
Abstract
The authors recently reported that sodium orthovanadate rescues cells from delayed neuronal death in gerbil hippocampus after transient forebrain ischemia through phosphatidylinositol 3-kinase-protein kinase B (Akt) pathway (Kawano et al., 2001). In the current study, they demonstrated that the activation of FKHR, a Forkhead transcription factor and a substrate for Akt, preceded delayed neuronal death in CA1 regions after transient forebrain ischemia. Adult Mongolian gerbils were subjected to 5-minute forebrain ischemia. Immunoblotting analysis with anti—phospho-FKHR antibody showed that phosphorylation of FKHR at serine-256 in the CA1 region decreased immediately after and 0.5 and 1 hour after reperfusion. The dephosphorylation of FKHR was correlated with the decreased Akt activity. Intracerebroventricular injection of orthovanadate 30 minutes before ischemia inhibited dephosphorylation of FKHR after reperfusion, and blocked delayed neuronal death in the CA1 region. Gel mobility shift analysis using nuclear extracts from the CA1 region prepared immediately after reperfusion revealed increases in DNA binding activity for the FKHR-responsive element on the Fas ligand promoter. The orthovanadate injection administered before ischemia inhibited its binding activity. Two days after reperfusion, expression of Fas ligand increased in the CA1 region and the orthovanadate injection inhibited this increased expression. These results suggest that the inactivation of Akt results in the activation of FKHR and, in turn, relates to the expression of Fas ligand in the CA1 region after transient forebrain ischemia.
Keywords
Transient forebrain ischemia results in delayed neuronal death of pyramidal neurons in the hippocampal CA1 region (Kirino, 1982). Although molecular mechanisms underlying the pathogenesis of delayed neuronal death is unclear at present, histologic and biochemical evidence suggests the involvement of apoptosis in dying cells after ischemia (MacManus et al., 1993;Nitatori et al., 1995). Activation of Akt (also called protein kinase B) protects cells from apoptosis (Datta et al., 1999). We recently reported that a decreased Akt activity is involved in ischemic-induced cell death (Kawano et al., 2001) and that an increased Akt activity accounts for neuroprotection in ischemic tolerance in gerbil hippocampus (Yano et al., 2001). Akt is phosphorylated on residues Thr-308 and Ser-473 before it is activated, and treatment with wortmannin, an inhibitor of phosphatidylinositol 3-kinase (PI3K), prevents this Akt activation (Alessi et al., 1996). Plasma membrane translocation is also an essential step in Akt activation. The activated Akt detaches from the plasma membrane and translocates to the nucleus (Andjelkovic et al., 1997;Meier et al., 1997). Ca2+/calmodulin-dependent protein kinase kinase can also phosphorylate Akt at Thr-308 with concomitant activation (Yano et al., 1998).
Several potential substrates for Akt that are related to cell survival include Bad, caspase-9, cyclic adenosine monophosphate-responsive element binding protein (CREB), nuclear factor-κB, and Forkhead transcription factors (Blume-Jensen et al., 1998;Brunet et al. 1999;Cardone et al., 1998;Datta et al., 1997;Del Peso et al., 1997;Du and Montminy, 1998;Kops et al., 1999;Romashkova and Makarov, 1999;Tang et al., 1999). Forkhead box transcription factor, class O (FOXO) is a mammalian homologue of DAF-16, which is known to regulate the lifespan of Caenorhabditis elegans (Lin et al., 1997) and includes subfamilies of forkhead transcription regulators such as AFX, FKHRL1, and FKHR. By stimulation with survival factors, FOXOs are phosphorylated by Akt and stay in the cytoplasm. Once survival factors are depleted, FOXOs are dephosphorylated and translocated into the nucleus (Biggs WH III et al., 1999;Brunet et al., 1999;Nakae et al., 2000;Rena et al., 1999;Takaishi et al., 1999). Furthermore, activation of FOXOs induces apoptosis (Brunet at al., 1999;Dijkers et al., 2000b;Nakamura et al., 2000;Tang et al., 1999). FOXO-induced cell death may be caused by expression of the Fas ligand and Bim, a Bcl-2 family member (Brunet et al., 1999;Dijkers et al., 2000a). FKHR, a FOXO family member, is phosphorylated on three sites (Thr-24, Ser-253, and Ser-316) in a PI3K-dependent manner (Nakae et al., 1999), and phosphorylation on all or a subset of these sites contributes to the inactivation of its transcriptional activity (Nakae et al., 2000). The phosphorylation of murine FKHR at Ser-253 (corresponding to Ser-256 in human FKHR) is required for phosphorylation of the other two sites (Nakae et al., 2000). It has not been documented whether the activation of FOXOs is involved in the mechanisms of ischemic cell death.
Fas (also called CD95, APO-1), a member of TNF receptor family, and Fas ligand (also called CD95-L, APO-1L) both play an important role in apoptosis (Nagata, 1997;Nagata and Golstein, 1995). Activation of Fas leads to formation of a death-inducing signaling complex that comprises Fas-associated death domain and procaspase-8. Procaspase-8 is proteolytically cleaved and consequently activates caspase pathways, thereby leading cells to apoptosis. The Fas/Fas ligand system, which was first documented in the immune system (Suda et al., 1993;Yonehara et al., 1989), is also important in CNS pathophysiology, such as Alzheimer disease (Nishimura et al., 1995), multiple sclerosis (Dowling et al., 1996), trauma (Beer et al., 2000;Li et al., 2000), and ischemia (Felderhoff-Mueser et al., 2000;Herdegen et al., 1998;Martin-Villalba et al., 1999;Matsushita et al., 2000;Northington et al., 2001;Rosenbaum et al., 2000;Tarkowski et al., 1999). Recently, it was shown that the Fas/Fas ligand system has a role in the mitochondrial apoptotic pathway by cleaving the Bcl-2 family member Bid (Li et al., 1998;Luo et al., 1998).
The present study investigated the dephosphorylation of FKHR and its concomitant activation after transient forebrain ischemia in gerbil hippocampus. The FKHR activation was correlated with the upregulation of Fas ligand. Orthovanadate treatments inhibited the activation of FKHR with expression of Fas ligand and, in turn, rescued cells from the delayed neuronal death.
MATERIALS AND METHODS
Induction of ischemia
Adult male Mongolian gerbils (60 to 80 g) were housed under constant environmental conditions (temperature, 22°C ± 2°C; humidity, 55% ± 5%; and a 12/12-hour light/dark cycle) in the Animal Research Center at Kumamoto University. Ischemia was induced as described previously (Kawano et al., 2001). The gerbils were anesthetized and maintained with 2% halothane in a mixture of 30% O2 and 70% N2O. The rectal temperature, which was monitored with a digital thermometer inserted 2 cm into the anus, was maintained at 37°C to 38°C throughout the operation by placing gerbils under a heat lamp and warming them with a blanket.
The bilateral common carotid arteries were surgically exposed and quickly occluded with aneurysm clips. After a 5-minute occlusion, the clips were removed to restore blood flow. After awakening from anesthesia, gerbils were returned to home cages and allowed to take food and water before the experiments were performed. Control animals were sham operated and underwent the same experimental procedures, except for occlusion of arteries. Ischemic and sham-operated gerbils were killed by decapitation at the indicated times and hippocampal CA1 regions were dissected from the brains. The Committee of Animal Experiments at Kumamoto University School of Medicine approved all animal experiments.
Administration of orthovanadate
Administration of orthovanadate was performed as described previously (Kawano et al., 2001). Animals were anesthetized and placed on a stereotaxic operation frame. After making one burr hole at the right parietal skull 2.0 mm posterior from bregma and 3.5 mm lateral, a Hamilton syringe was inserted into the ventricle at a depth of 2.5 mm from the cortical surface. Two microliters 5 mmol/L sodium orthovanadate (Sigma, St. Louis, MO, U.S.A.) was injected into the ventricle at a flow rate of 0.4 μL/min on the right side. Thirty minutes later, animals were subjected to 5-minute forebrain ischemia as described previously. Sodium orthovanadate (5 mmol/L) was dissolved in phosphate-buffered saline (PBS).
Immunohistochemical studies
For immunohistochemical studies using anti-Fas ligand antibody (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), gerbils were anesthetized 2 days after 5-minute ischemia with pentobarbital (50 mg/kg, intraperitoneally) and perfused through the left ventricle of the heart with ice-cold PBS followed by 4% paraformaldehyde in 0.1 mol/L phosphate buffer (PB). For immunohistochemical studies using anti—phospho-FKHR (Ser-256) antibody (Cell Signaling technology, Beverly, MA, U.S.A.), animals were perfused immediately after 5-minute ischemia with PBS containing 30 mmol/L sodium pyrophosphate and 50 mmol/L NaF, followed by 4% paraformaldehyde in 0.1 mol/L PB containing 30 mmol/L sodium pyrophosphate and 50 mmol/L NaF to prevent dephosphorylation. The brains were removed and postfixed overnight at 4°C in the same fixative solution. Coronal brain sections (30-μm thick) at the level of the hippocampus were prepared using a vibratome. The sections were incubated by floating in the following media: 0.1 mol/LM PB containing 0.01% Triton X-100 (30 minutes); PBS with 3% bovine serum albumin (BSA; blocking solution, 1 hour); polyclonal anti-FKHR antibody (diluted 1:200, overnight; Santa Cruz Biotechnology); and polyclonal anti—phospho-FKHR antibody (diluted 1:100) (Upstate Biotechnology, Lake Placid, NY, U.S.A.) in blocking solution. The sections were then labeled for 2 hours with biotinylated antirabbit IgG in TNB buffer, followed by both streptavidin-HRP and Alexa 488-labeled antisheep IgG (Molecular Probes, Eugene, OR, U.S.A.) in PBS (diluted 1:200). Sections were then labeled with tetramethylrhodamine tyramide for 10 minutes using the TSA-Direct kit (NEN Life Science Products, Boston, MA, U.S.A.). After several washes with PBS, the sections were mounted on glass slides with coverslips and analyzed using a confocal laser microscope (Fluoview; Olympus, Tokyo, Japan). To normalize immunoreactivity with anti—phospho-FKHR antibody, we analyzed hippocampal slices without changing the confocal laser intensity and laser aperture. Furthermore, sections were double-stained with anti—phospho-FKHR and anti—FKHR antibodies. We confirmed that the intensity of immunoreactivity with anti-FKHR antibody was the same between control and ischemic hippocampal slices. Similar methods were used for immunofluorescence staining with anti-Fas ligand antibody (Santa Cruz Biotechnology).
Gel electrophoresis and immunoblotting
Gerbils were killed by decapitation immediately after reperfusion or at the indicated times after reperfusion. After brains were removed and rinsed once with cold PBS, CA1 regions of the hippocampus were dissected and removed in cold PBS under a microscope. Each tissue sample was stored in a test tube and kept at −80°C before use. Frozen tissues were homogenized with a hand homogenizer and sonicated with a Sonifier-250 (Branson, Danbury, CT, U.S.A.) at 0°C in 0.2 mL homogenization buffer containing 50 mmol/L Tris-HCl (pH 7.5), 0.5% Triton X-100, 4 mmol/L EGTA, 10 mmol/L EDTA, 0.5 mol/L NaCl, 1 mmol/L Na3VO4, 30 mmol/L sodium pyrophosphate, 50 mmol/L NaF, 50 μg/mL leupeptin, 25 μg/mL pepstatin A, 50 μg/mL trypsin inhibitor, and 1 mmol/L dithiothreitol. Insoluble materials were removed by a 10-minute centrifugation at 15,000 g. After determining the protein content in each supernatant fraction using Bradford's solution, each sample containing equivalent amounts of protein was applied to 10% acrylamide denaturing gel (SDS-PAGE) (Laemmli, 1970). After electrophoresis, proteins were transferred for 2 hours at 70 V to an Immobilon PVDF membrane (Millipore Corporation, Bedford, MA, U.S.A.) (Towbin et al., 1979). Blotting membranes were incubated for 1 hour in PBS containing 3% bovine serum albumin (blocking solution) at room temperature and incubated overnight at 4°C in a 1:200 dilution of anti—phospho-FKHR antibody (Upstate Biotechnology) or a 1:1000 dilution of anti-FKHR antibody in blocking solution (Upstate Biotechnology). After several washes with PBS containing 0.1% Tween-20, the membranes were incubated for 1 hour with horseradish peroxidase (HRP)-conjugated antirabbit antibody (Amersham Pharmacia Biotech, Arlington Heights, IL, U.S.A.) or antisheep IgG antibody (Leinco Technologies, Ballwin, MO, U.S.A.) diluted 1:10000. The membranes were then processed with enhanced chemiluminescence Western blotting detection reagents (Amersham Pharmacia Biotech). The images were scanned and analyzed semiquantitatively using the NIH image (public domain software developed at the US National Institutes of Health and available at http://rsb.info.nih.gov/nih-image). Similar methods were used for immunoblotting with anti-Fas ligand antibody (Transduction Laboratories, Lexington, KY, U.S.A.) and anti α-tubulin antibody (Sigma).
Preparation of subcellular fractions
Subcellular fractions were prepared from sham-operated animals and from ischemic animals immediately after reperfusion. Frozen tissues of the CA1 regions were homogenized with a hand homogenizer and sonicated with a Sonifier-250 (Branson) at 0°C in 0.6 mL homogenization buffer containing 10 mmol/L Tris-HCl (pH 7.5), 0.32 mol/L sucrose, 0.5 mmol/L EDTA, 25 mmol/L sodium pyrophosphate, 100 mmol/L β-glycerophosphate, 200 nmol/L calyculin A, 50 μg/mL leupeptin, 25 μg/mL pepstatin A, 50 μg/mL trypsin inhibitor, and 1 mmol/L dithiothreitol. The homogenates were centrifugated at 1,000 g at 4°C for 10 minutes. The pellets were resuspended and used as a crude nuclear fraction. The supernatants were further centrifugated at 100,000 g at 4°C for 30 minutes to obtain the cytosolic fraction. The protein concentration in the crude nuclear and cytosolic fractions was determined with the Bradford method.
Preparation of nuclear extracts and gel mobility shift assay
Gerbils were intraventricularly administrated vehicle or orthovanadate and, 30 minutes later, underwent 5-minute transient forebrain ischemia as described previously. Sham-operated or ischemic animals were decapitated immediately after reperfusion. After brains were removed and rinsed once with cold PBS, CA1 regions of the hippocampus were dissected and removed in cold PBS under a microscope. Each tissue sample was stored in a test tube and kept at −80°C before use. Nuclear extracts were prepared as described by Schreiber et al. (1989). The gel mobility shift assay was performed as described by Yano et al. (1996). The probes used in the gel shift assays were the complementary oligonucleotides corresponding to FKHR responsive elements within the promoter of the Fas ligand gene (5′-AATAGATCTTAAATAAATAGATCTTTA-3′) (Brunet et al., 1999) and were annealed. The 5′ ends of probe DNAs were labeled with [γ-32P]ATP by T4 polynucleotide kinase. The standard binding reaction was performed in a 25-μL mixture containing 10 mmol/L HEPES (pH 7.8), 50 mmol/L KCl, 1 mmol/L EDTA, 5 mmol/L MgCl2, 10% glycerol, 25 mmol/L dithiothreitol, 10 μg/mL pepstatin, 10 μg/mL leupeptin, 5 mmol/L sodium orthovanadate, 3 μg poly (dI-dC), 1 fmol 32P-labeled probe (≈1 × 104 cpm), and nuclear extracts (5 μg protein). In competition analysis, 1 picomole of the competitor oligonucleotide was mixed before addition of 32P-labeled probe. For gel supershift analysis, extracted nuclear proteins were incubated with 10 μg polyclonal antibodies against the FKHR, FKHRL1, and AFX proteins (Santa Cruz Biotechnology) for 1 hour before the addition of a 32P-labeled probe. After 30 minutes at room temperature, the samples were loaded onto a 4% polyacrylamide gel made in a buffer containing 89 mmol/L Tris, 89 mmol/L boric acid, and 2.5 mmol/L EDTA. After electrophoresis, the gel was dried and autoradiographed on x-ray films at −80°C.
Statistical analysis
All values reported here are expressed as mean ± SD. Overall statistical significance for differences among groups was tested by one-way analysis of variance, followed by multiple comparisons between each group or between the control group and other groups using the Dunnett-type multiple comparison test. P < 0.05 was considered significant.
RESULTS
Changes in phosphorylation of FKHR after transient forebrain ischemia with or without orthovanadate pretreatment
Seven days after transient forebrain ischemia without drug treatments, severe neuronal losses in the CA1 region were observed with a cell survival of less than 10% compared with sham-operated animals. Intraventricular injection with orthovanadate 30 minutes before transient ischemia protected neurons from delayed neuronal death, resulting in 80% cell survival (Kawano et al., 2001). Five-minute ischemia resulted in dephosphorylation of Akt by 30% of the control levels in the CA1 region immediately after reperfusion (0 hours), and an intraventricular injection of orthovanadate prevented the dephosphorylation of Akt (Kawano et al., 2001).
Because Akt phosphorylates FOXOs such as FKHR and FKHRL1 as downstream targets in cell-survival signaling (Brunet et al., 2001;Brunet et al. 1999;Kops et al. 1999;Tang et al., 1999), we examined changes in the phosphorylation of FKHR after transient forebrain ischemia. Phosphorylation of FKHR-Ser-256 by Akt is essential for its activation (Brunet et al., 2001;Nakae et al., 2000).
Immunoblotting analysis with anti—phospho-FKHR—Ser-256 antibody revealed that ischemia induced the dephosphorylation of FKHR-Ser-256 in the CA1 region immediately after and 0.5 and 1 hour after reperfusion without producing changes in FKHR protein levels (Figs. 1A and 1C). The FKHR—Ser-256 phosphorylation returned to the basal level within 2 hours after reperfusion. An intraventricular injection of orthovanadate 30 minutes before ischemia prevented the dephosphorylation of FKHR-Ser-256 (Figs. 1B and 1C), while levels of FKHR protein remained unchanged. The FKHR—Ser-256 dephosphorylation was closely correlated with the dephosphorylation of Akt—Ser-473 reported previously, which implies that decreased Akt activity accounts for the decreased FKHR phosphorylation. However, the FKHR phosphorylation recovered to the basal level more slowly than Akt phosphorylation (data not shown). Because dephosphorylation of FKHR is associated with translocation into the nucleus as describe previously, we investigated changes in localization of FKHR in the CA1 regions after subcellular fractionation. In sham-operated animals, FKHR mainly localized in the cytosolic fractions (Fig. 1D). As expected, FKHR significantly translocated to the nuclear fractions immediately after reperfusion (Fig. 1D).
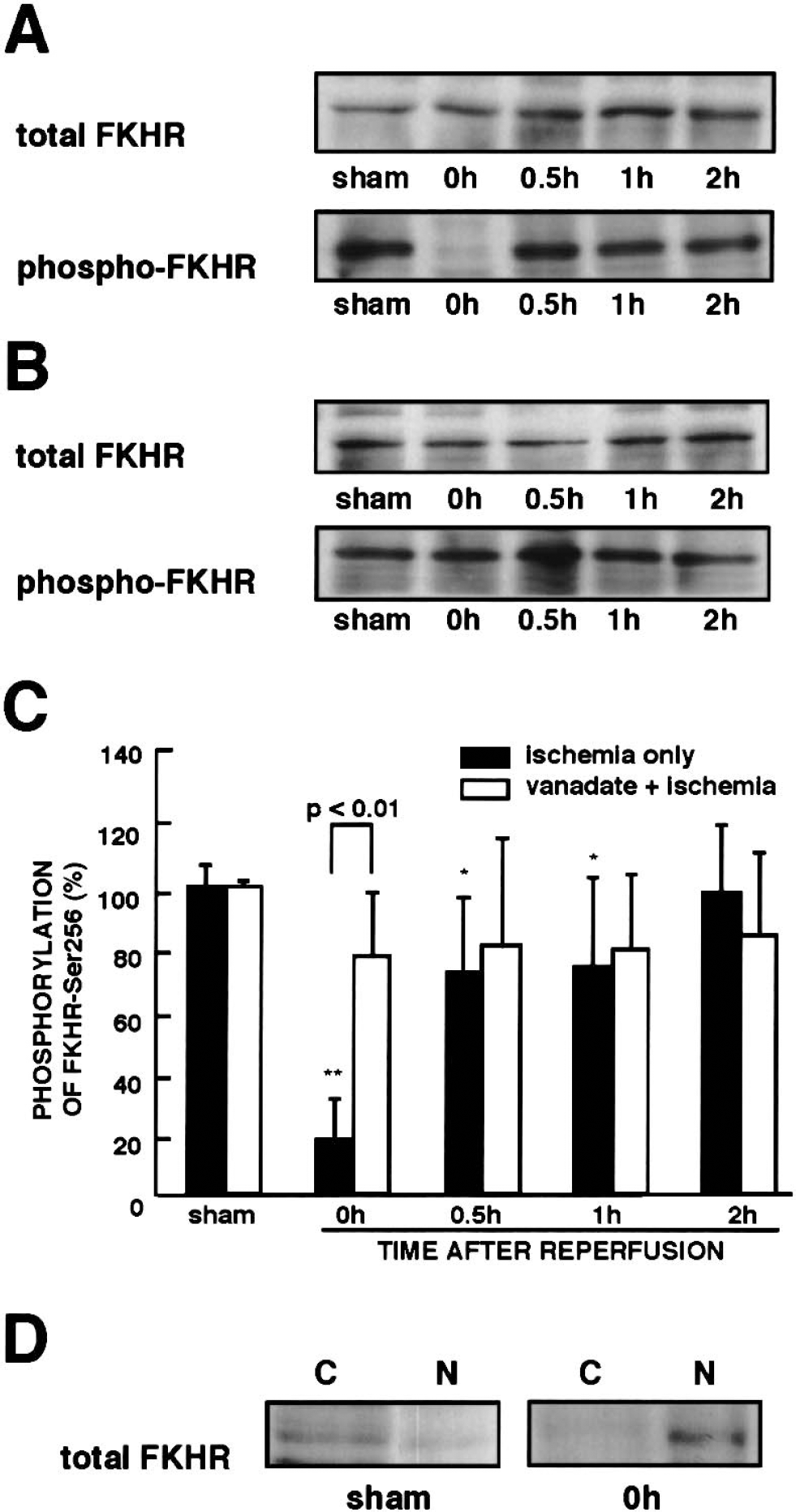
Changes in FKHR—Ser-256 phosphorylation in the CA1 region after reperfusion.
Immunohistochemical localization of FKHR after ischemia
To confirm dephosphorylation and translocation of FKHR after ischemia in the hippocampal CA1 regions, slices from sham-operated and ischemic animals were double-stained with conventional FKHR antibody and phospho-specific antibody for FKHR—Ser-256. In sham-operated animals, immunoreactivity for the phospho-specific antibody showed that phosphorylated FKHR was localized predominantly in the cytoplasm of the pyramidal neurons in the CA1 regions (Figs. 2A and 2E). After transient ischemia, the immunoreactivity markedly decreased in the cytoplasm (Fig. 2B). In contrast, immunoreactivity against conventional FKHR antibody was relatively weak in the CA1 regions in the sham-operated animals (Fig. 2C). However, the transient ischemia caused an apparent accumulation of FKHR into the nuclei of the CA1 pyramidal neurons (Fig. 2D). The merged immunofluorescent images clearly showed that the dephosphorylation of FKHR—Ser-256 is associated with the translocation of FKHR in the pyramidal neurons (Figs. 2E and 2F).
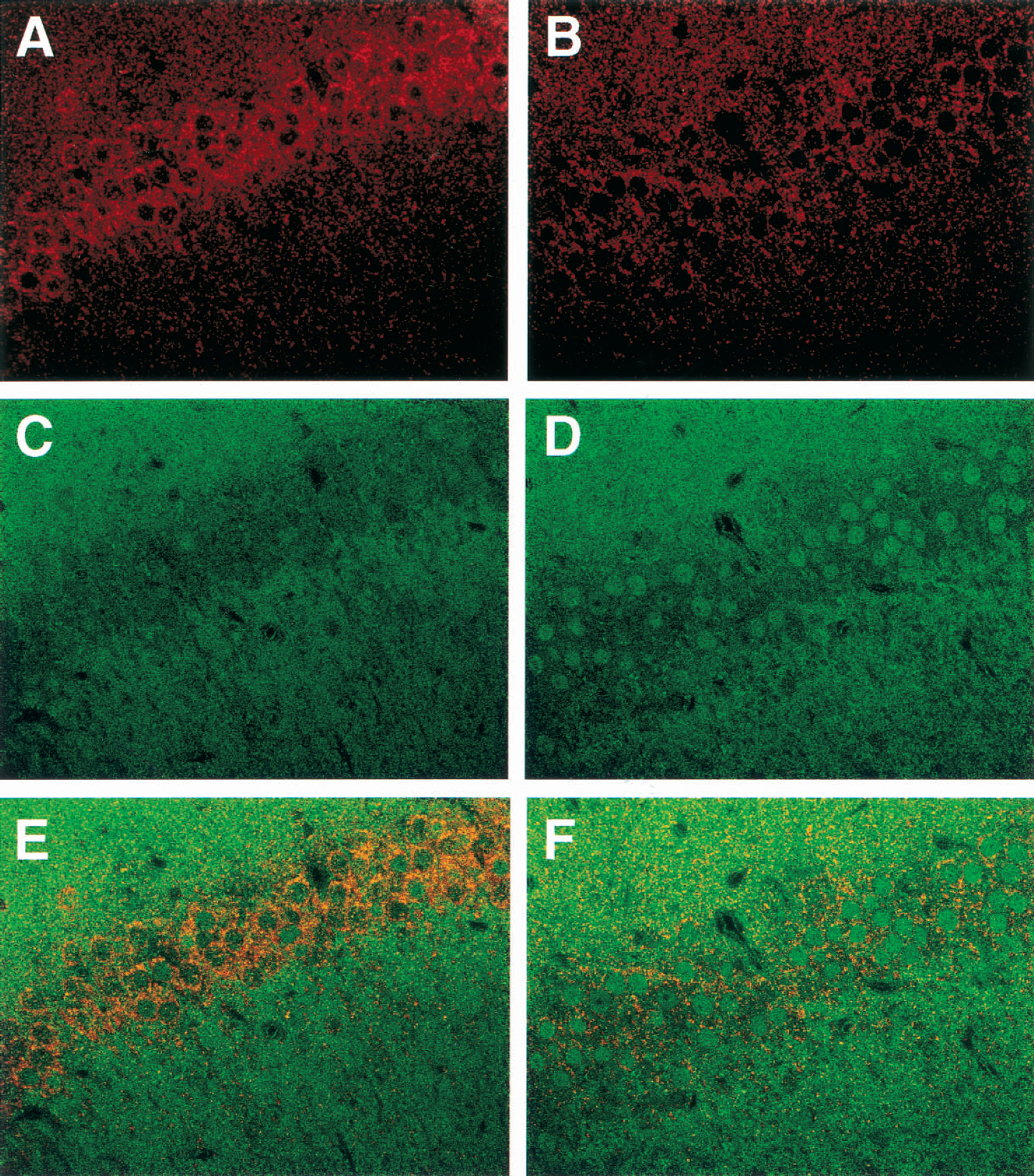
Immunohistochemical localization of phosphorylated FKHR and FKHR in the hippocampal CA1 region after ischemia. Gerbils were perfused and fixed immediately after reperfusion with 4% paraformaldehyde with or without ischemia. Hippocampal slices were obtained from sham-operated gerbils (
DNA binding activities of the Forkhead-responsive element within the Fas ligand promoter after ischemia
The Forkhead consensus binding sites have been characterized (Kaufmann et al., 1995;Pierrou et al., 1994), and the consensus element for FOXO binding, termed the Forkhead-responsive element, has been reported within the promoter of the Fas ligand gene (Brunet et al., 1999). To determine whether FKHR localized into the nuclei has a DNA-binding ability, we performed gel mobility shift analysis using the Forkhead-responsive element oligonucleotide probe derived from the promoter of the Fas ligand gene. Ischemia induced an increase in the binding activity to the Forkhead-responsive element in nuclear extracts from the CA1 regions (arrow in Fig. 3). Orthovanadate treatments reduced this binding activity to the basal levels observed in sham-operated animals (Fig. 3). An excessive amount of nonlabeled oligonucleotide probe competed with the specific binding in the nuclear extracts. Supershift experiments were performed to determine whether FKHR or the other homologues are involved in the DNA-binding complexes. Supershift analysis showed that the DNA-binding complexes (shown by arrow) were shifted by antibodies against FKHR, FKHRL1, and AFX (shown by arrowhead). Although the supershift by the antibodies revealed that at least these three FOXO family members are included in the DNA-binding complexes, the smear-patterned supershift with the antibodies suggests that the affinity and/or the specificity of the antibodies for each FOXO is relatively low.
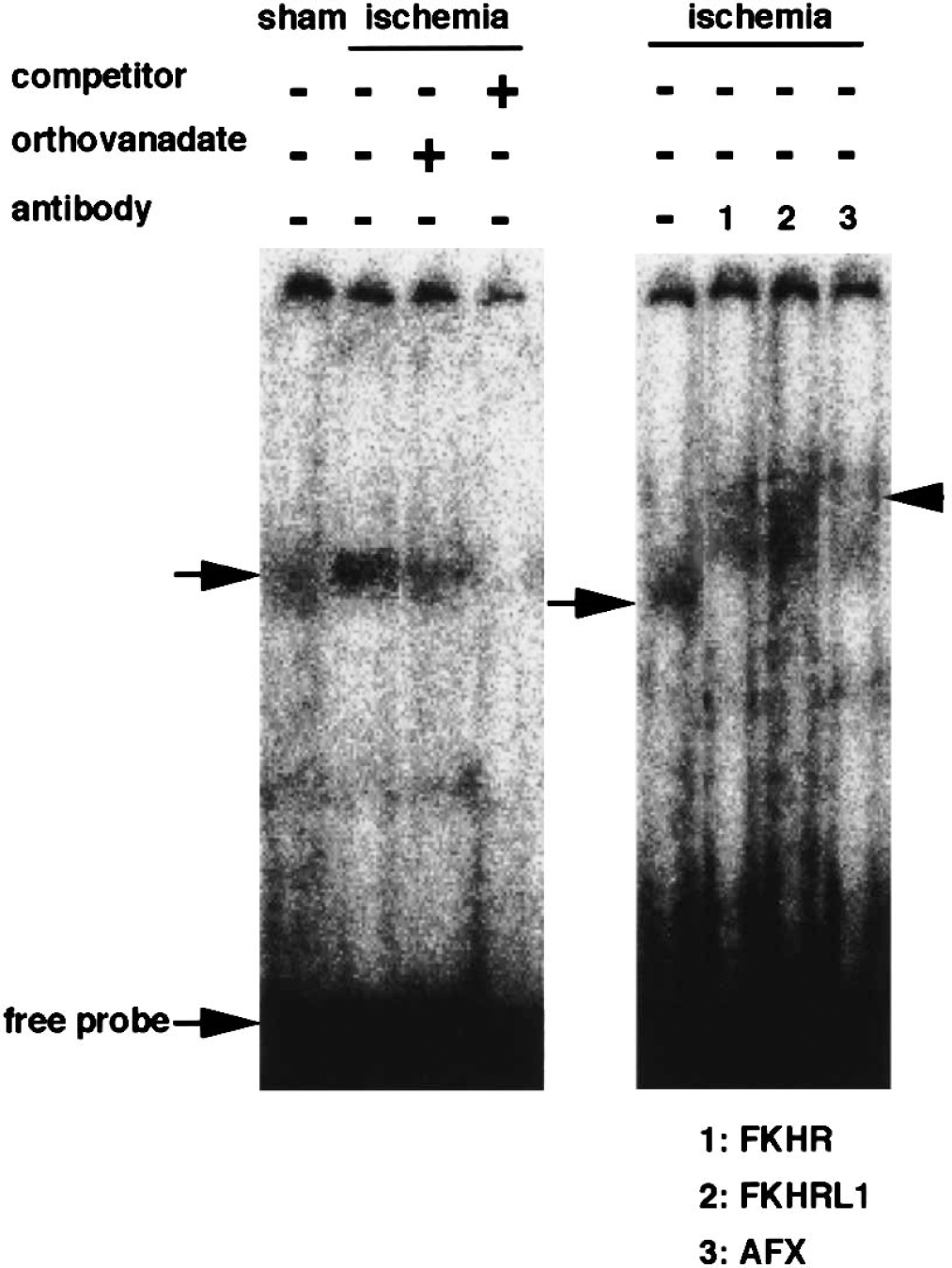
Gel mobility shift assay using the Forkhead-responsive (FKHR) element in the promoter of the Fas ligand gene. Nuclear extracts were obtained from the hippocampal CA1 region of sham-operated animals (sham) or animals exposed to 5-minute ischemia immediately after reperfusion (ischemia). Gel mobility shift analysis showing the nuclear protein binding to the consensus element for FOXOs within the promoter of the Fas ligand gene in hippocampal nuclear extracts of sham-operated animals or animals exposed to 5-minute ischemia with or without pretreatment with orthovanadate. When indicated, the excess amount of nonlabeled competitor was added during incubation. For supershift experiments, antibodies to FKHR, FKHRL1, or AFX were added to the extracts before adding the 32P-labeled probe. Similar results were obtained in two additional experiments.
Expression of Fas ligand after transient forebrain ischemia
FOXOs are known to induce the expression of cell death-related genes, such as Fas ligand and Bim, a proapoptotic Bcl-2 family member (Brunet et al. 1999;Dijkers et al., 2000a). We next examined the expression of Fas ligand protein after ischemia in the CA1 regions. Immunoblotting analysis revealed expression of the Fas ligand in the CA1 regions stimulated 2 days after reperfusion (Fig. 4A), and orthovanadate treatment prevented the ischemia-induced expression of Fas ligand (Fig. 4B). Immunoblotting analysis with α-tubulin showed that an equal amount of protein was loaded in each lane. Densities of Fas ligand protein bands on the immunoblot were analyzed semiquantitatively (data not shown). The expression of Fas ligand 2 days after reperfusion increased significantly (P < 0.05) compared with sham-operated animals. In the hippocampal CA1 regions obtained from sham-operated animals, immunoreactivities against anti-Fas ligand antibody were faint, a finding that is in agreement with previous reports (Rosenbaum et al., 2000) (Fig. 4C). The immunoreactivities for Fas ligand increased in the CA1 pyramidal neurons 2 days after reperfusion (Fig. 4D).
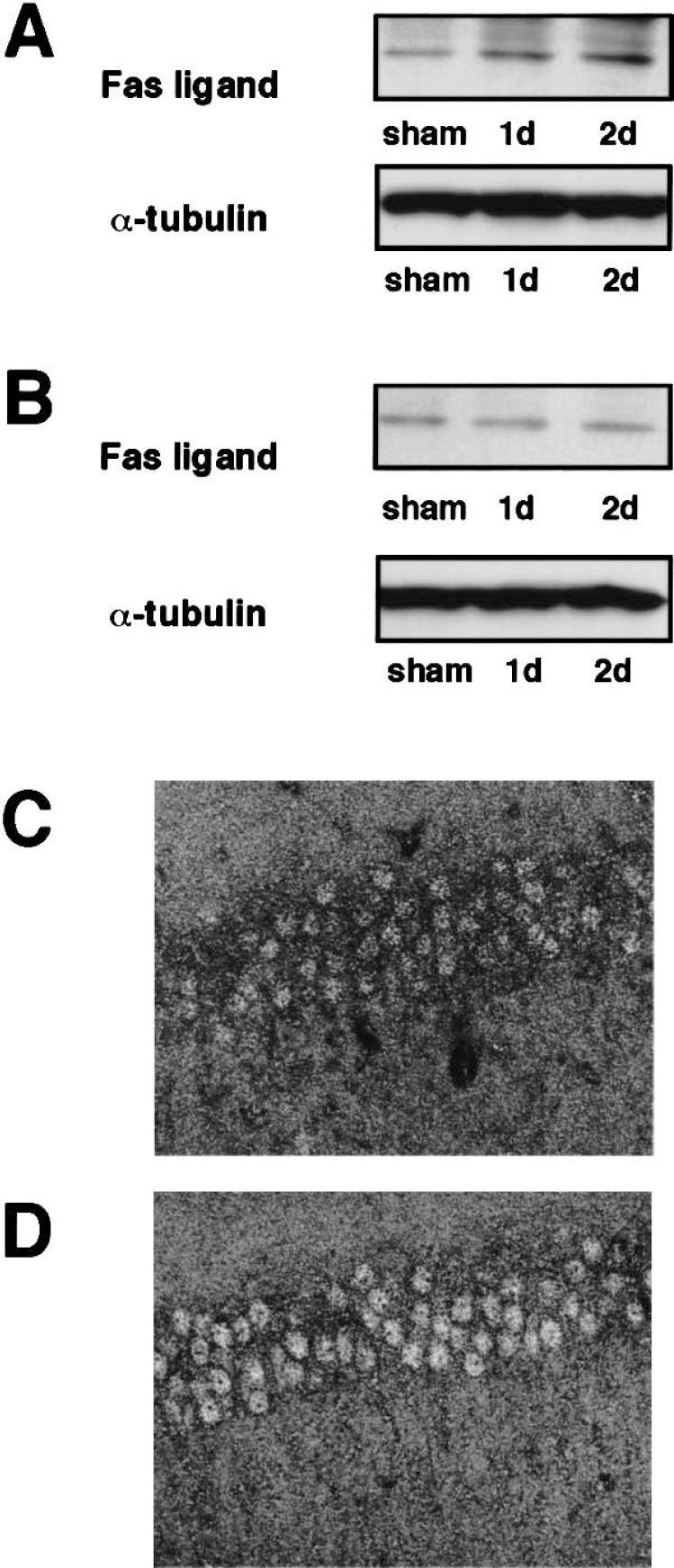
Expression of Fas ligand in the CA1 region after transient forebrain ischemia and its inhibition by orthovanadate treatment.
DISCUSSION
Accumulating evidence suggests that the PI3K-Akt pathway plays an important role in cell survival that is promoted by treatment with growth factors, neurotrophic factors, and cytokines. In a previous study, we documented that orthovanadate has neuroprotective effects on delayed neuronal death after ischemia through the PI3K-Akt pathway (Kawano et al., 2001). Brief exposure to ischemic insults caused activation of Akt and thereby induced ischemic tolerance (Yano et al., 2001). However, downstream targets for Akt that underlie neuroprotection have not been identified in the neurons. Several potential targets for Akt were evident in the cytoprotective action in nonneuronal cells and neuronal cells. For example, Bad, a proapoptotic Bcl-2 family member, caspase-9, CREB, and IκB kinase are substrates for Akt and are involved in the signaling of apoptosis (Brunet et al., 1999;Cardone et al., 1998;Datta et al., 1997;Del Peso et al., 1997;Du and Montminy, 1998;Kops et al., 1999;Ozes et al., 1999;Tang et al., 1999). FOXOs are also phosphorylated by Akt, thereby inhibiting its nuclear translocation (Biggs WH III et al., 1999;Brunet et al., 1999;Nakae et al., 2000 l Takaishi et al., 1999). The Akt-induced phosphorylation of FOXOs suppresses their transcriptional activities that are induced by apoptosis (Brunet et al., 1999;Nakae et al., 2000;Tang et al., 1999). The current study focused on phosphorylation of FKHR, a member of the FOXOs, after ischemia and in orthovanadate-induced neuroprotection.
We showed that FKHR is activated with concomitant dephosphorylation immediately after reperfusion, and that the dephosphorylation of FKHR was accompanied by an increase in its DNA-binding activity on the FKHR-responsive element of the Fas ligand promoter. Furthermore, orthovanadate treatments prevented the dephosphorylation of FKHR concomitant with a reduction of its DNA-binding activity. We also confirmed the apparent translocation to the nuclei from the cytoplasm in the immunohistochemical studies and immunoblotting analysis after subcellular fractionation using a conventional anti-FKHR antibody (Figs. 3 and 1D, respectively). Taken together, the results suggest that FKHR is a potential target for Akt in the pyramidal neurons of the hippocampal CA1 region, and that FKHR dephosphorylation was due to a reduction in Akt activity after ischemia.
Several studies suggest that apoptosis is underlying the delayed neuronal death in ischemic brain injuries (MacManus et al., 1993;Nitatori et al., 1995). The Fas/ Fas ligand system in apoptosis has fundamental roles in the immune system (Nagata, 1997), and current studies have shown the involvement of Fas/Fas ligand signaling in CNS pathophysiology (Beer et al., 2000;Dowling et al., 1996;Li et al., 2000;Nishimura et al., 1995). For example, Fas ligand mRNA expression increased in the cerebral cortex after permanent middle cerebral artery occlusion (MCAO) in adult rats (Harrison et al., 2000), and Fas ligand mRNA and its protein were expressed in the cerebral cortex after reversible MCAO in adult rats (Herdegen et al., 1998;Martin-Villalba et al., 1999;Rosenbaum et al., 2000). Furthermore lpr mice expressing mutant, nonfunctional Fas had significantly smaller infarct sizes after MCAO compared with their wild-type counterparts (Rosenbaum et al., 2000). In the present study, we also confirmed that the activation of FKHR was associated with an increased expression of Fas ligand. Interestingly, the Fas ligand was present in the nuclei and perinuclear regions in the hippocampal CA1 regions. The immunohistochemical localization was consistent with that of previous reports (Cheema et al., 1999;Herdegen et al., 1998;Martin-Villalba et al., 1999;Rosenbaum et al., 2000). Orthovanadate also inhibited the ischemia-induced Fas ligand expression. However, the induction of the Fas-ligand was relatively weak in the immunoblotting analysis and immunohistochemical study. Because the dephosphorylation of FKHR was transient and occurred immediately after reperfusion, further experiments are needed to confirm the relevance of moderate induction of Fas ligand in the delayed neuronal death in the gerbil ischemic model. The activation of Fas is accompanied by the recruitment of procaspase-8 into the death-inducing signaling complex and activation of caspase-8 (Medema et al., 1997). Activation of FOXOs also accounted for the expression of another apoptotic protein, Bim, which is a Bcl-2 family member (Dijkers et al., 2000a). To address the relevance of FKHR activation and Fas signaling in delayed neuronal cell death, the activation of caspase-8 and the induction of Bim are being investigated in our current ischemic model.
Treatment with orthovanadate suppressed Fas ligand expression and FKHR activation, and thereby rescued the pyramidal neurons from delayed neuronal death. Orthovanadate, a protein tyrosine phosphatase inhibitor, is also known as insulin mimetics. The treatment with orthovanadate stimulated mitogen-activated protein kinase (MAPK) and PI3K-Akt—signaling pathways in vitro (D'Onofrio et al. 1994;Hadari et al. 1992;Zhao et al. 1996) and in vivo (Kawano et al., 2001). The neuroprotective effect of orthovanadate was totally blocked by pretreatment with a combination of inhibitors of PI3K and MAPK (Kawano et al., 2001). Thus, further experiments are required to define downstream targets not only for Akt, but also for MAPK in orthovanadate-induced neuroprotection.
Footnotes
Acknowledgments
The authors thank Drs. Y. Takeuchi (Kumamoto University) and N. Ogata (Kumamoto Rosai Hospital) for technical support and critical comments regarding the manuscript.