβ25–35-induced mitochondrial dysfunction including Endo G and Smac release was confirmed by the observations that specific bad knockdown with a siRNA strategy resulted in a reduction in Aβ25–35-induced Endo G and Smac release and CEC death. Endonuclease G is an effector enzyme in the apoptotic cascade, cleaving chromatin on translocation from mitochondria to nucleus (Susin et al, 1999; Li et al, 2001). Smac, released to the cytoplasm, relieves IAP protein inhibition of caspases, resulting in the propagation of the apoptotic cascade (Du et al, 2000).
Together, these results establish an Aβ25–35-activated death signaling cascade in CECs involving Akt inactivation and resulting in a series of cellular events. The downstream pathway involves Bad activation and translocation from the cytoplasm to mitochondria, and binding to Bcl-xL, resulting in disruption of the mitochondrial membrane and release of cytochrome c, Smac, and Endo G. These events triggered by mitochondrial dysfunction are likely the underlying mechanism of Aβ25–35-induced CEC apoptosis. Understanding the death signaling processes activated by Aβ will contribute to future development of strategies to prevent vascular degeneration in cerebral amyloid angiopathy.