
Abstract
Poly(ADP-ribose) (PAR) is a polymer synthesized by poly(ADP-ribose) polymerases (PARPs) and metabolized into free adenosine diphosphate (ADP)-ribose units by poly(ADP-ribose) glycohydrolase (PARG). Perturbations in PAR synthesis have been shown to play a key role in brain disorders including postischemic brain damage. A single parg gene but two PARG isoforms (110 and 60 kDa) have been detected in mouse cells. Complete suppression of parg gene causes early embryonic lethality, whereas mice selectively lacking the 110 kDa PARG isoform (PARG−/−110) develop normally. We used PARG−/−110 mice to evaluate the importance of PAR catabolism to postischemic brain damage. Poly(ADP-ribose) contents were higher in the brain tissue of PARG−/−110 than PARG+/+110 mice, both under basal conditions and after PARP activation. Distal middle cerebral artery occlusion caused higher increase of brain PAR levels and larger infarct volumes in PARG−/−110 mice than in wild-type counterparts. Of note, the brain of PARG−/−110 mice showed reduced heat-shock protein (HSP)-70 and increased cyclooxygenase-2 expression under both control and ischemic conditions. No differences were detected in brain expression/activation of procaspase-3, PARP-1, Akt, HSP-25 and interleukin-1β. Our findings show that PAR accumulation worsens ischemic brain injury, and highlight the therapeutic potential of strategies capable of maintaining PAR homeostasis.
Introduction
Poly(ADP-ribose) polymerases (PARPs) are nicotinamide adenine dinucleotide (NAD)-consuming enzymes forming poly(ADP-ribose) (PAR) and involved in maintenance of nuclear homeostasis. Among them, the DNA damage-activated PARP-1 shows the highest poly(ADP-ribosyl)ating activity. Poly(ADP-ribose) consists of long, branched and negatively charged polymers of ADP-ribose units whose polymerization onto chromatin-interacting proteins including PARP-1 significantly affects their functioning through electric repulsion and steric hindrance (D'Amours et al, 1999; Smith, 2001; Herceg and Wang, 2001). A large body of evidence shows that PAR formation increases during brain ischemia and participates in stroke pathogenesis (for a review see Chiarugi, 2005b). Several mechanisms can underlie the neurotoxic role of PAR in ischemic brain injury (Chiarugi, 2005a). For instance, bulk PAR neoformation causes NAD depletion that in turn prompts adenosine triphosphate (ATP) depletion and cellular energy failure (Szabo and Dawson, 1998; Ha and Snyder, 2000; Skaper, 2003). In addition, PAR can trigger mitochondrial release of apoptosis-inducing factor (AIF) and ensuing neuronal death (Hong et al, 2004). Finally, a key role for PAR in transcription of genes involved in neurotoxicity/neuroprotection has been reported (Ullrich et al, 2001; Chiarugi, 2002; Ha et al, 2002; Chiarugi and Moskowitz, 2003; Park et al, 2004). However, despite remarkable neuroprotection conferred by suppression of PAR neosynthesis during brain ischemia, underlying mechanisms still wait to be clearly established (Moroni et al, 2001; Chiarugi, 2002; Goto et al, 2002). Intriguingly, ischemic neuroprotection by PARP-1 suppression in the mouse seems to be restricted to males (Hagberg et al, 2004; McCullough et al, 2005).
In contrast with the increasing number of PARPs involved in poly(ADP-ribosyl)ation (Ame et al, 2004), poly(ADP-ribose) glycohydrolase (PARG) is the only enzyme capable of degrading PAR into ADP-ribose identified so far (Davidovich et al, 2001; Bonicalzi et al, 2005). Poly(ADP-ribose) glycohydrolase is ubiquitously expressed in mammalian cells and cleaves PAR into free ADP-ribose units owing to its exoglycosidic activity. Nuclear localization and exportation signals in PARG amino-acid sequence allow its nucleocytoplasmic shuttling and efficient PAR catabolism (Bonicalzi et al, 2003). Although little is known about the pathophysiologic role of PARG, its involvement in cell cycle regulation (Ohashi et al, 2003), mitotic spindle assembly (Chang et al, 2004), development (Hanai et al, 2004; Koh et al, 2004), differentiation (Di Meglio et al, 2003), and cell death (Affar et al, 2001; Ying et al, 2001; Hanai et al, 2004) has been reported.
Given the cytotoxic potential of PAR, attention has been devoted to elucidating the role of PARG in disease pathogenesis. The complete suppression of the parg gene causes early embryonic lethality (Koh et al, 2004), thereby precluding studies in adult animals. Conversely, the recently developed mice selectively lacking the 110 kDa isoform of PARG (PARG−/−110) are viable and apparently develop normally, despite being more susceptible to genotoxic shock and endotoxin treatment (Cortes et al, 2004). To further understand the role of PARG in the brain and its relevance to stroke pathogenesis, in this study we investigated basal and ischemia-induced cerebral PAR contents as well as sensitivity to ischemic brain injury in PARG−/−110 mice. In these animals, we also evaluated constitutive and ischemia-induced brain expression of several neuroprotective or neurotoxic proteins, as well as the extent of PARP-1-dependent brain energy derangement.
Materials and methods
Induction of Distal Middle Cerebral Artery Occlusion and Determination of Infarct Size
Development and genotyping of PARG−/−110 mice has been reported previously (Cortes et al, 2004). In this study, we used homozygous knockout and wild-type mice. They derived originally from heterozygous intercrosses (i.e., heterozygous PARG110 mice with a 129/Sv/Ola background were intercrossed and homozygous mice were then kept in sister–brother breeding for five generations). Permanent distal middle cerebral artery occlusion (MCAO) was induced in age-matched male PARG−/−110 mice and corresponding wild-type littermates (n = 8, per group). Animals (25 to 30 g) were anesthetized with 4% isoflurane and maintained on 1.5% isoflurane in air. Rectal temperature was monitored and maintained between 36.5 and 37.5°C with a homeothermic blanket. A 1 cm vertical scalp incision was made between the right eye and ear. The temporalis muscle was bisected and a 2 mm burr hole was made at the junction of the zygomatic arch and squamous bone. The distal MCA was exposed and permanently occluded by cauterization above the rhinal fissure. Physiologic parameters such as rectal temperature, mean arterial blood pressure and pH, PaO2 as well as PaCO2 did not differ between strains before, during, and 1 h after ischemia. After surgery, mice were kept at 37°C for 1 h in an incubator and then placed in their cage until being killed. Mice were killed 14 h (Western blotting and immunohistochemistry) or 24 h (infarct determination) after MCAO, and their brains snap frozen in N2 vapor for cryostat sectioning. For infarct determination, hematoxylin- and eosin-stained coronal sections (15 μm) were imaged by using the Image 3.0 ProPlus analysis software. Infarct areas were calculated subtracting the area of intact tissue in the ipsilateral hemisphere from the area of the contralateral hemisphere to minimize the error that is introduced by edema, which distorts and enlarges the infarcted tissue and surrounding white matter. Infarct volumes were calculated multiplying the infarct area by the distance among sections as described previously (Swanson and Sharp, 1994).
Western Blotting and Immunohistochemistry
The ischemic and controlateral healthy cortex of mice subjected to 14 h MCAO as well as control and methyl-N-nitroso guanidine (MNNG)-exposed brain slices were collected in eppendorf tubes and stored at −80°C. Samples were homogenized in lysis buffer (50 mmol/L Tris, pH 7.4, 1 mmol/L EDTA, 1 mmol/L phenylmethylsulfonyl fluoride, 4 μg/mL aprotinin and leupeptin, 1% sodium dodecyl sulfate (SDS)), proteins were isolated and measured according to standard techniques, and 20 to 40 μg of protein/lane were loaded. After 4% to 20% SDS-polyacrylamide gel electrophoresis (PAGE) and blotting, membranes (Hybond-ECL, Amersham, UK) were blocked with phosphate-buffered saline (PBS) containing 0.1% Tween-20 and 5% skimmed milk (TPBS/5% milk) and then probed overnight with primary antibodies (1:1000 in TPBS/5% milk). The anti-PAR monoclonal antibody (10H) was from Alexis (Vinci, Italy), the anti-inducible nitric oxide synthase (iNOS) and anti-interleukin (IL)-1β were polyclonal antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and the anti-cyclooxygenase-2 (COX-2) polyclonal antibody was from Cayman Chemicals (Ann Arbor, MI, USA). Membranes were then washed with TPBS and incubated 1 h in TPBS/5% milk containing the corresponding peroxidase-conjugated secondary antibody (1:2000). After washing in TPBS, enhanced chemiluminescence (Amersham, UK) was used to visualize the peroxidase-coated bands. Densitometric data were obtained using the NIH image analysis system (Bethesda, MD, USA) and expressed as the mean ± standard error of mean (s.e.m.). For immunohistochemistry, frozen brains of PARG+/+110 and PARG−/−110 mice subjected to 14 h MCAO were cut with a cryostat, sections mounted on slides, and ethanol-fixed. Slides were preincubated 1 h with PBS/Triton X-100 0.3% (PBST) containing 20 mg/mL bovine serum albumin (BSA) and then overnight with the above-mentioned anti-PAR monoclonal antibody (1:100 in PBST/5 mg/mL BSA). A subsequent incubation (1 h) with a Cy2-conjugated anti-mouse secondary antibody (1:200 in PBST/5 mg/mL BSA) allowed visualization of the binding. A subsequent incubation (2 h) with the anti-neuronal nuclei monoclonal antibody (NeuN) (Chemicon, Tamecula, CA, USA) or anti-glial fibrillary acidic protein (GFAP) (Sigma, Saint Louis, MO, USA) antibodies was performed for double labeling. Binding was revealed with the corresponding Cy3-conjugated secondary antibodies and an inverted Nikon fluorescence microscope. Specificity of immune detection was established by omission of the primary antibody.
Semiquantitative Real-Time Polymerase Chain Reaction
Total RNA (1 μg) extracted from the brain parietal cortex was reverse transcribed and the DNA mixture subjected to polymerase chain reaction (PCR) using the following oligonucleotide primers: PARG110, 5′-CCACCTCGTTTGT TTTCA-3′ (sense) and 5′-CCAACATCTGGCAAAGGA-3′ (antisense); PARG60, 5′-GGACCTCCTCTCAAAGAGACA-3′ (sense) and 5′-CTGGGGAAAAAAGGACGAAA-3′ (antisense); and β-actin, 5′-GACCTGACAGACTACCTC-3′ (sense) and 5′-AGACAGCACTGTGTTGGC-3′ (antisense). Numbers of cycles (94°C 30 secs, 58°C 30 secs, 72°C 1 min for PARG60; 94°C 30 secs, 52°C 30 secs, 72°C 1 min for PARG110 and β-actin) selected after determining the linear working range for the reaction were 25, 27, 30, and 33 for PARG isoforms and 20 for β-actin. PCR products were separated on 1.8% agarose gels.
Measurement of Nicotinamide Adenine Dinucleotide, Adenosine Triphosphate and Adenosine Diphosphate-Ribose in Mouse Brain Slices
Poly(ADP-ribose) glycohydrolase+/+110 and PARG−/−110 mice were anesthetized with isofluorane and decapitated. The cerebellum was removed and brains were immersed in ice-cold PBS and cut with a vibratome to obtain coronal slices of 200 μm. Only coronal sections containing both the cortex and striatum were used, whereas those corresponding to the anterior or posterior regions of the brain were discarded. Slices were preincubated for 1 h in a 24-multiwell plate containing 200 μL of serum-free Dulbecco's modified Eagle's medium (DMEM), at 37°C in a water-saturated 5% CO2/95% air atmosphere, and then exposed to 1 mmol/L MNNG (directly dissolved in DMEM) for different times. As for NAD measurement, slices were transferred in eppendorf tubes containing 100 μL HClO4 1 N, sonicated, and the solution neutralized with an equal volume of KOH 1 N. After the addition of 200 μL bicine 100 mmol/L (pH 8) and centrifugation (13,000g/3 mins), 100 μL of the extract was mixed with an equal volume of bicine buffer containing 23 μL/mL ethanol, 0.17 mg/mL 3-[4,5-dimethylthiazol-2-yl]-2,5-di-phenyltetrazolium bromide (MTT), 0.57 mg/mL fenazine ethosulfate, and 10 μg alcohol dehydrogenase. The mixture was kept at room temperature for 20 mins and then absorbance at 550 nm measured. A standard curve allowed quantification of NAD. Adenosine triphosphate was measured in 50 μL of the neutralized slice extract by means of the ATPlite luminescence detection kit (Perkin-Elmer, Zaventem, Belgium). Adenosine diphosphate-ribose was quantified by high-performance liquid chromatography (HPLC) and ultraviolet (UV) detection kit (Perkin-Elmer model LC 90). Briefly, MNNG-treated slices were transferred in eppendorf tubes containing 100 μL HClO4 (50 mmol/L), sonicated, boiled, and neutralized with 100 μL KOH (50 mmol/L). The extract was centrifuged (12,000 g/5 mins) and 100 μL of the supernatant injected in a C18 5 μm, 4.6 × 250 mm, Nucleosil HPLC column. The mobile phase was 100 mmol/L phosphate buffer, pH 7.5, containing 13 mmol/L tetrabutylammonium bromide and 1% acetonitrile at 1 mL/min flow rate. Absorbance was measured at 260 nm.
Results
Transcription of Poly(ADP-Ribose) Glycohydrolase Isoforms in Poly(ADP-Ribose) Glycohydrolase110 Wild-Type and Knockout Mouse Brains
Although a single parg gene is present in mouse genome, mouse embryonic fibroblasts express two PARG isoforms of 110 and 60 kDa because of the presence of an alternative translation start codon in exon 4 (Figure 1A). Consistently, deletion of exons 2 and 3 in parg gene generates mouse lacking PARG110 but still expressing PARG60 (Cortes et al, 2004). To investigate whether transcription of PARG60 also occurs in the brain of adult PARG−/−110 mice, we used complementary deoxyribonucleic acid (cDNA) derived from the brain cortex of wild-type and knock out animals and forward and reverse primers amplifying sequences in exon 2 or 13 of parg. Both couples of primers amplified sequences of the predicted molecular weight in cDNA deriving from PARG+/+110 brain cortex. Conversely, only primers binding to exon 13 gave an amplification product when cDNA from PARG−/−110 mouse cortex was used (Figure 1Ba), suggesting that PARG60 was expressed in the brain of PARG−/−110 mice. In principle, disruption of exons 2 and 3 in PARG gene might favor transcription of PARG60. Given that anti-PARG antibodies are not currently available, we compared the amount of amplification products of exon 13 (obtained with different number of cycles comprised in the linear working range of the reaction to warrant semiquantitative analysis) by using cDNA from the cortex of PARG+/+110 and PARG−/−110 mice. As shown in Figure 1Bb, the amount of amplification products did not differ between strains, suggesting that PARG60 expression is not altered in PARG110 null mice.
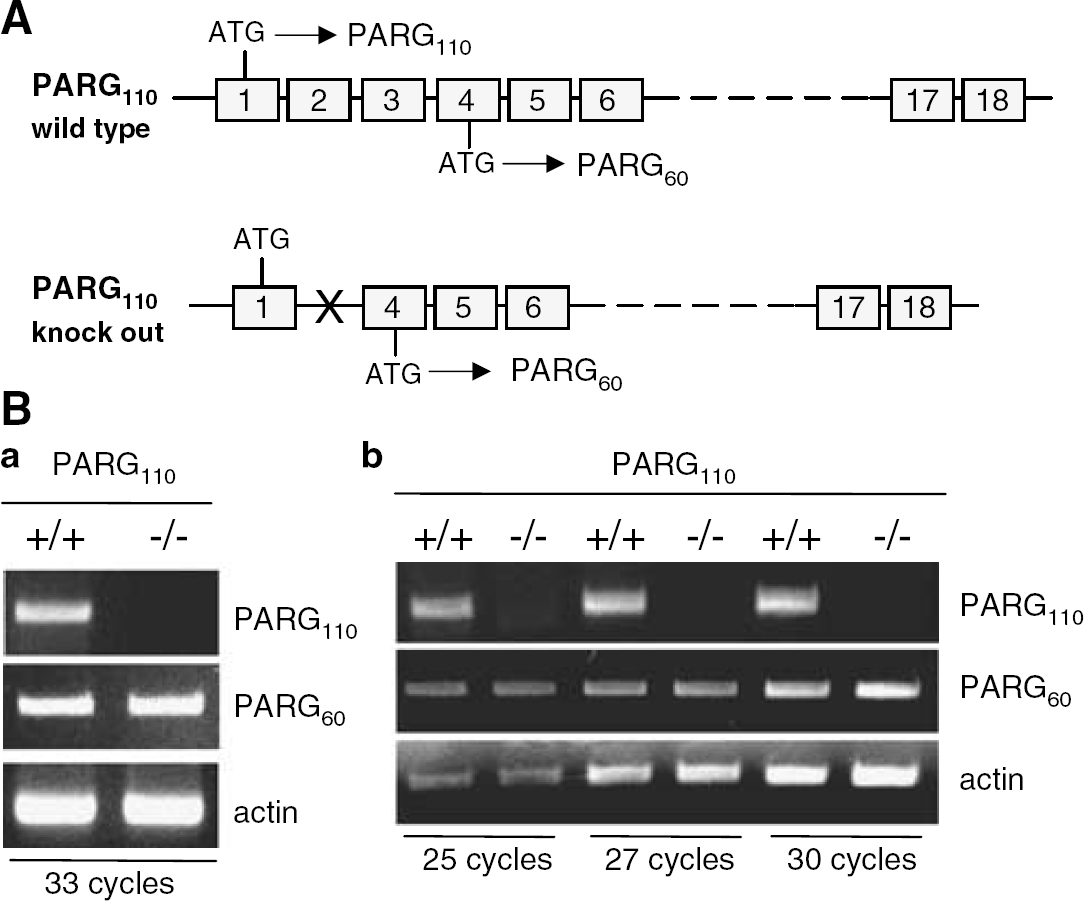
Transcription of poly(ADP-ribose) glycohydrolase (PARG) isoforms in the mouse brain. (
Effect of Poly(ADP-Ribose) Glycohydrolase110 Deletion on the Brain Tissue Response to Poly(ADP-Ribose) Polymerase-1 Hyperactivation
To study the effects of PARG110 deletion on brain PAR levels and energy dynamics, we evaluated PAR, NAD, ATP, and ADP-ribose contents in brain slices (50 μm) of PARG−/−110 and PARG+/+110 mice under basal conditions or after exposure to MNNG, a widely used PARP-1 activator (Ha and Snyder, 1999; Yu et al, 2002). Although it is possible that both constitutive PARP-1 activity and polymer contents are altered during slice preparation, Figure 2Aa shows that basal PAR level was higher in PARG−/−110 than in PARG+/+110 brain slices. MNNG exposure caused a rapid (5 mins) and transient (30 mins duration) increase of PAR content in both wild-type and PARG−/−110 brain tissue. At each time point after MNNG exposure, brain slices obtained from PARG−/−110 mice had higher PAR contents than those obtained from PARG+/+110 animals (Figure 2Ab). Basal NAD contents were 5.9 ± 1.1 and 7.4 ± 0.9 nmol/mg protein in PARG+/+110 and PARG−/−110 brain slices, respectively, and were not significantly affected by MNNG exposure (Figure 2Ba). Basal ATP levels were 16.6 ± 2.3 and 18.3 ± 3 nmol/mg protein in PARG+/+110 and PARG−/−110 brain slices, respectively. Adenosine triphosphate contents decreased to about 55% at 15 and 30 mins after MNNG exposure in slices from both mouse strains (Figure 2Bb). The basal ADP-ribose contents were 5.6 ± 1.3 and 6.2 ± 0.4 nmol/mg protein in PARG+/+110 and PARG−/−110 brain slices, respectively. On exposure to MNNG, ADP-ribose contents increased equally (analysis of variance + Tukey's post hoc test) and time-dependently in PARG+/+110 and PARG−/−110 slices (Figure 2Bc).
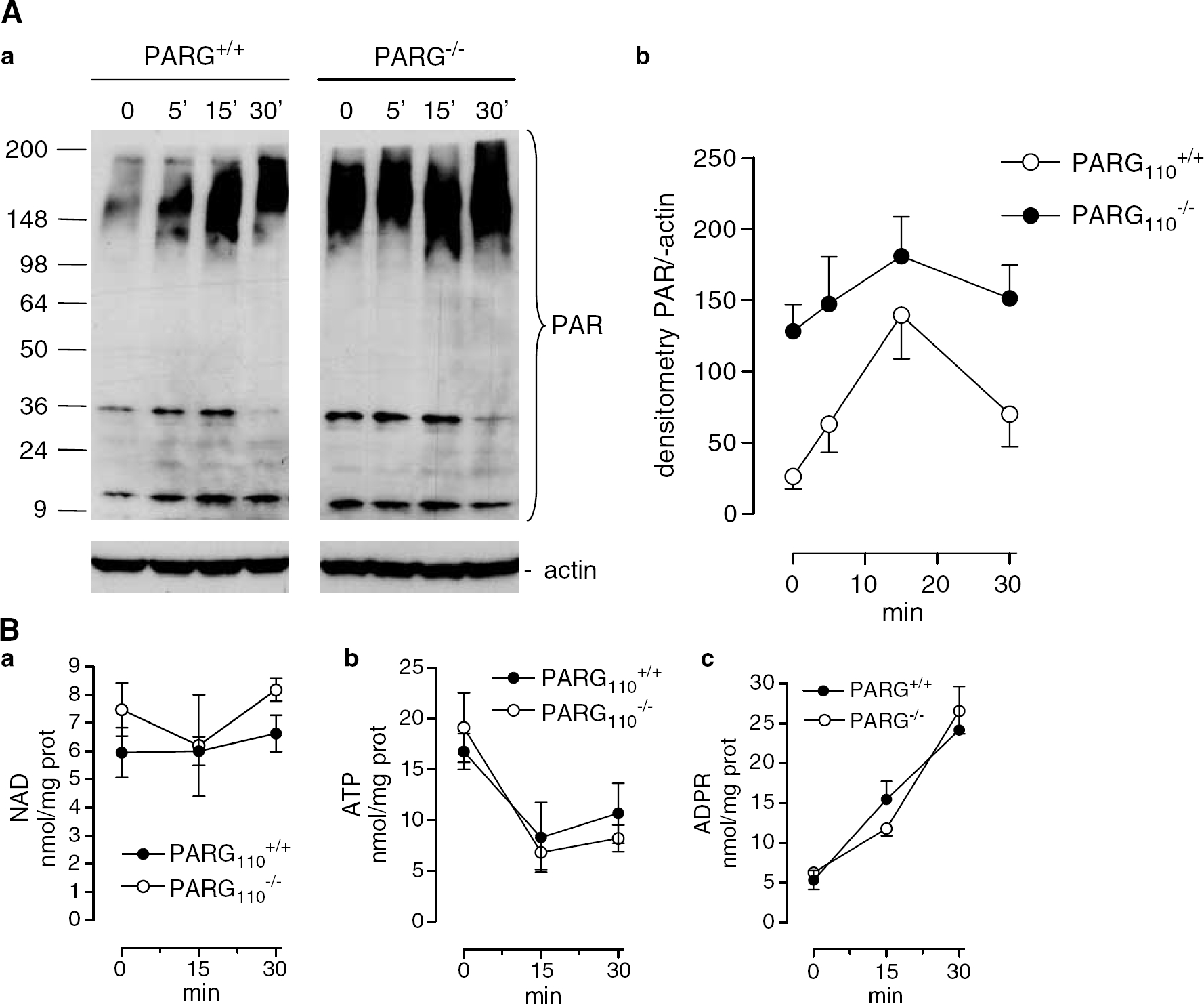
Effect of methyl-N-nitroso guanidine (MNNG) on contents of poly(ADP-ribose) (PAR), adenosine triphosphate (ATP) and adenosine diphosphate (ADP)-ribose in brain slices of poly(ADP-ribose) glycohydrolase (PARG)+/+110 and PARG−/−110 mice. (
Poly(ADP-ribose) Glycohydrolase110 Deletion Increases the Vulnerability to Ischemic Brain Injury
The finding that PAR accumulated in the brain tissue of PARG−/−110 mice, along with the relevance of the polymer to stroke pathogenesis, prompted us to evaluate the sensitivity of these animals to the ischemic brain injury. In line with lack of phenotype or developmental abnormalities in PARG−/−110 animals (Cortes et al, 2004), there were no macroscopic differences in size and histology (hematoxylin/eosin staining) between PARG+/+110 and PARG−/−110 mouse brains (Figure 3C and not shown). To investigate whether PARG110 plays a role in brain ischemic injury, PARG+/+110 and PARG−/−110 mice were subjected to distal MCAO. Rectal temperature, mean arterial blood pressure, pH, PaO2, and PaCO2 did not differ between strains before, during, and 1 h after ischemia (not shown). Twenty-four hours after MCAO, PARG−/−110 mice had a larger infarct volume than wild-type animals (11.8 ± 1.3 versus 6.7 ± 1 mm3, respectively; n = 8 per group; *P < 0.05, **P < 0.01, Student's t-test; Figure 3A). In keeping with the ischemic model we used, most of the infarct was in the cortex. The striatum was minimally injured in two out of eight wild-type and three out of eight PARG−/−110 mice (Figures 3A to C).
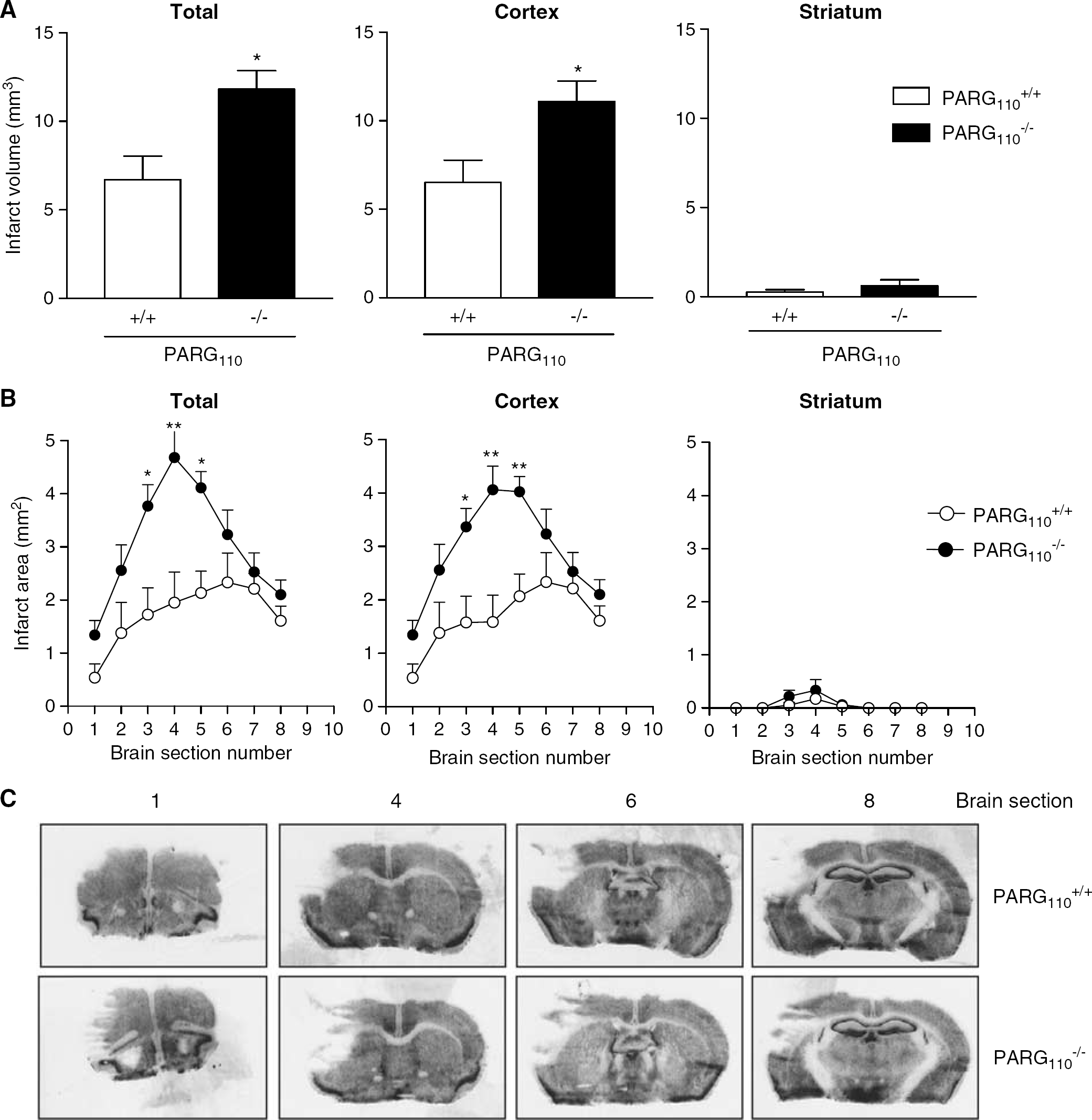
Deletion of poly(ADP-ribose) glycohydrolase(PARG)110 worsens ischemic brain injury. Infarct volumes (
Poly(ADP-Ribose) Glycohydrolase110 Deletion Increases Basal and Ischemia-Induced Cerebral PAR Contents
Figure 4 shows that PAR contents in the brain cortex were higher in PARG−/−110 than wild-type animals both under basal conditions and after MCAO. The figure also shows that the pattern of poly(ADP-ribosyl)ated proteins in the two mouse strains consisted of a smear of 200 to 140 kDa, plus strong (∼58 and 25 kDa) and faint (∼15, 36, and 110 kDa) bands. Size and density of the 58, 25, and 15 kDa bands increased in the ischemic cortex of both strains. Densities of the 110 and 36 kDa bands (classically related to PARP-1 and histone H1, respectively; D'Amours et al, 1999; Rouleau et al, 2004) were not consistently increased in ischemic tissues.
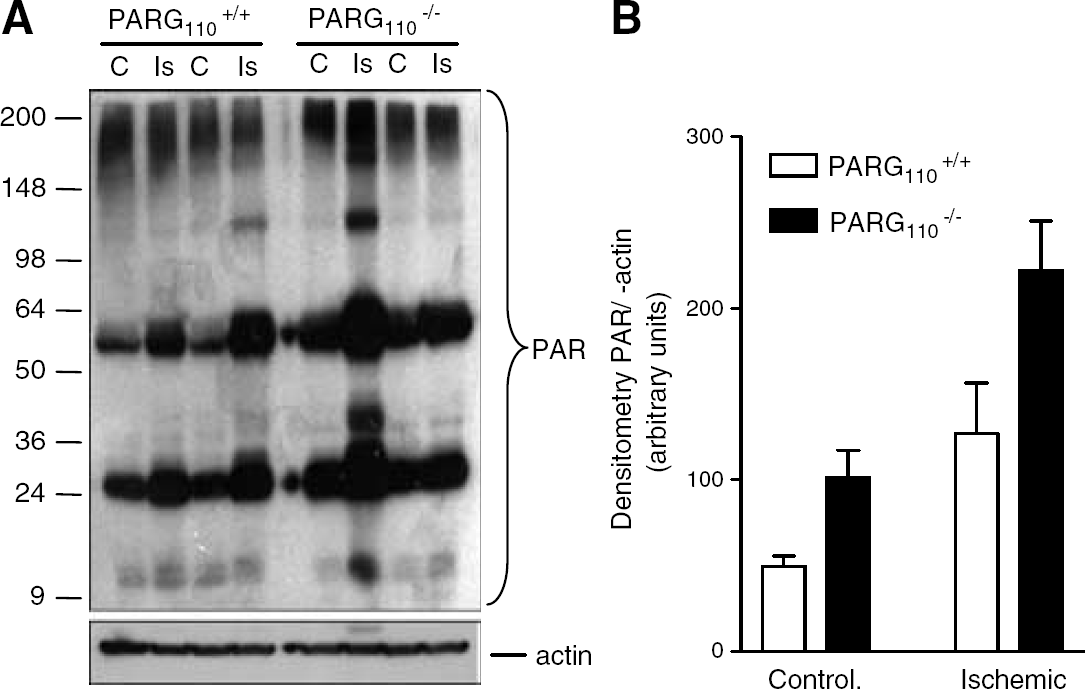
Poly(ADP-ribose) (PAR) contents in the cortex obtained from control (controlateral) and ischemic sites of poly(ADP-ribose) glycohydrolase (PARG)+/+110 and PARG−/−110 mice. (
In the controlateral cortex of PARG+/+110 mice, immunoreactivity of PAR was weak and homogeneously scattered, whereas some densely stained cells were present in the peri-infarct zone (Figure 5A). In keeping with Western blotting, PAR immunoreactivity was higher in the controlateral cortex of PARG−/−110 mice compared with that of PARG+/+110 animals. Of note, numerous densely stained cells were found in the peri-infarct area of PARG−/−110 mouse brains (Figure 5A). Poly(ADP-ribose) immunoreactivity appeared perinuclear in the controlateral cortex of PARG+/+110 and PARG−/−110 mice. On the contrary, PAR mostly localized in the nucleus of cells in the ischemic cortex of both mouse strains (Figure 5B). Double labeling experiments revealed that these nuclei were positive for the neuronal marker NeuN in the ischemic cortex of both PARG+/+110 and PARG−/−110 mice. Conversely, PAR immunoreactivity was almost completely absent in GFAP-positive astrocytes of the ischemic tissue (Figure 6).
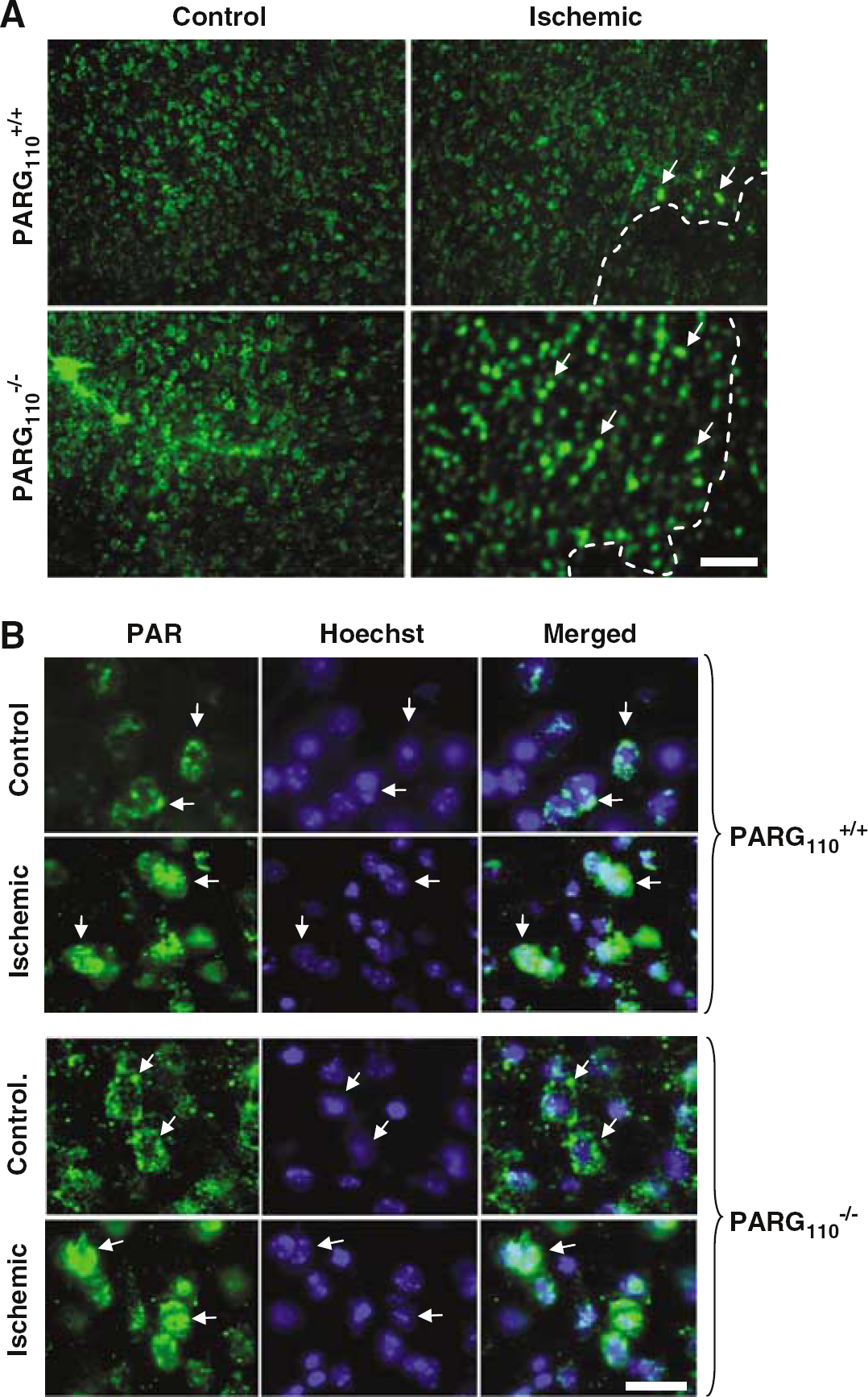
Immunohistochemical visualization of poly(ADP-ribose) (PAR) in control and ischemic cortex of poly(ADP-ribose) glycohydrolase (PARG)+/+110 and PARG−/−110 mice. (
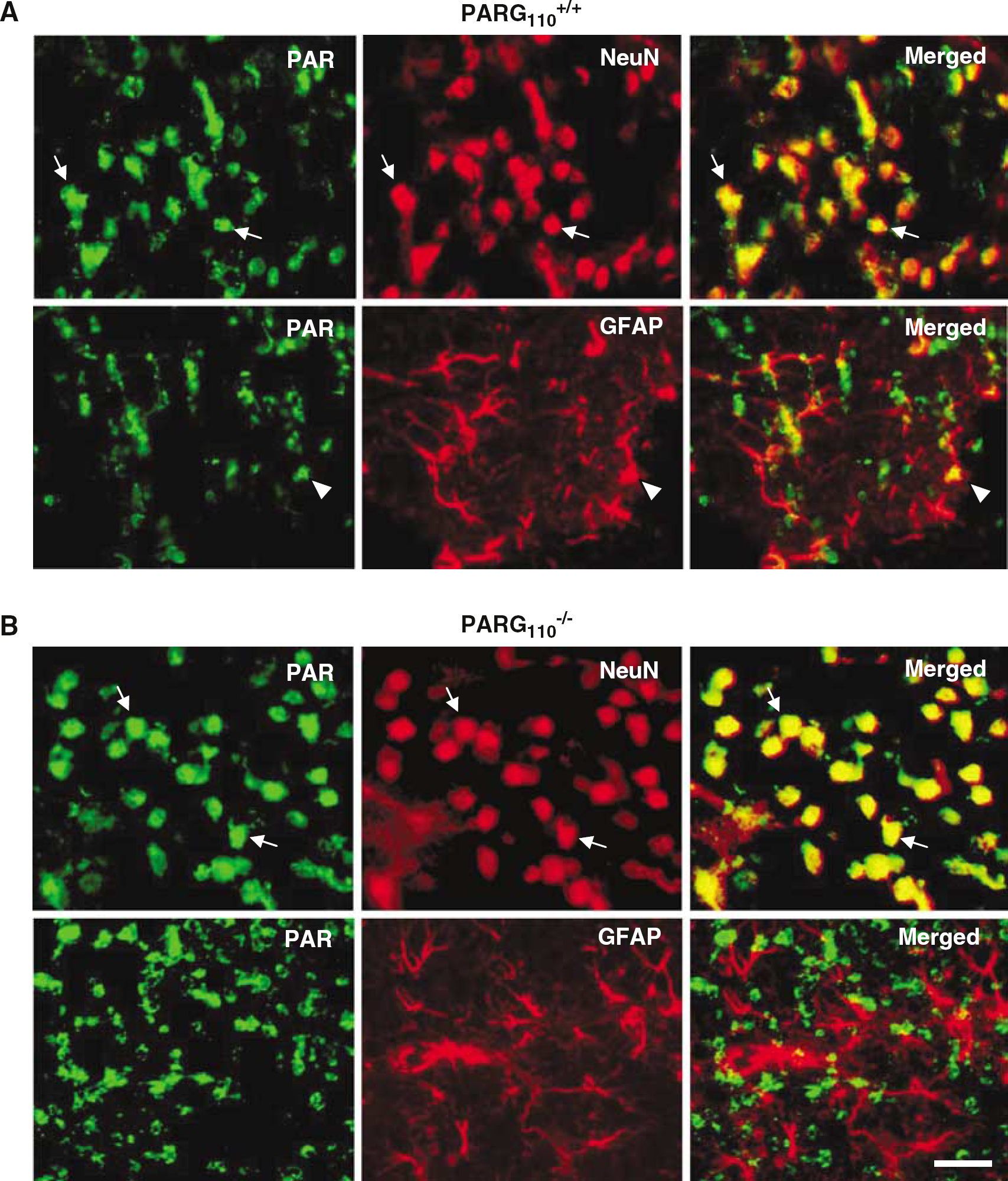
Cellular localization of poly(ADP-ribose) (PAR) in the ischemic cortex of poly(ADP-ribose) glycohydrolase (PARG)+/+110 and PARG−/−110 mice 14 h after middle cerebral artery occlusion (MCAO). In the ischemic cortex of wild-type mice, PAR immunoreactivity (green) almost completely colocalizes (yellow, arrows) with neuronal nuclei monoclonal antibody-positive nuclei (red), suggesting neuronal origin. Conversely, sporadic glial fibrillary acidic protein-positive astrocytes (red) have a PAR-positive nucleus (yellow, arrowhead). A similar cellular distribution of PAR is present in the ischemic cortex of PARG−/−110 mice (arrows). One experiment representative of two is shown. Bar = 30 μm.
Effect of Poly(ADP-Ribose) Glycohydrolase110 Deletion on Expression/Activation of Neuroprotective or Neurotoxic Proteins in Control and Ischemic Mouse Brain
The brain's gene expression profile plays an important role in postischemic brain damage (Read et al, 2001; Bates et al, 2001; Weinstein et al, 2004; Stenzel-Poore et al, 2004), and PAR finely modulates transcriptional activation (Ziegler and Oei, 2001; Chiarugi, 2002; Kraus and Lis, 2003; Rouleau et al, 2004). On this basis, and also considering that PARG is emerging as a regulator of gene expression (Tsai et al, 1992; Panda et al, 2002; Rapizzi et al, 2004), we compared in PARG+/+110 and PARG−/−110 mice the expression and/or activation profiles of several proteins involved in ischemic brain injury (Figure 7). In the light of the relevance of apoptosis to stroke pathogenesis (Lo et al, 2003) and that of PAR to apoptotic cell death (Hong et al, 2004), we evaluated expression and activation of caspase-3 within the ischemic brain of both mouse strains. Brain expression of procaspase-3 did not differ between PARG+/+110 and PARG−/−110 animals, and reduction of procaspase-3 levels (an index of caspase-3 activation) was similar in the ischemic cortex of both mouse strains (Figure 7A and Ba). Deletion of PARG110 did not affect basal expression of PARP-1. Consistent with analogous caspase-3 activation, PARP-1 cleavage (a hallmark of caspase-3 activity) was similar in the ischemic cortex of PARG+/+110 and PARG−/−110 mice. Because Akt phosphorylation promotes neuronal survival and ischemic preconditioning (Datta et al, 1999; Yano et al, 2001), we wondered whether different activation levels of Akt were present in the brain of PARG−/−110 mice. On the contrary, Akt phosphorylation in the controlateral and ischemic cortex did not differ between PARG+/+110 and PARG−/−110 mice. Heat-shock proteins (HSP) participate to ischemic neuroprotection (Weinstein et al, 2004) and are regulated by poly(ADP-ribosyl)ation (Tulin and Spradling, 2003; Zingarelli et al, 2004). Interestingly, HSP-70 expression levels were lower in the controlateral and ischemic cortex of PARG−/−110 mice compared with the corresponding one of wild-type littermates (Figures 7A and Bb). No differences in HSP-25 expression were found between mouse strains under control and ischemic conditions. Finally, considering the relevance of PAR to neuroinflammation (Ullrich et al, 2001; Chiarugi and Moskowitz, 2003; Park et al, 2004), and the pathogenetic role of the inflammatory response to ischemic brain injury (Barone and Feuerstain, 1999; Iadecola and Alexander, 2001), we next evaluated the expression of iNOS, IL-1β, and COX-2 in control and ischemic cortices of PARG+/+110 and PARG−/−110 mice. Importantly, basal and ischemia-induced COX-2 expression was higher in PARG−/−110 compared with PARG+/+110 animals (Figures 7A and Bc). Conversely, iNOS expression was not detected in control or ischemic brain of both animal strains, whereas IL-1β levels in controlateral and ischemic cortex was very weak and did not differ between strains.
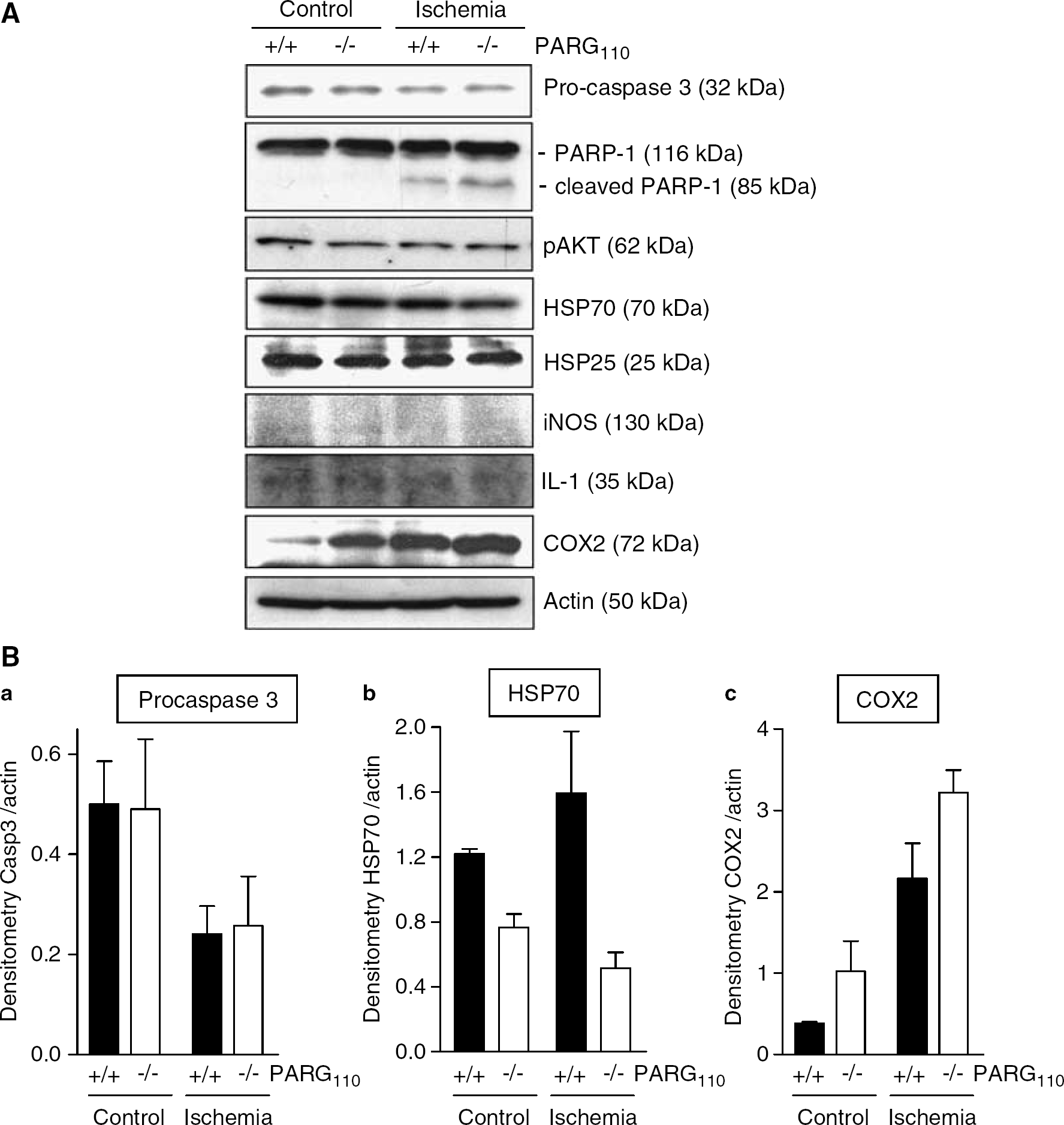
Expression of neuroprotective or neurotoxic proteins in control and ischemic brain cortex of poly(ADP-ribose) glycohydrolase (PARG)+/+110 and PARG−/−110 mice 14 h after middle cerebral artery occlusion (MCAO). (
Discussion
We report here that mice null for PARG110 have increased PAR content and increased brain vulnerability to ischemic stroke. These findings provide evidence that PARG activity protects from ischemic brain injury, and point to PAR catabolism as a key determinant of ischemic neurodegeneration.
In eukaryotic cells, several PARG isoforms originating from a single parg gene have been identified (Davidovich et al, 2001). In human cells, alternative splicing of PARG mRNA generates three variants: PARG111, PARG102, and PARG99 (Meyer-Ficca et al, 2004; Bonicalzi et al, 2005). In mouse cells, alternative transcription initiation of parg gene gives rise to PARG110 and PARG60 (see Figure 1A and Cortes et al, 2004 for further details). We show here that transcripts for both PARG110 and PARG60 are present in the brain of PARG+/+110 mice, whereas only transcripts for PARG60 are present in cells obtained from PARG−/−110 animals. These findings, along with evidence that ADP-ribose increases on PAR formation in PARG−/−110 brain tissue, suggest that transcripts for PARG60 are translated into a functional PARG in the brain of PARG−/−110 mice. Yet, whether PARG60 in PARG−/−110 mice maintains the same functional properties of PARG60 of wild-type animals requires further investigation. It also waits to be clarified why a ∼3-fold increase of PARG activity has been reported in the mitochondrial fraction of embryonic fibroblasts of PARG−/−110 mice, and PARP-1 auto-poly(ADP-ribosyl)ation levels are reduced in PARG−/−110 cells (Cortes et al, 2004). Together, these events could contribute to the phenotype of mice null for the 110 kDa isoform of PARG.
PAR accumulation and lethality at embryonic day 3.5 in mice with complete inactivation of the parg gene (Koh et al, 2004) indicate an essential role of PARG during mammalian development, and underscore the toxic effects of increased polymer contents. Conversely, PARG−/−110 mice have normal development, suggesting that, under basal condition, PARG110 activity is somehow redundant. Poly(ADP-ribose) glycohydrolase60 as well as other putative PAR degrading enzymes such as phosphodiesterases (Davidovich et al, 2001; Rouleau et al, 2004) might partly compensate for lack of PARG110. Deletion of PARG110, however, does not increase transcript levels for PARG60, suggesting that this type of compensation, if any, is not because of changes in PARG60 expression. However, the observation that basal PAR contents were higher in the brain of PARG−/−110 than wild-type mice implies that PARG110 participates in PAR degradation within the brain. Of note, 110 kDa PARG is the dominant PARG isoform normally present in the nucleus. Accordingly, we found that nuclear PAR contents were higher in the ischemic brain of PARG−/−110 than PARG+/+110 mice, suggesting that PARG110 is mainly responsible for nuclear PAR degradation on cerebral ischemia. Although nuclear localization of mouse PARG60 has not yet been shown, evidence for a residual PARG activity in the nuclear fraction of PARG−/−110 fibroblasts (Cortes et al, 2004) suggests that PARG60 migrates into the nucleus. This concept is further corroborated by the presence of multiple putative nuclear localization signals codified by sequences downstream to exon 3 in the human PARG gene, and by the notion that significant portions of the PARG sequence are well conserved from rodents to humans (Bonicalzi et al, 2005).
Cerebral PAR contents in PARG−/−110 mice under basal conditions were similar to those induced by ischemia in wild-type animals (Figure 4). Possibly, chronic exposure to high PAR contents in PARG−/−110 mice has lessened the cells' sensitivity to the detrimental effects of polymer accumulation. Accumulation of PAR in the nucleus of PARG−/−110 cortical neurons and their increased vulnerability after ischemia indicate that PARG110 is pivotal in maintaining PAR homeostasis in the injured brain. Because PARG-dependent PAR catabolism leads to ADP-ribose, we measured its content in the brain of PARG+/+110 and PARG−/−110 mice. Unexpectedly, we found that ADP-ribose did not differ between the two mouse strains. Considering that ADP-ribose may have a fast enzymatic degradation (Oei and Ziegler, 2000), it is possible that a simple measurement of its steady-state levels is not sufficient to evaluate the rate of its formation.
In keeping with the emerging role of PAR in transcriptional regulation (Ziegler and Oei, 2001; Chiarugi, 2002; Hassa and Hottiger, 2002; Kraus and Lis, 2003; Rouleau et al, 2004), we report here that polymer accumulation in the brain was associated with reduced HSP-70 and increased COX-2 expression. Notably, in line with the present finding that PAR accumulation correlates with decreased levels of HSP-70, the expression of this protein is facilitated in PARP-1−/– mice (Zingarelli et al, 2004). The augmented expression of COX-2 in the brain of PARG−/−110 mice is in agreement with previous finding showing that these animals are particularly sensitive to septic shock and have higher serum levels of proinflammatory mediators compared with wild-type counterparts after lipopolysaccharide (LPS) challenge (Cortes et al, 2004). Also, it has been recently reported that PARG inhibition causes COX-2 expression (Rapizzi et al, 2004). Notably, reduced expression of HSP-70 in the brain increases vulnerability to ischemic injury (Lee et al, 2001), whereas increased COX-2 activity worsens postischemic brain damage (Barone and Feuerstain, 1999; Iadecola and Alexander, 2001). It is possible therefore that changes in the expression of these two proteins contribute to the increased infarct volume found in PARG−/−110 mice after MCAO. The reduced PARP-1 automodification levels found in PARP-1−/– mice might have altered the functional properties of PARP-1 as a homeostatic transcriptional regulator, thereby causing transcriptional derangement and increased sensitivity to ischemic brain injury.
A large body of evidence suggests that PARP-1 hyperactivity and PAR formation play a role in postischemic brain damage by triggering NAD/ATP depletion and energy failure (Szabo and Dawson, 1998; Ha and Snyder, 2000; Chiarugi, 2002; Skaper, 2003). It has been reported, however, that ATP but not NAD contents are reduced in the ischemic brain (Paschen et al, 2000). Likewise, we report that procedures able to increase PAR formation in the brain tissue caused ATP but not NAD reduction. Selective ATP depletion may be because of ATP utilization in DNA repair processes and/or PARP-1 activity-dependent mitochondrial impairment (Alano et al, 2004; Hong et al, 2004; Cipriani et al, 2005). Furthermore, evidence that ATP depletion was similar in PARG+/+110 and PARG−/−110 mice suggests that impairment of PAR catabolism does not significantly affect the brain's energy dynamic during hyper-poly(ADP-ribosyl)ation. Under these conditions, it was unknown whether brain synthesizes ADP-ribose. The present finding that ADP-ribose rapidly accumulates in the brain tissue on PARP-1 activation, along with knowledge that it is a precursor of ATP (Oei and Ziegler, 2000), may be of pathophysiologic significance.
It has been reported that PARG inhibitors protect neural cells from oxidative stress, excitotoxicity and brain ischemia (Ying et al, 2001; Lu et al, 2003). Possibly, acute drug actions have a different outcome than congenital PARG suppression. For instance, pharmacological inhibition of PARG leads to increased PARP-1 auto-poly(ADP-ribosyl)ation while PARG−/−110 cells have a reduced PARP-1 automodification (Cortes et al, 2004). This may result in opposing effects on PARP-1 activity and on cell resistance to noxious stimuli. It is also worth noting that PARG inhibitors may block all PARG isoforms, whereas genetic inactivation of a single PARG isoform may have a less pronounced impact on polymer catabolism. It is also worth noting, however, that specificity as well as efficacy of current chemical inhibitors of PARG have been recently challenged (Falsig et al, 2004), thereby indicating that caution should be exercised when interpreting data obtained by pharmacologically targeting PARG activity. Finally, in contrast with our findings, two studies have recently reported the detrimental role of PARG110 in the gut (Cuzzocrea et al, 2005) and renal ischemia (Patel et al, 2005). Unfortunately, in these studies it has not been reported whether PAR accumulates in the gut or kidney after ischemia. In the light of the fact that inhibition of PAR neoformation protects from postischemic damage in the gut, kidney, and brain, at present we are unable to explain the apparent opposite role of PARG110 in peripheral and brain ischemia. Yet, our result are in keeping with the detrimental effects of parg targeting in Drosophila (Hanai et al, 2004) and mouse embryo (Koh et al, 2004).
In conclusion, our findings emphasize the relevance of PAR catabolism to protein expression profile within the brain and ischemic brain injury. Strategies aimed at preventing deregulation of PAR homeostasis might prove useful for therapeutic purposes.