
Abstract
The prolonged expression of the leucine zipper fos/jun immediate early genes (IEG) has been correlated with neuronal death after cerebral ischemia. In this study, the expression of six zinc finger IEG was examined using in situ hybridization in adult rats after middle cerebral artery occlusion (MCAO) with the suture model. NGFI-A, NGFI-B, NGFI-C, egr-2, egr-3, and Nurr1 mRNA were all induced throughout the ipsilateral cortex at 1 hour to 12 hours after MCAO. The cortical induction for most of the genes was greatest in the anterior cingulate and the anterior cerebral artery (ACA) and middle cerebral artery (MCA) transition zone. All of the zinc finger IEG were induced at 1 hour in all regions of hippocampus. NGFI-A and NGFI-B were induced in ipsilateral thalamus. Within areas of infarction, the basal IEG mRNA expression, and expression of the housekeeping gene cyclophilin A mRNA, decreased below control levels by 12 hours after the ischemia. Immediate early gene expression outside areas of infarction returned to control levels in most brain regions by 24 hours except for egr-3, which continued to be induced in the MCA/ACA transition zone for 24 hours, and NGFI-A, which continued to be expressed in specific regions of the thalamus for 72 hours. The induction of these IEG in the cortex is likely caused by ischemia-induced cortical spreading depression, with the hippocampal and thalamic IEG induction being caused by activation of efferent cortical pathways to these regions. The prominent induction of NGFI-B, NGFI-C, egr-2, and egr-3 in the anterior cingulate cortex, the ACA/MCA transition zone, and medial striatum could reflect the ischemic regions around MCA infarcts. The prolonged NGFI-A expression observed in thalamus in this study, and in CA1 of hippocampus after global ischemia in the gerbil in a previous study, suggests that the prolonged NGFI-A expression could be the result of or the cause of the delayed cell death. Prolonged NGFI-A expression, like c-fos and c-jun, seems to provide a marker for slowly dying neurons.
The fos/jun immediate early genes (IEG) are induced in brain by a variety of physiological and pathological stimuli. Transcription factors containing zinc finger motifs constitute a less well characterized immediate early protein family. The zinc finger domain is probably the most common DNA binding structure (Berg and Shi, 1996). Several protein structures have the capability to bind zinc, with more than 10 structurally diverse sub-classes of putative zinc binding domains having been reported. The zinc finger, originally characterized from the Xenopus transcription factor IIIA (Miller et al., 1985), is comprised of an antiparallel β-sheet followed by an α-helix stabilized by zinc ion, which is attached to two cysteine and two histidine residues (Berg et al., 1996; Gibson et al., 1988). The Cys2-His2 zinc finger present in the early growth response (egr-) family members NGFI-A (also termed krox-24, zif268, TIS8 and egr-1), NGFI-C (also termed egr-4 and pAT133), egr-2 (also termed krox-20), and egr-3 recognizes and activates transcription from the consensus sequence GC-GGGGGCG (G(S)G) (Madden and Rauscher, 1993; Berg and Shi, 1996). The zinc finger domain of the immediate early protein NGFI-B (also termed Nur77 and TIS1) and Nurr1 (also RNR-1, NOT and TINUR) differs from the classical zinc finger and is similar to the Cys2-Cys2 or Cys8 zinc finger found in the steroid/thyroid receptor superfamily (Berg and Shi, 1996). Like the members of the steroid/thyroid receptor superfamily, NGFI-B and Nurr1 also have a predicted ligand binding domain, but because the ligands are currently unknown, they are referred to as orphan receptors.
After sublethal stimuli, IEG induction is usually rapid and transient, returning to basal levels within a few hours (Honkaniemi et al., 1992; Honkaniemi et al., 1994; Honkaniemi et al., 1995). In contrast, after lethal injury the expression of some fos/jun family members continues for days in cells destined to die. For example, prolonged expression of some fos/jun family members occurs in the vulnerable CA1/CA3 regions of hippocampus in neurons destined to die after kainic acid-induced status epilepticus or cerebral ischemia (Popovici et al., 1990; Schreiber et al., 1993; Dragunow et al., 1993; Taniguchi et al., 1994; Smeyne et al., 1993). This neuronal death seems to be at least partially apoptotic as judged by the fragmentation of genomic DNA into internucleosomal pieces and the presence of typical apoptotic bodies in the dying neurons (Linnik et al., 1993; MacManus et al., 1993; Pollard et al., 1994; Filipkowski et al., 1994). Programmed cell death requires new protein synthesis and the cell death after seizures and ischemia can be decreased by the protein synthesis inhibitor cycloheximide (Linnik et al., 1993; Schreiber et al., 1993). The prolonged IEG expression after seizures and ischemia has been suggested to contribute to cell death by inducing transcription of genes that promote apoptosis.
The possible role of the zinc finger IEG in the cell death that occurs after global ischemia has also been examined (Honkaniemi and Sharp, 1996). After 5 minutes of global ischemia in gerbil, only NGFI-A was expressed for more than 24 hours hours in CA1 pyramidal neurons. These neurons die and show signs of apoptotic DNA fragmentation 2 to 3 days after the ischemia, suggesting that prolonged NGFI-A expression might be correlated with the apoptotic death of these neurons.
To further study the possible connection between ischemic cell death and prolonged zinc finger IEG expression, we examined the time course and distribution of NGFI-A, NGFI-B, NGFI-C, egr-2, egr-3, and Nurr1 expression after permanent middle cerebral artery (MCA) occlusions. After MCA infarctions, there is slow atrophy and cell death in thalamus and substantia nigra that may be apoptotic (Fujie et al., 1990; Iizuka et al., 1990; Ciricillo et al., 1994). We postulated that prolonged expression of one or several of the zinc finger IEG might occur in these regions where cells died slowly after the infarction. We also examined the expression of the housekeeping gene cyclophilin A to compare the ischemia-induced IEG changes with general transcriptional levels.
MATERIALS AND METHODS
Animal treatments
Focal ischemia was produced using the intravascular suture model of Longa et al. (1989). Animals were anesthetized with 3% isoflurane in 70% N2O/30% O2. The animals were intubated and the anesthesia maintained with mechanical ventilation using 1.5% isoflurane in a N2O/O2 mixture. The rectal temperature of the animals was monitored and the body temperature was maintained at 37 ± 0.5°C using a heating blanket. The right common carotid artery was exposed from a midline cervical incision and the occipital, superior thyroidal, maxillary, and lingual branches of the external carotid artery were coagulated. The pterygopalatine artery was ligated with a 4-0 silk suture. The external carotid artery was transected and a 3-0 nylon monofilament suture, with its tip rounded by heating, was inserted into the internal carotid artery through the external carotid artery stump and advanced 22 mm beyond the common carotid artery bifurcation. The suture was sewn in place, the incision wound was closed, and the animals were extubated. No seizure activity was observed during the 2- to 4-hour monitoring period after the operation. The animals were killed 1 hour (n = 4), 4 hours (n = 4), 12 hours (n = 4), 1 day (n = 5) or 3 days (n = 4) after the operation. The ischemic hemisphere appeared pale. Swelling of the ischemic hemisphere was observed in animals killed at 12 hours or later. The brains were rapidly removed and frozen in 2-methylbutane at −25°C.
In situ hybridization
Coronal sections (14-µm thick) were cut with a cryostat and mounted onto Fisherbrand Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA, U.S.A). At least four sections at the level of the striatum and the hippocampus were hybridized with each oligonucleotide probe for each subject. The oligonucleotide probes were directed against rat NGFI-A (bases 355 to 399, Genbank accession number M18416; Milbrandt, 1987), mouse NGFI-B (bases 181 to 225, accession number J04113; Hazel et al., 1988), rat NGFI-C (bases 417 to 460, accession number M65008; Crosby et al., 1991), mouse egr-2 (bases 1597 to 1641, accession number X06746; Chavrier et al., 1988), mouse egr-3 (bases 188 to 232, accession number S40835; Patward-han et al., 1991), and mouse Nurr1 (bases 733 to 777, accession number S53744; Law et al., 1992). Searches of the Genbank database revealed no significant homology of these oligonucleotide sequences with any other previously characterized gene. These oligonucleotides detect single bands of appropriate size during Northern blot testing (Honkaniemi et al., 1995). The oligonucleotides were labeled with 35S-dATP (DuPont-NEN Research Products, Boston, MA, U.S.A.) using terminal deoxynucleotidyltransferase (Gibco BRL). The sections were air dried at room temperature and hybridized at 42°C for 12 to 18 hours with a mixture of 4 × SSC, 50% formamide, 1 × Denhardt's solution, 1% sarcosyl, 0.02 mol/L phosphate buffer (pH 7.0), 10% dextran sulfate, 500 µg/mL heat-denatured salmon sperm DNA, 200 mmol/L dithiothreitol, and 1 × 107 cpm/mL of the labeled probe. After hybridization, the sections were washed 4 times for 15 minutes each in 1 × SSC at 55°C and thereafter cooled to room temperature for 1 hour. The sections were dipped in distilled water and subsequently, in 75% and 90% ethanol and were then air dried at room temperature. The sections were covered with Kodak SB5 film (Eastman Kodak, Rochester, NY, U.S.A.) and exposed for 1 to 2 weeks. The films were developed with Kodak GBX developer and fixer. Some sections were dipped in Kodak NTB2 emulsion, dried, and developed 2 to 4 months later.
RESULTS
Controls
The expression of the zinc finger IEG in untreated, control rat brain (Figs. 1A and 1B) was similar to that described in our previous studies (Honkaniemi et al., 1995). NGFI-A, NGFI-B, NFI-C, and egr-3 were expressed throughout the cortex, layers CA1 to CA3 and dentate subfields of the hippocampus, and in the caudate-putamen. In contrast, Nurr1 expression was limited to medial habenula, layers V and VI of the cortex, and CA 1 to CA3 subfields of the hippocampus. Egr-2 was nearly undetectable in control brain, possibly being expressed slightly above background in the cortex.
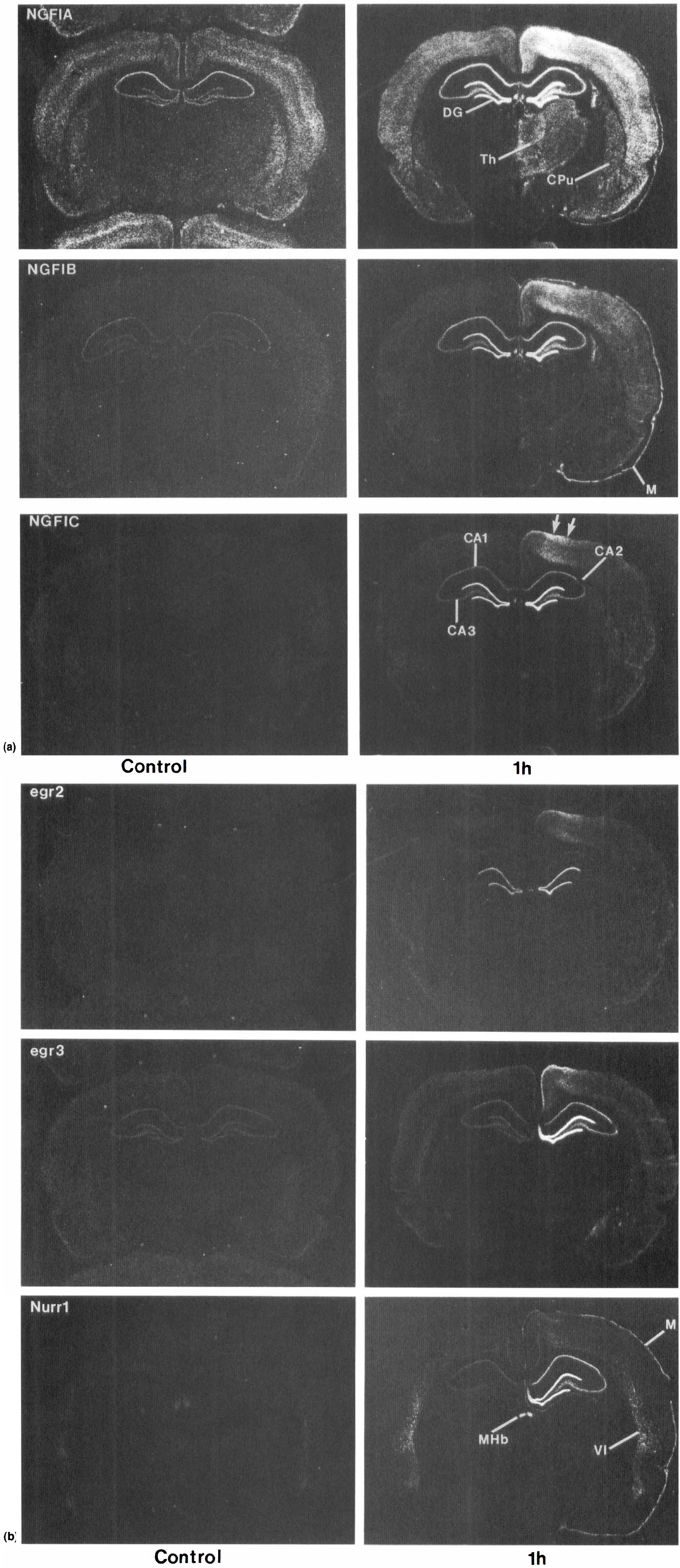
Coronal sections at the level of the hippocampus showing the expression of NGFI-A, NGFI-B, NGFI-C (A), egr-2, egr-3, and Nurr (B) zinc finger immediate early gene mRNA in the brains of control adult rats and 1 hour after permanent middle cerebral artery occlusions (MCAO) produced using the suture model. Note induction of all genes in the hippocampus and neocortex. DG, dentate gyrus; CA1, CA1 pyramidal layer of hippocampus; CA2, CA2 pyramidal layer of hippocampus; CA3, CA3 pyramidal layer of hippocampus; Th, thalamus; CPu, caudate putamen; M, meninges; Mhb, medial habenula. VI, layer 6 of neocortex.
Areas of infarction
Within 1 hour after ischemia, the genes were induced in many brain regions (Figs. 1 and 2; Table 1). In general, the rat focal ischemia model used produced a reproducible IEG induction with relatively little variation (Table 2). The major variations involved unilateral or bilateral IEG induction in the hippocampus and thalamus. Furthermore, there was some variation whether the areas of infarction involved either the entire striatum (see NGFI-A at 1 hour in Fig. 2A) or just the lateral striatum (see NGFI-B at 1 hour in Fig. 2A). The cortical infarction included dorsolateral and lateral cortex at the level of the striatum (Figs. 2A and 2B), with little cortical infarction at the level of the hippocampus (Figs. 1A and 1B). In the areas of striatal and cortical infarction, there was either no mRNA expression (NGFI-C, egr-2, egr-3, Figs. 2A and 2B), or a decrease in the mRNA expression below control levels (NGFI-A, NGFI-B, Figs. 2A, 2B) by 12 hours after the ischemia.
Summary of the IEG induction 1 hour after MCAO

MCAO, middle cerebral artery occlusion. +++, high induction; ++, moderate induction; +, weak induction; —, no induction.
Summary of the different patterns of the IEG induction 1 to 12 hours after MCAO
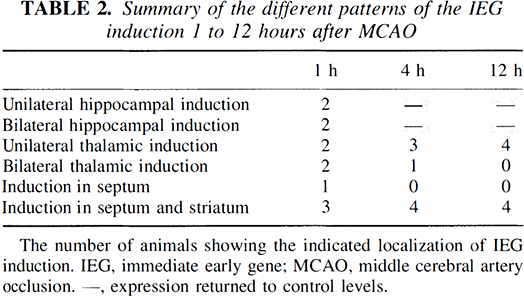
The number of animals showing the indicated localization of IEG induction. IEG, immediate early gene; MCAO, middle cerebral artery occlusion. —, expression returned to control levels.
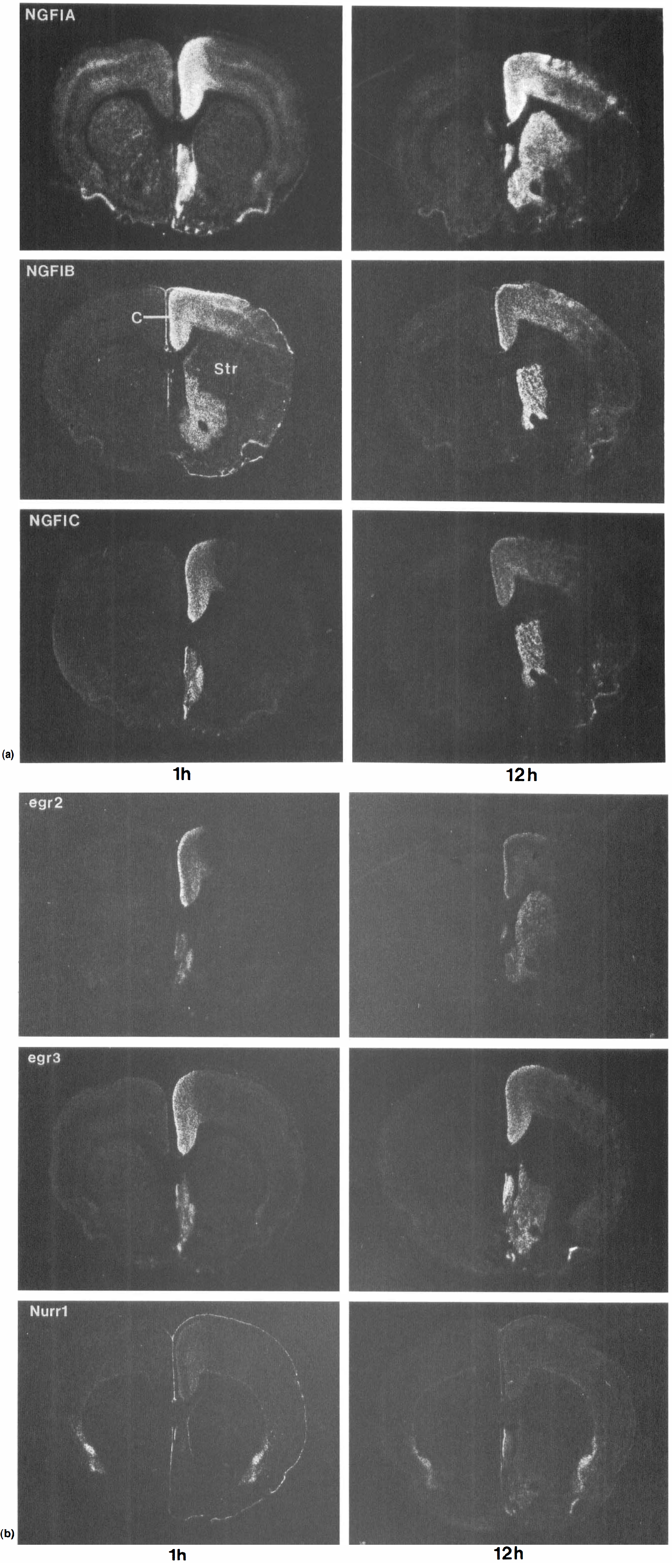
Coronal sections of adult rat brains at the level of the striatum showing the expression of NGFI-A, NGFI-B, NGFI-C (A), egr-2, egr-3, and Nurr1 (B) mRNA at 1 hour and 12 hours after MCAO. Note the induction of all the genes in cortex, medial striatum, and septum. Str, striatum.
Cortex
NGFI-A, NGFI-B, NGFI-C Figs. 1A and 2A) and to lesser extent egr-2, egr-3, and Nurr1 (Figs. 1B, 2B) were induced in cortex ipsilateral to the MCA occlusion. NGFI-A (Fig. 1A) and to some extent egr-2 and Nurr1 (Fig. 1B) were also induced in the contralateral cortex. The ipsilateral and contralateral cortical induction peaked at 1 hour (Figs. 1A, 1B, 2A, and 2B), remained the same or declined somewhat by 12 hours (Figs. 2A and 2B), returned to baseline by 24 h for most genes (not shown) and even decreased below baseline for some genes at 3 d (Fig. 4). Cortical induction was greatest in the anterior cingulate cortex for NGFI-A, NGFI-B, NGFI-C, egr-2, egr-3, and to lesser extent for Nurr1 (Figs. 2A and 2B). There was less induction in posterior cingulate cortex (Figs. 1A and 1B) except for NGFI-A mRNA, which was expressed at high levels in anterior (Fig. 2A) and posterior cingulate (Fig. 1A). mRNA for all of the genes were induced to some degree in piriform cortex, though the induction was the greatest for NGFI-A (Fig. 1A).
Induction of several mRNA was particularly prominent at the MCA/anterior cerebral artery (ACA) transition zone at the level of the hippocampus (Fig. 1A, arrows at 1 hour for NGFI-C; also see NGFI-B, 1 hour; egr-2 and egr-3 at 1 hour in Fig. 1B). This IEG induction was most prominent at 1 hour, declined by 4 hours, and reached control levels by 12 to 24 hours for all of the genes except for egr-3. Egr-3 continued to be expressed in the ACA/MCA transition zone for 24 hours (two arrows in Fig. 3).
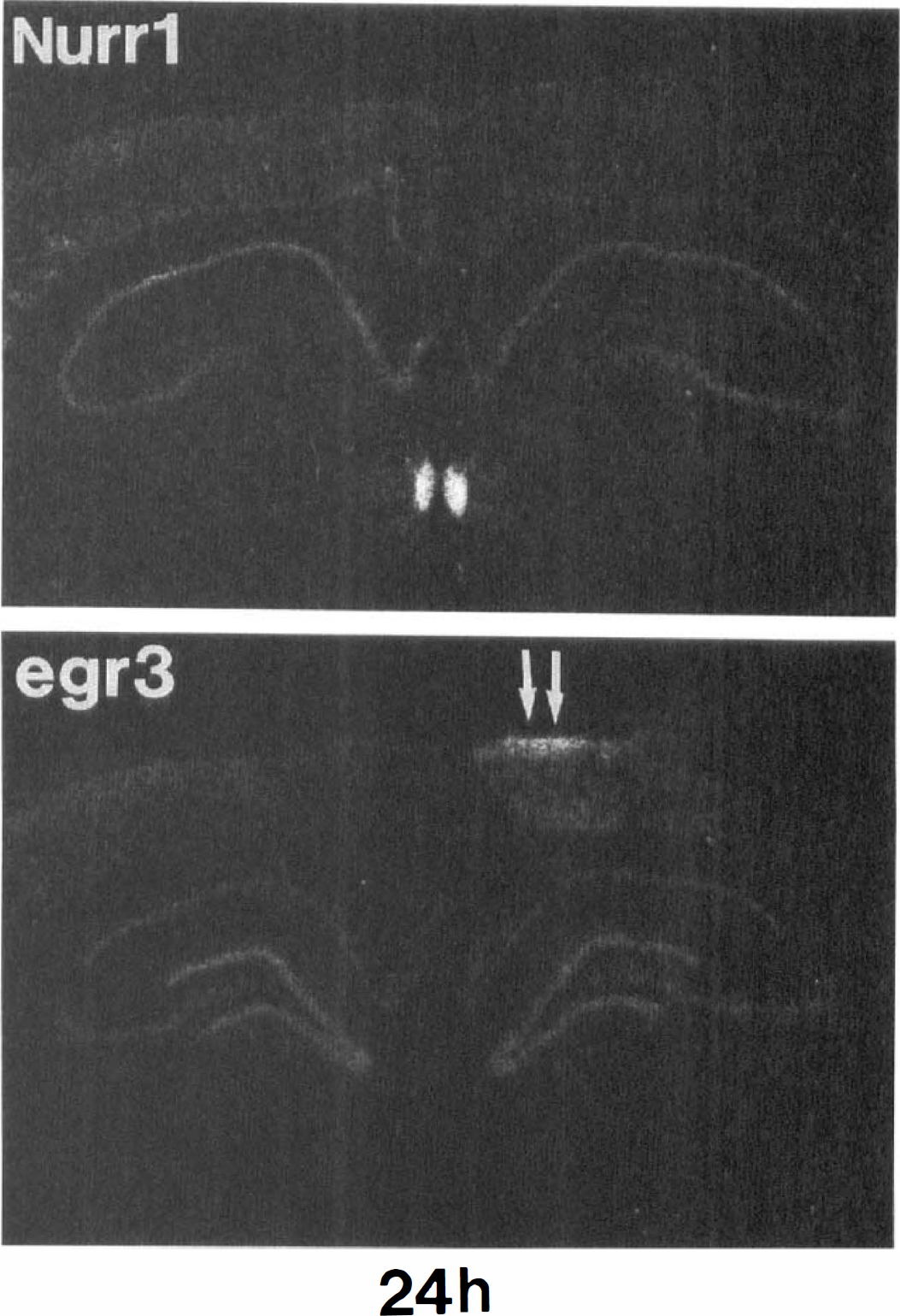
The expression of Nurr1 and egr-3 mRNA in adult rat brain 24 hours after MCAO. Arrows point at the middle cerebral artery and anterior cerebral artery boundary zone showing prolonged expression of egr-3, but not Nurr1.
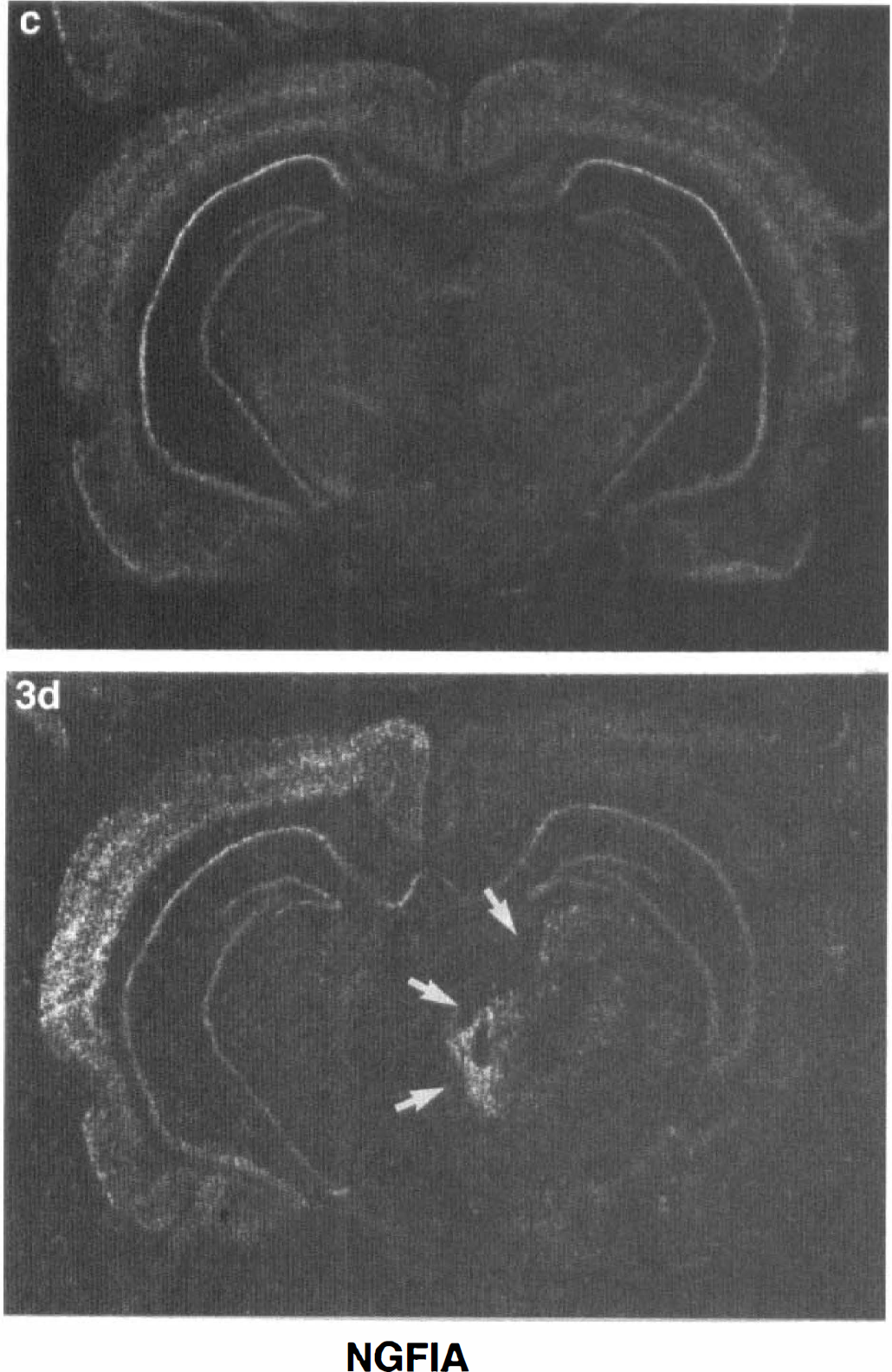
The expression of NGFI-A mRNA in the brains of a control adult rat (
Hippocampus
At 1 hour after the permanent MCA occlusions, all of the zinc finger genes examined were maximally induced in hippocampus (Figs. 1A and 1B). The greatest induction for each of the IEG occurred in the dentate gyrus, most likely in the dentate granule cell neurons (Figs. 1A and 1B). NGFI-A, NGFI-B, NGFI-C (Fig. 1A), egr-3, and Nurr1 Fig. 1B) were also induced in CA1 and CA3 pyramidal neurons. The induction of the genes in CA1 and CA3 neurons was confirmed by cellular in situ hybridization for each of the genes on emulsion dipped in situ hybridization slides (not shown). Egr-2 was markedly induced in dentate and slightly induced in CA1 and CA3 pyramidal neurons at 1 hour after the ischemia (Fig. 1B).
There was bilateral activation of the IEG in hippocampus for two of the animals at 1 hour after the MCA occlusions (see NGFI-A, NGFI-B, NGFI-C in Fig. 1A; and egr-2 in Fig. 1B; Table 2). When there was bilateral hippocampal activation for one of the genes, there was bilateral activation for all of the genes. In Fig. 1, the 1 hour sections for NGFI-A and NGFI-B are taken from the same animal, and the 1 hour sections for NGFI-C and egr-2 are taken from the same animal.
In two other animals at 1 hour after the MCA occlusions, there was unilateral activation of the genes in hippocampus, ipsilateral to the MCA occlusion (see egr-3 and Nurr1 in Fig. 1B). In Fig. 1 the 1 hour sections for egr-3 and Nurr 1 were taken from two different animals. When there was unilateral activation in hippocampus for one of the genes, there was unilateral activation for all of the genes. Whether unilateral or bilateral, the dentate induction returned to control levels by 4 hours. The CA1 to CA3 induction returned to control levels by 4 hours.
Thalamus
NGFI-A and to lesser extent NGFI-B were induced in the thalamus ipsilateral to the MCA occlusion (Fig. 1A). In four animals, the IEG induction also occurred in the contralateral thalamus (Table 2). The mRNA were maximal at 1 hours and involved all the thalamic nuclei. This induction persisted for 4 hours and reached baseline in most regions of thalamus by 24 hours (not shown). However, at 24 and 72 hours (Fig. 4) after MCA occlusions, NGFI-A expression was persistently elevated in specific thalamic nuclei, including the parafasicular nucleus (Fig. 4, lower two arrows).
Striatum, septum, meninges
All of the genes were also induced in the ipsilateral caudate-putamen (Figs. 1 and 2). Striatal induction occurred mainly in medial, noninfarcted striatum. In one animal, the striatal induction involved just the septum (Table 2). Nurr1 was induced in the striatum at midstriatal levels, but not in the midhippocampal levels unlike the other IEG. The zinc finger mRNA were detectable in striatum by 1 hour but appeared to peak for many of the genes at 12 hours (Figs. 2A and 2B). They declined to baseline levels by 24 hours (not shown). All of the genes were also induced in the septum (Figs. 2A and 2B). NGFI-A, NGFI-B, and Nurr1 (Figs. 1 and 2) were notably induced in the meninges 1 hour after ipsilateral MCA occlusion. The gene induction in the meninges markedly declined by 12 hours in all animals.
Cyclophilin
The expression of the housekeeping gene cyclophilin was examined in order to compare the IEG induction with general levels of transcription. As expected, cyclophilin A was not induced any time after ischemia. In the core of the infarct, the expression of cyclophilin A mRNA decreased below control values. A slight decrease was seen by 4 hours in the striatum and by 12 hours the cyclophilin mRNA levels were well below control levels in the striatum and in the ipsilateral cortex in the MCA distribution (not shown).
DISCUSSION
Zinc finger immediate early gene induction in cortex–comparison to fos
The cortical zinc finger IEG induction occurred throughout the entire cortex outside the infarction. The ipsilateral NGFI-A induction in the cortex has been shown previously (Kinouchi et al., 1994a). The cortical induction throughout the hemisphere is quite similar to the ipsilateral induction of c-fos and junB after MCA occlusion (Kinouchi et al., 1994b). The unifying mechanism of this whole hemispheric IEG induction is most likely cortical spreading depression produced by middle cerebral artery occlusion (MCAO), (Rother et al., 1996; Kinouchi et al., 1994b). This is supported by studies showing that prior administration of MK801, which blocks cortical spreading depression, also blocks the diffuse fos induction in cortex after MCA occlusions (Kinouchi et al., 1994c; Gass et al., 1992; Collaco-Moraes et al., 1994).
However, the highest regions of IEG induction were localized in the ACA/MCA borderzone in the parietal cortex, anterior cingulate cortex, medial striatum, nucleus accumbens, and septum. This induction is not likely due to spreading depression. Instead, it is likely due to ischemia in the distribution of the anterior and middle cerebral arteries–in areas where blood flow decreased but infarctions did not occur after the intravascular suture occlusions. Fos/Jun expression is similarly high in regions around a cortical injury (Sharp et al., 1990) and at the borders of cortical infarcts (Gass et al., 1992). It seems likely that these regions are affected by the ischemia, but the impact is not severe enough to cause the immediate death of cells. Instead, the cells, being able to transcribe, respond to the ischemia by synthesizing novel mRNA including IEG and heat shock genes (States et al., 1996). However, it is unclear whether the IEG induction contributes to cell death or is part of the cellular survival response.
Possible mechanisms of hippocampal induction
A notable finding after MCA infarctions was the induction of all the IEG in all sectors of the hippocampus, especially in the dentate gyrus. Similar zinc finger IEG induction has been noted in hippocampal neurons after penetrating brain injury (Honkaniemi et al., 1995) and global ischemia (Honkaniemi and Sharp, 1996) and it is comparable to that previously described for the fos/jun family members (Taniguchi et al., 1994; Jorgensen et al., 1989; Kindy et al., 1991; Dragunow et al., 1993; Kinouchi et al., 1994b). The mechanisms of the hippocampal IEG induction are unknown. The acute induction could be caused by trans-synaptic activation of the hippocampus (Sloviter and Lowenstein, 1992) from the entorhinal cortex. It is possible that ischemia-induced spreading depression depolarizes entorhinal cortical neurons that project to hippocampus, and this activates hippocampal neurons and induces the IEG. Similar cortical spreading depression and activation of cortical afferents to thalamus could account for acute induction of NGFI-A and NGFI-B (current study; Kinouchi et al., 1994a), as well as similar induction of c-fos and junB (Kinouchi et al., 1994 b, 1994c ), throughout the entire ipsilateral thalamus.
Hippocampal ischemia might provide an alternative mechanism of zinc finger lEG induction. In a previous study, half of the animals examined had both HSP70 induction and evidence of DNA fragmentation in CA1 neurons after permanent MCAO (States et al., 1996). Histological evidence of dying CA1 neurons after MCA occlusions has also been detected (Czurko and Nishino, 1993; Benyo et al., 1995; Zhu and Auer, 1995). Though unilateral, permanent MCA occlusion is not known to decrease hippocampal blood flow, the combination of MCA occlusion and increasing intracranial pressure markedly decreases hippocampal blood flow and energy charge (Ginsberg et al., 1977). Though it seems unlikely that intracranial pressure increases within 1 hour after MCA occlusion–which could then decrease hippocampal blood flow–a systematic study of hippocampal blood flow or intracranial pressure has not been performed using the suture stroke model.
Relationship of zinc finger and other immediate early genes to apoptosis
A number of IEG are induced during apoptosis. C-fos, c-jun and NGFI-A are induced in sympathetic neurons that undergo apoptosis after NGF deprivation (Estus et al., 1994; Ham et al, 1995). NGFI-B is induced in T-cell hybridomas and thymocytes undergoing apoptosis (Liu et al., 1994; Woronick et al, 1994) and Nurr1 is expressed in apoptotic T-cells (Okabe et al., 1995). Sympathetic neurons overexpressing c-jun undergo apoptosis (Estus et al, 1994; Ham et al., 1995) and fibroblasts overexpressing c-fos die via apoptosis after serum deprivation (Smeyne et al., 1993).
Apoptotosis can be inhibited by attenuating the effects of IEG. C-jun-dominant, negative mutant and neutralizing antibodies against c-Jun and the Fos family members rescue sympathetic neurons from NGF deprivation (Estus et al., 1994; Ham et al., 1995). NGFI-B-dominant, negative mutant and NGFI-B antisense treatment inhibit apoptosis in T-cells (Liu et al., 1994; Woronick et al., 1994). In vivo, continuous prolonged expression of c-fos (Popovici et al., 1990; Schreiber et al., 1993; Taniguchi et al., 1994), c-fos promoter driven LacZ (Smeyne et al., 1993), c-jun (Dragunow et al, 1993), and NGFI-A (Honkaniemi and Sharp, 1996) precedes neuronal death after kainic-acid induced status epilepticus and hypoxic-ischemic brain injury. This data has suggested that the prolonged IEG expression might contribute to the cell death. However, there are no studies to show that blocking the prolonged IEG expression in vivo (e.g. using antisense) attenuates the cell death. Furthermore, the brains of mice bearing null mutations of NGFI-B (Lee et al, 1995) develop normally without obvious abnormalities. Apoptosis does not seem to differ between NGFI-B knock-outs (Lee et al, 1995) or c-fos/c-jun double knock-outs (Roffler-Tarlov et al, 1996) compared with wild-type animals. Though NGFI-B is highly expressed in the lymphoid system, the spleen and the thymus of animals with a NGFI-B null-mutation develop normally, and thymic and peripheral T-cell death rate is not affected by this mutation (Lee et al., 1995). The apoptotic death of the motoneurons in the spinal cord after sciatic nerve transection is not influenced by the absence of c-fos (Roffler-Tarlov et al., 1996). Based on these data, it is unclear whether any of the IEG actively mediate apoptotic neuronal death in vivo.
Significance of persistent zinc finger immediate early gene expression in cerebral ischemia
After a nonlethal stimulus, the expression of the zinc finger IEG generally declines to control levels by 6 hours (Honkaniemi et al., 1994). In the present study, prolonged IEG expression lasting for 12 hours was observed in the cingulate cortex and septal nuclei, regions that are outside the MCA territory and are not known to sustain injury with the suture model. We have performed DNA nick-end labeling (TUNEL) from the sections used in the present study. TUNEL-stained cells were not detected in the cingulate cortex or in the septum after permanent MCA occlusions (States et al., 1996). These findings of prolonged IEG expression in the cingulate cortex and septum, which survive the insult, do not support the role of prolonged IEG expression in mediating neuronal death in these regions.
Only egr-3 and NGFI-A showed prolonged expression lasting 1 and 3 days, respectively. The egr-3 expression was localized in the ACA/MCA transition zone. The fate of cells in this region is uncertain (States et al., 1996), and hence it is difficult to correlate egr-3 expression with cell death or survival.
NGFI-A continued to be expressed for more than 72 hours in specific regions of the thalamus. Thalamus is known to atrophy which is probably caused by slow neuronal degeneration after permanent MCAO (Fujie et al., 1990; Iizuka et al., 1990). Interestingly, c-jun mRNA is also expressed for prolonged periods of time in specific nuclei of the thalamus after MCAO (Soriano et al., 1996). NGFI-A was also the only IEG showing prolonged expression in CA1 hippocampal neurons destined to die after 5 minutes of global ischemia in the gerbil (Honkaniemi and Sharp, 1996). These results suggest that prolonged NGFI-A expression correlates with slow neuronal degeneration after ischemic injury. This prolonged NGFI-A expression could be a result of the cells dying, or could be contributing to the cell death by inducing apoptotic target genes. The present experiments suggest that prolonged NGFI-A expression could prove to be a marker of cells that are dying slowly.
The expression of glial fibrillary acidic protein is also induced in thalamus after MCAO (Soriano et al., 1996). Though the same study found the c-jun immunoreactivity induced in the thalamus after MCAO occurred mainly in apoptotic, TUNEL-stained neurons, it is possible that the prolonged c-jun and NGFI-A expression in thalamus is localized, in part, to reactive glia. It is known that the expression of multiple IEG can be stimulated in vitro in both glia and neurons by serum and growth factors (Mill-brandt, 1987; Wu et al., 1989; Mohn et al., 1991; Hisanaga et al., 1992, 1993).
Taken together, these results expand the IEG induced by ischemia from the widely studied Fos/Jun family members to the less well-characterized zinc finger IEG and thus widen the spectrum of target genes involved. However, whether the zinc finger IEG contribute or protect from ischemic injury remains to be established.