
Abstract
Autophagy is a homeostatic process for recycling of proteins and organelles, induced by nutrient deprivation and regulated by oxygen radicals. Whether autophagy is induced after traumatic brain injury (TBI) is not established. We show that TBI in mice results in increased ultrastructural and biochemical evidence of autophagy. Specifically, autophagosomal vacuoles and secondary lysosomes were frequently observed in cell processes and axons in ipsilateral brain regions by electron microscopy, and lipidated microtubule-associated protein light chain 3, a biochemical footprint of autophagy referred to as LC3 II, was increased at 2 and 24 h after TBI versus controls. Since oxygen radicals are believed to be important in the pathogenesis of TBI and are essential for the process of starvation-induced autophagy in vitro, we also sought to determine if treatment with the antioxidant γ-glutamylcysteinyl ethyl ester (GCEE) reduced autophagy and influenced neurologic outcome after TBI in mice. Treatment with GCEE reduced oxidative stress and partially reduced LC3 II formation in injured brain at 24 h after TBI versus vehicle. Treatment with GCEE also led to partial improvement in behavioral and histologic outcome versus vehicle. Taken together, these data show that autophagy occurs after experimental TBI, and that oxidative stress contributes to overall neuropathology, in part by initiating or influencing autophagy.
Introduction
Autophagy has long been recognized as an important physiologic adaptation to starvation (Neely and Mortimore, 1974; Seglen and Gordon, 1982). Classic, starvation-induced autophagy is initiated by either nutrient and amino-acid deprivation or by administration of glucagon and cAMP. Phosphorylation of preautophagosomal membranes, conjugation of Atg12-Atg5 and Atg8/microtubule-associated protein light chain 3 (LC3 I) complexes, and development of mature autophagosomal vacuoles are regulated by class III phosphatidylinositol 3-kinase and the Bcl-2 family member Beclin-1 (Zeng et al, 2006). Modification of LC3 I (the mammalian homologue of yeast Atg8) with covalent attachment of phosphatidylethanolamine results in LC3 II, a process essential for macroautophagy referred to as an ‘LC3 shift’ or ‘LC3 lipidation’ (Kouno et al, 2005). The autophagosomes then engulf regions of cytoplasm containing long-lived proteins or organelles such as mitochondria—‘mitophagy.’
Recently, with the development of Atg transgenic mice and other molecular tools, a role for autophagy has been uncovered in nonstarvation-related central nervous system disease (Hara et al, 2006; Koike et al, 2005; Komatsu et al, 2006; Mizushima and Hara, 2006; Wang et al, 2006) and in normal development (Levine and Klionsky, 2004; Yue et al, 2003). Disrupted autophagy has been implicated in amyloidopathies related to toxic accumulation of proteins (Larsen and Sulzer, 2002), and transgenic mice lacking the Atg7 gene, essential for autophagy, show neurodegeneration (Komatsu et al, 2006). Control of autophagy may differ in nonstarvation conditions, involving kinases other than phosphatidylinositol 3-kinase during oxidative neuronal injuries (Zhu et al, 2007). Recently, reactive oxygen species were found to be essential for starvation-induced autophagy to proceed (Scherz-Shouval et al, 2007). Reactive oxygen species specifically regulate the activity of Atg4, the cysteine protease responsible for the cleavage of the C terminus of LC3 I (Atg8), a reaction essential for its lipidation during the formation of autophagosomes in vitro (Tanida et al, 2004).
Whether oxygen radicals are essential for autophagy after acute brain injury is not known; however, oxidative stress is believed to be important in the pathogenesis of both ischemic and traumatic brain injury (TBI) (Hamm et al, 1996; Mikawa et al, 1996). Hence, we sought to determine whether autophagy occurred after TBI in mice in vivo, and if treatment with the antioxidant γ-glutamylcysteinyl ethyl ester (GCEE) reduced autophagy and influenced neurologic outcome after TBI in mice.
Materials and methods
Mouse Traumatic Brain Injury Model
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. Mice had free access to food and water before and after surgery. The controlled cortical impact (CCI) model for mice was used as described previously (Satchell et al, 2003). Adult male mice (12 weeks of age, 25 to 35 g) were anesthetized with 2% isoflurane (Anaquest, Memphis, TN, USA), N2O and O2 (2:1) using a nose cone. The mouse was positioned in a stereotaxic frame and a brain temperature probe (0.009 in; Physitemp Corp., Clifton, NJ, USA) was inserted into the left frontal cortex through a burr hole. A bone flap was made on the parietal cortex using a dental drill, and mice were subjected to CCI using a pneumatic cylinder with a 5 cm stroke using a vertically driven 3-mm metal tip at a velocity of 6 m/sec and a depth of penetration of 1.2 mm. After CCI or sham surgery, the bone flap was replaced and the scalp incision was sutured. Anesthesia was discontinued, and mice were allowed to awaken and then were returned to their cages.
Mouse Study Protocols
For ultrastructural analysis of injured brain for autophagy, naive mice and mice at 2 and 24 h after CCI (n = 3 per group) were killed by perfusion fixation with 2% paraformaldehyde/0.01% glutaraldehyde in phosphate-buffered saline. For the evaluation of LC3 after CCI, naive mice and mice at 2, 24, and 48 h after CCI (n = 4 per group) were killed by perfusion with ice-cold saline and Western blot analysis was performed. For the evaluation of the acute effects of GCEE, sham vehicle, and vehicle and GCEE-treated mice 24 h after CCI were used for determination of total antioxidant reserves using a chemiluminescence assay (n = 4 per group) or for LC3 II formation using Western blot analysis (n = 7 per group). For the evaluation of the effects of GCEE on neurologic outcome, sham vehicle, sham GCEE treated, CCI vehicle, and CCI GCEE-treated mice (n = 15 per group) underwent Morris-water maze testing on days 14 to 20, and then were killed by perfusion fixation with 2% paraformaldehyde for histologic analysis.
Treatment with γ-Glutamylcysteinyl Ethyl Ester
γ-Glutamylcysteinyl ethyl ester is an antioxidant that can serve as a cysteine donor for replenishment of reduced glutathione (GSH). Mice in the treatment protocols were randomized to receive a single injection of 150 mg/kg of GCEE (Bachem Bioscience Inc., King of Prussia, PA, USA) diluted in 100 μL of normal saline or an equal volume of saline vehicle intraperitoneally 10 mins after CCI. This dose was based on the report by Drake et al (2003) showing that GCEE can preserve mitochondrial GSH levels under conditions of nitrosative stress. This dose is comparable to the human dose of the related compound N-acetylcysteine when used for the treatment of acetaminophen overdose.
Transmission Electron Microscopy
Regions of interest including the ipsilateral cortex and cortical white matter, dorsal hippocampus, and thalamus, and the contralateral cortex and cortical white matter were grossly dissected under a microscope into ~1-mm tissue blocks, and then fixed in cold electron microscope (EM) grade 2.5% glutaraldehyde in 0.1 mol/L phosphate-buffered saline (pH 7.3), rinsed in phosphate-buffered saline, postfixed in 1% OsO4 with 0.1% K3Fe(CN)6, dehydrated through a graded series of EtOH, and embedded in Epon (dodecenyl succinic anhydride, nadic methyl anhydride, Scipoxy 812 Resin, and dimethylaminomethyl; Energy Beam Sciences, East Granby, CT, USA). Ultrathin sections (65 nm) were cut and then stained with 2% uranyl acetate and Reynold's lead citrate and examined on a Jeol 1011 transmission EM. Sections were qualitatively analyzed for the presence of autophagosomal vacuoles by an observer unaware of experimental condition (MS).
Western Blot Analysis
To identify biochemical changes consistent with autophagy in injured brain, a 3-mm coronal section encompassing the contusion was obtained as described (Satchell et al, 2003) and shown in Figure 3C. The ipsilateral parietal cortex including injury site, dorsal hippocampus, and dorsal thalamus was homogenized in 10 vol. lysis buffer (20 mmol/L Hepes (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)-KOH, 10 mmol/L NaCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA (ethylene glycol bis(β-aminoethylether)-N,N,N',N',-tetraacetic acid), 250 mmol/ L sucrose, 1 μmol/L dithiothreitol, 1 mmol/L phenylmethylsulfonyl fluoride, 2 μg/mL aprotinin). Cellular proteins were subfractionated to isolate organelles including autophagosomes using differential centrifugation as described (Clark et al, 2007). Briefly, lysates were centrifuged at 1,025g for 15 mins at 4°C, with the pellets containing nuclei (P1). The S1 fractions were centrifuged at 735g at 4°C for 10 mins, the resulting supernatants further centrifuged at 10,000g at 4°C for 15 mins to pellet autophagosomes, mitochondria, and small organelles (P2). Samples were stored at −80°C in 10% glycerol. Protein concentration was determined using a Bradford-based protein assay.
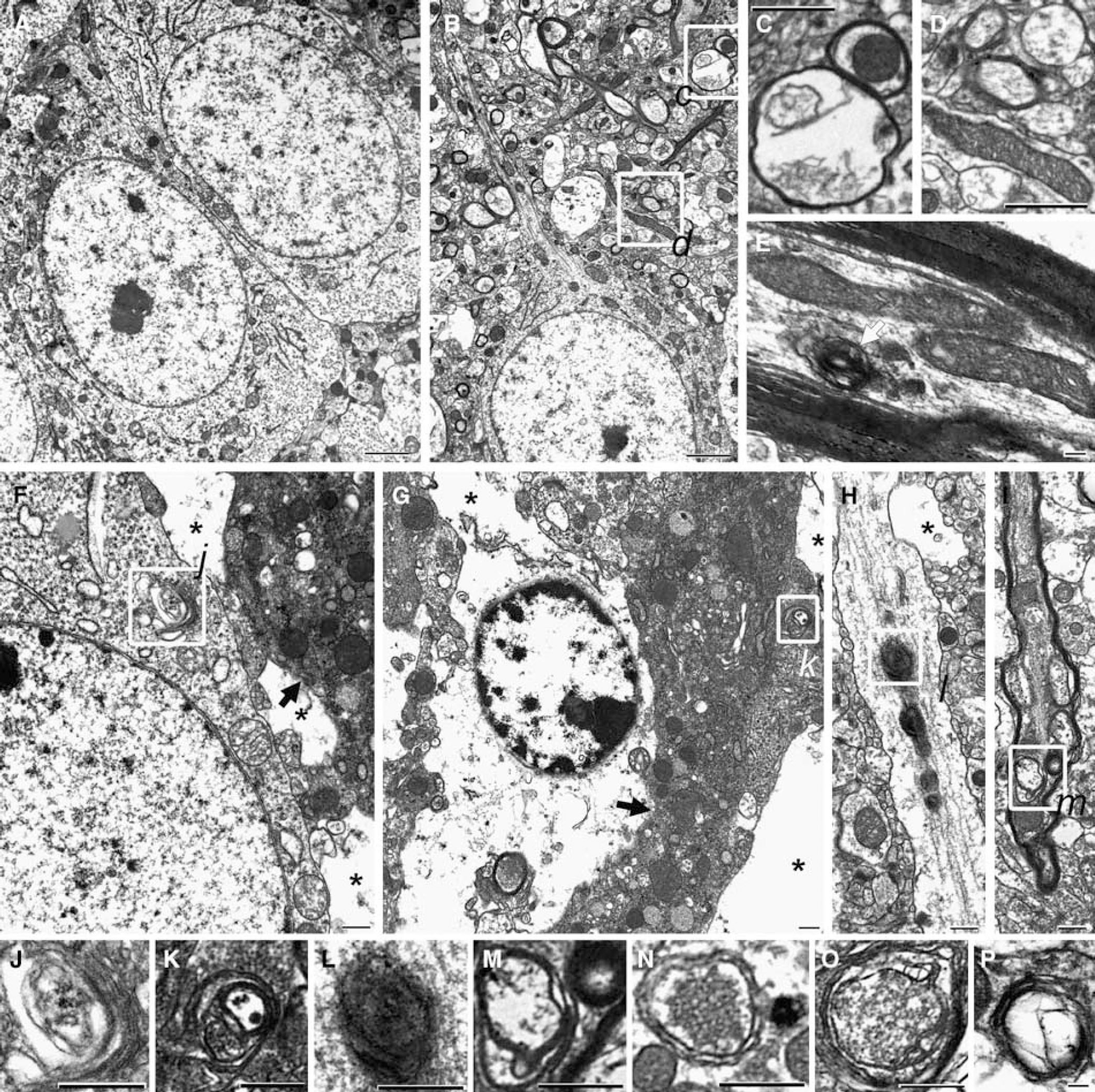
Ultrastructural evidence of autophagy 2 h after controlled cortical impact (CCI) injury in adult male mice. Transmission electron micrographs from naive mice (
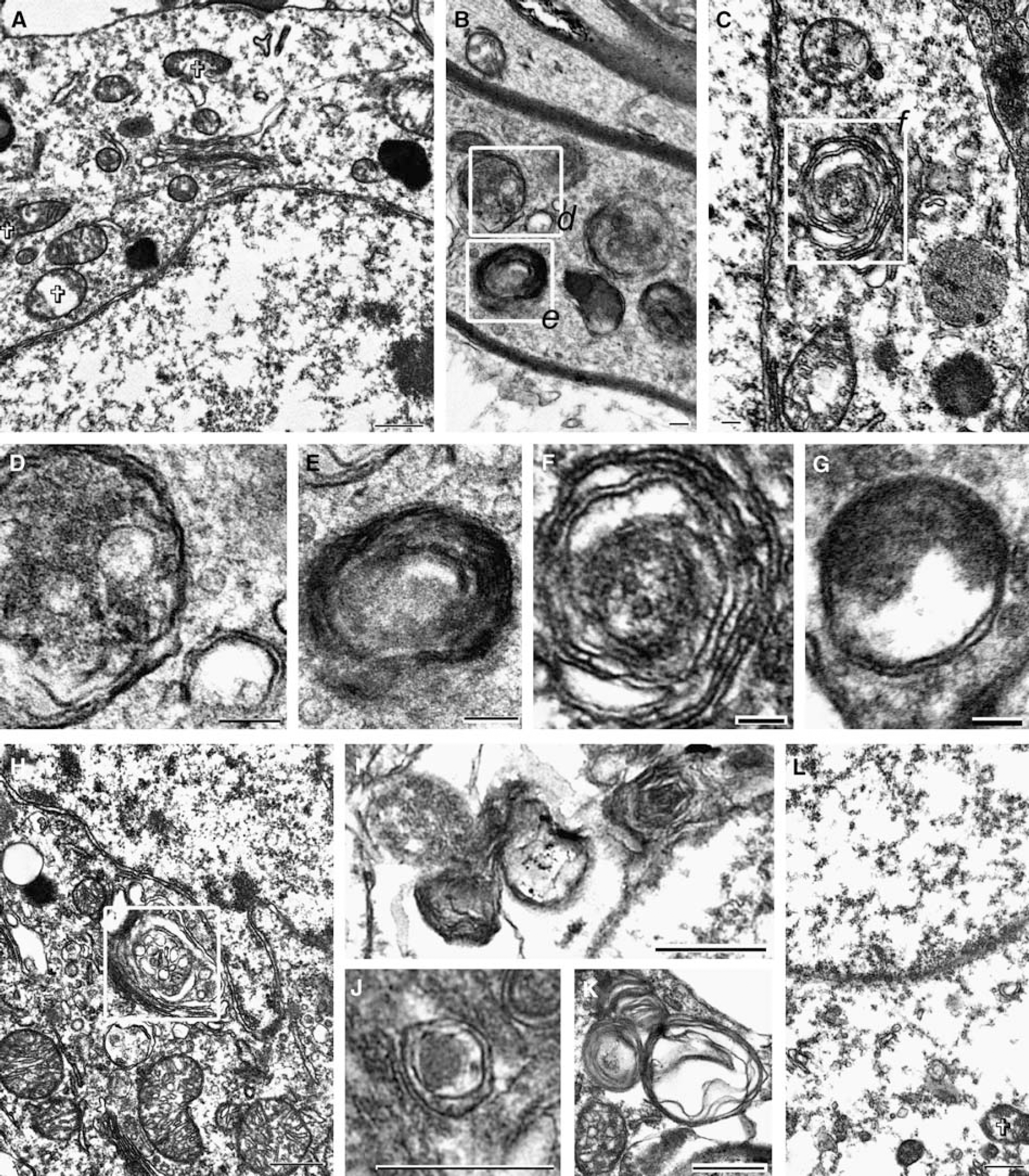
Ultrastructural evidence of autophagy 24 h after controlled cortical impact (CCI) injury in adult male mice. Transmission electron micrographs from mice 24 h after CCI (representative of 3 per group). (
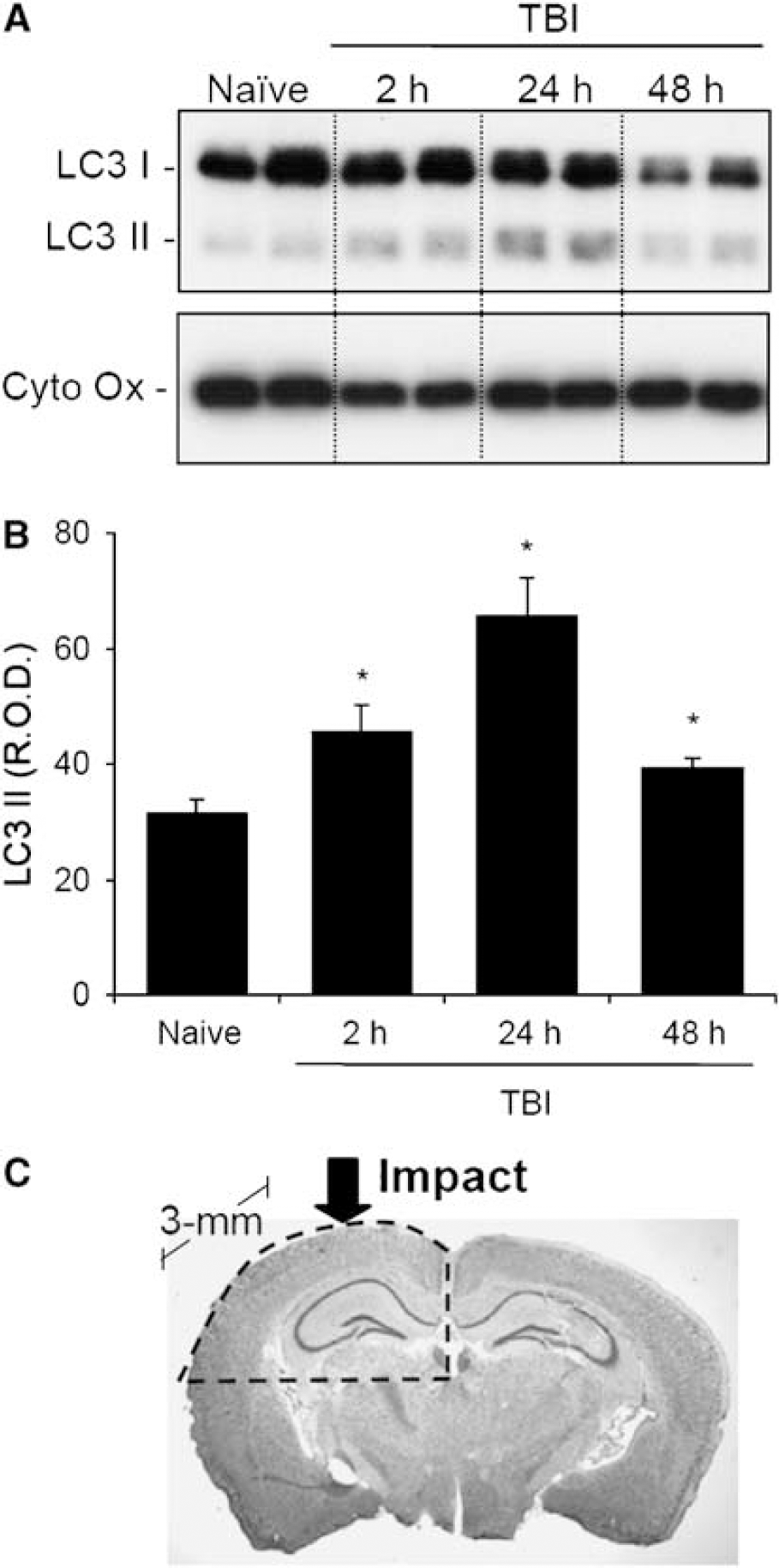
Temporal increase in LC3 II after CCI in mice. A 3-mm coronal section containing the dorsal hippocampus, thalamus, and overlying cortex was sampled for each mouse. (
Proteins were separated electrophoretically, and transferred to a polyvinylidene fluoride membrane (Amersham, Arlington Heights, IL, USA) overnight. The transferred membranes were incubated in a 1:1,000 dilution of a monoclonal antibody, which recognizes both LC3 I and II (Clone 51-11; MBL International, Woburn, MA, USA) at room temperature for 1 h. Membranes were washed in phosphate-buffered saline containing 0.1% Tween 20, then incubated in the appropriate secondary antibody (1:3,000) for 1 h, then incubated in commercial enhanced chemiluminescence reagents (NEN Life Science Products, Boston, MA, USA,) and exposed to X-ray film. The relative optical densities (RODs) for LC3 II were semiquantified, by subtracting the gel background ROD from the ROD of the LC3 II protein band. Membranes were then stripped and probed for cytochrome oxidase (Molecular Probes, Carlsbad, CA, USA) as a loading control for the P2 subfraction.
Chemiluminescence Measurement of Total Antioxidant Reserve
Total antioxidant reserves in brain homogenates from the ipsilateral parietal cortex including injury site and dorsal hippocampus were assayed as described (Tyurin et al, 2000). Briefly, oxidation of luminol (400 mmol/L) by 2,2′-azobis-2-amidinopropane hydrochloride-derived peroxyl radicals in 50 mmol/L Na-phosphate buffer (pH 7.4, 37°C) was measured by monitoring chemiluminescence (Luminescent Analyzer 633, Coral Biomedical, San Diego, CA, USA). The observed delay in the chemiluminescence response resulting from the interaction of 2,2′-azobis-2-amidinopropane hydrochloride-derived peroxyl radicals with endogenous antioxidants within tissue samples was determined for each sample. In the view of the known rate of peroxyl radical generation by 2,2′-azobis-2-amidinopropane hydrochloride, the amount of peroxyl radicals scavenged by endogenous antioxidants within each sample was calculated as the total antioxidant reserve for that sample.
Assessment of Spatial Memory Acquisition
Spatial memory acquisition was assessed using the Morris-water maze for mice by observers unaware of experimental group as described previously (Satchell et al, 2003). Briefly, a white pool (83-cm diameter, 60-cm deep) filled with 24°C water to 29-cm depth was situated in a room with several extramaze cues located on the walls. A 10-cm round goal platform located 1 cm below the surface of the water was placed 15-cm from the wall of the goal quadrant. A video tracking system mounted above the pool (Chromotrack 3.0; San Diego Instruments, San Diego, CA, USA) recorded the swimming movements of the mice. To ensure recovery from motor deficits, testing was performed on days 14 to 20 after CCI. Each mouse was subjected to a series of 4 trials/day. For each trial, mice were randomized to one of the four starting locations and placed in the pool. Mice were given a maximum of 120 secs to find the submerged platform. Mice were placed in a 37°C incubator for 4 mins between trials. Performance in the Morris-water maze was quantified by the latency to find the platform and also the rate of learning over the trial period, expressed as change in latency per day, since this was shown to be more sensitive in detecting injury effects in mice (Zohar et al, 2003). To exclude differences in motor performance between groups during Morris-water maze testing, swim speeds were determined for each mouse on days 19 and 20. To exclude differences in visual acuity between groups during Morris-water maze testing, visible platform testing was performed on days 19 and 20.
Histologic Analysis
Morphometric image analysis (MCID; Imaging Systems, St Catherines, Ontario, Canada) was used to determine contusion, ventricular, and hemispheric volumes at 21 days after CCI by an observer blinded to experimental group. Coronal sections (20 μm) were cut at 0.5-mm distances from the anterior to the posterior brain and mounted on glass slides. At each 0.5-mm interval, four sections were obtained and stained with cresyl violet. The areas of the lesion, left lateral ventricle, and injured and non-injured hemispheres from each section were determined. From these data, lesion, ventricular, and hemispheric volumes were calculated. Occult tissue loss was calculated as the difference between the uninjured (right) and injured (left) remaining hemisphere tissue volumes, and is a reflection of brain tissue loss apart from the lesion cavity.
Surviving neurons in the CA1 and CA3 hippocampal regions from the dorsal hippocampus directly below the impact were counted for each mouse at high magnification (× 400) by an observer blinded to treatment group. Neurons with well-defined nuclei and cell bodies were counted in two sections separated by 20 μm, and then averaged for each region. The total number of surviving neurons in the CA1 and CA3 hippocampus was determined in both ipsilateral and contralateral hemispheres for each mouse brain. For consistency, CA1 neurons were counted from the position superior to the medial aspect of the dentate gyrus laterally to the confluence of CA2 (typically 4 fields/section), and CA3 neurons were counted from the lateral aspect of the dentate gyrus to the bend of CA3 (1 to 2 fields/section). CA1 and CA3 hippocampal neuron density (neurons/ × 400) was also calculated for each mouse. Since neuron density paralleled total neuron survival, the latter was used for statistical analysis. Results are expressed as the percentage of neurons in the injured/uninjured hemisphere in the CA1 and CA3 hippocampal regions.
Statistical Analysis
Data are expressed as mean±s.e.m. Between group differences in Western blot, antioxidant reserve, rate of learning, swim speed, and hippocampal neuron survival data were analyzed using analysis of variance (ANOVA) with Student's—Newman—Keuls post hoc tests. Morris-water maze data were analyzed using repeated measures of ANOVA with Tukey's post hoc tests. Between-group differences in lesion volume and occult tissue loss were analyzed using a t-test. Analysis was performed using Sigma Stat software (Systat Software Inc., San Jose, CA, USA). Analysis of electron micrographs was descriptive.
Results
Ultrastructural and Biochemical Evidence for Autophagy after Traumatic Brain Injury in Mice
The gold standard for detection of autophagy is ultrastructural evidence of autophagosomal vacuoles by EM (Mizushima, 2004). Autophagosomal vacuoles were rarely observed in electron micrographs from naive mice in any of the regions examined (Figures 1A to 1D). Occasional multilamellar bodies were detected (0 to 2 per ultrathin section) within axons in naive mice (Figure 1E). In contrast, in brains from mice at 2 and 24 h after CCI, autophagosomal vacuoles, secondary lysosomes, double-membrane structures, and multilamellar bodies were readily detected in every section examined (n = 3 per group) from the ipsilateral cortex, cortical white matter, hippocampus, and thalamus (Figures 1 and 2). Occasional autophagosomal vacuoles were also observed in contralateral structures after CCI (Figure 2C). With rare exception, autophagosomal vacuoles were seen in cell processes and axons, although they were occasionally detected in perinuclear areas (Figures 1J and 2H). These autophagosomal vacuoles could be discriminated from axons or cell processes in coronal section, which had dissolution of intraluminal contents (examples of the latter in Figures 1C and 1D) by size (~100 to 500 nm in diameter), clear double membranes, and location within a cell process or myelinated axon. In brains from mice at 2 and 24 h after CCI, electron dense and compacted cells ultrastructurally consistent with dying neurons were seen; autophagosomal vacuoles were rarely observed in these cells (Figures 1F and 1G). Electron lucent astrocytes and swollen astrocytes foot processes were frequently observed, but autophagosomal vacuoles were not detected in astrocytes or oligodendrocytes, after CCI (examples shown in Figures 1G and 2L). In brains from mice 24 h after CCI, many disrupted mitochondria were also observed (Figure 1F).
To semiquantify the amount of autophagosomal vacuole formation after CCI relative to naive controls, we examined injured brain for evidence of an LC3 shift from an apparent ~18 to ~16 kDa position (Ichimura et al, 2000) using Western blot analysis. In pilot studies, LC3 II was minimally detected in whole-cell lysates from injured brain (data not shown). We found using an in vitro model of nutrient deprivation in primary neurons that the P2 fraction was enriched with LC3 II versus the P1 or S1 fractions, so this fraction was used for these in vivo studies. Ipsilateral cortices and hippocampi were combined to increase the protein yield of the subcellular compartment potentially containing autophagosomes. After CCI, increased LC3 II was observed at 2, 24, and 48 h, peaking at 24 h, versus naive mice (Figure 3; n = 4 per group; P < 0.05).
Treatment with γ-Glutamylcysteinyl Ethyl Ester Preserves Antioxidant Reserves and Reduces LC3 II Formation after Traumatic Brain Injury
Since in addition to starvation autophagy can be induced by mitochondrial oxidative stress (Chu, 2006), and reactive oxygen species are essential for starvation-induced autophagy to proceed in vitro (Scherz-Shouval et al, 2007), we determined whether the antioxidant cysteine-donor GCEE could reduce autophagy. γ-Glutamylcysteinyl ethyl ester penetrates the blood—brain barrier and preserves mitochondrial levels of GSH under conditions of nitrosative stress in vivo (Drake et al, 2002, 2003). First, the antioxidant capacity of GCEE in vivo after CCI was tested. A single intraperitoneal dose of GCEE (150 mg/kg) administered 10 mins after CCI preserved total antioxidant reserves in injured brain was measured 24 h after CCI versus vehicle-treated mice (256±11 versus 153±28 nmol/mg protein, respectively; P < 0.05, n = 4 per group; Figure 4A). Next, the capacity for GCEE to reduce autophagy, determined by measuring the relative amount of LC3 II in injured brain, was evaluated. γ-Glutamylcysteinyl ethyl ester treatment reduced the relative protein abundance of LC3 II at 24 h after CCI versus vehicle-treated mice (P < 0.05, n = 7 per group; Figure 4B).
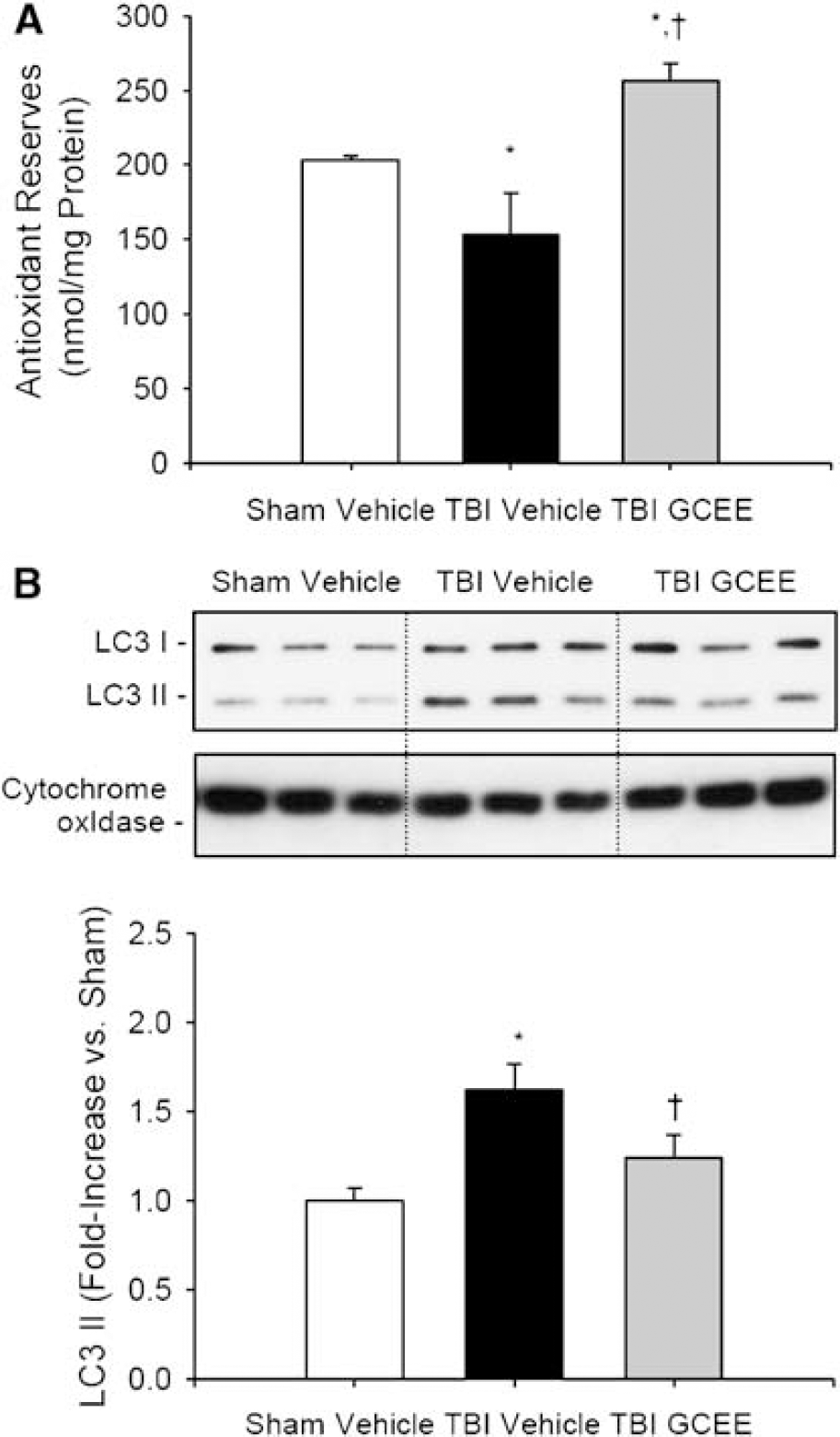
Biochemical effects of GCEE after CCI in mice. A 3-mm coronal section containing the dorsal hippocampus, thalamus, and overlying cortex was sampled for each mouse. (
Treatment with γ-Glutamylcysteinyl Ethyl Ester Improves Behavioral and Histologic Outcome after Traumatic Brain Injury
We next sought to determine if the reduction in oxidative stress and autophagy with GCEE treatment was associated with improved neurologic outcome after CCI. The same treatment regimen, GCEE (150 mg/kg), or vehicle intraperitoneally 10 mins after CCI was used. After CCI, worsening performance in the Morris-water maze, defined as a longer latency to find the hidden platform, was observed in vehicle-treated mice but not in GCEE-treated mice (P < 0.05 CCI vehicle versus all other groups, n = 15 per group; Figure 5A). In addition, GCEE-treated mice maintained a daily rate of learning similar to sham-injured mice, whereas vehicle-treated mice showed a reduced daily rate of learning in the Morris-water maze (13.0±2.3 versus 5.6±2.8 secs/ trial, respectively; P < 0.05; Figure 5B). There was no difference in swim speed between the GCEE and vehicle-treated groups after CCI (36.9±3.5 versus 39.0±2.8 cm/sec, respectively; P > 0.05), suggesting that the differences observed in the Morris-water maze performance were not due to differences in motor function between groups.
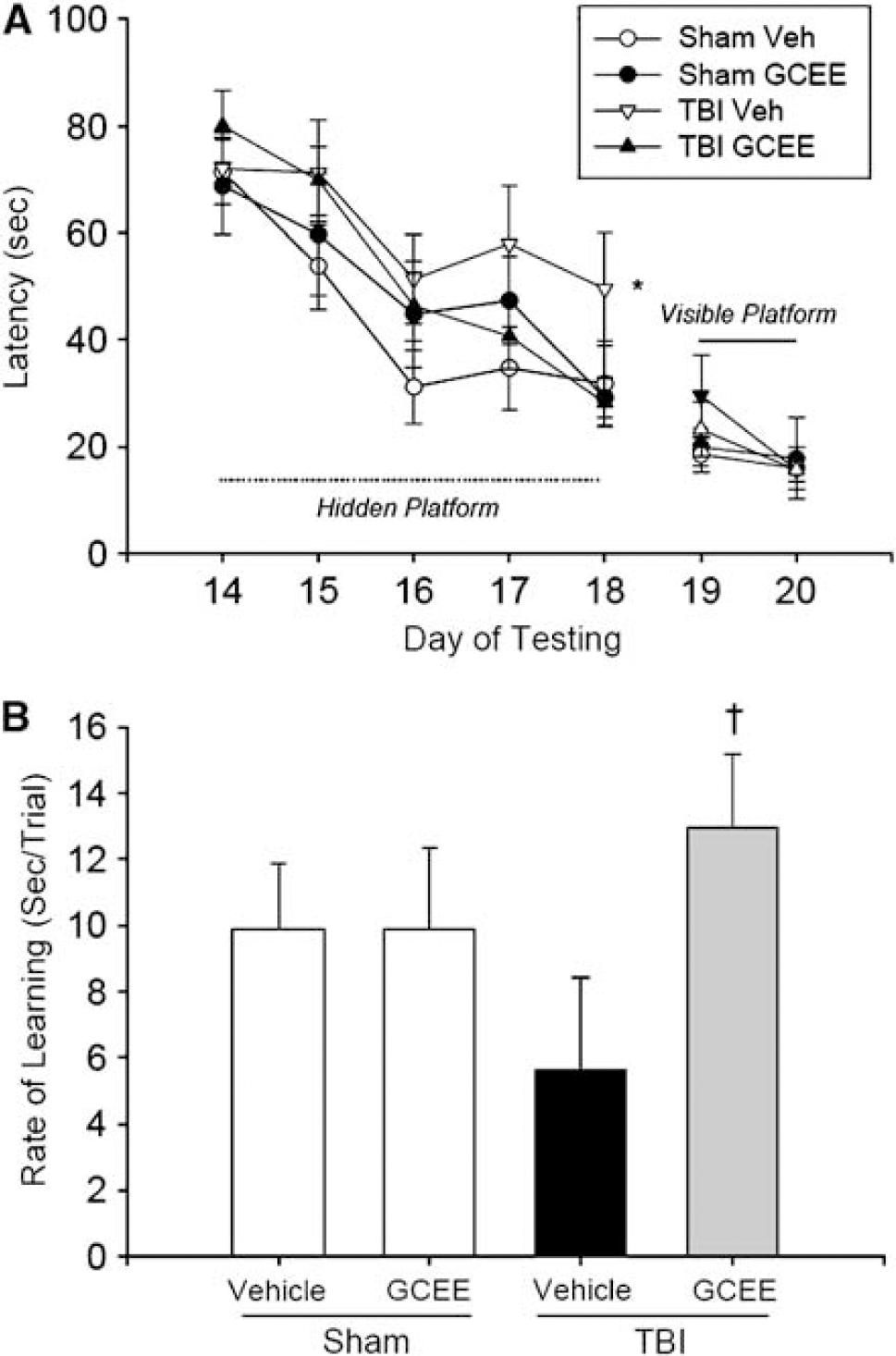
Effect of GCEE on Morris-water maze performance after CCI in mice. (
Treatment with GCEE (150 mg/kg intraperitoneally) versus vehicle 10 mins after CCI increased CA1 (41.2±4.0 versus 24.5±3.8% contralateral hemisphere, respectively; P < 0.05; Figures 6A and 6B) and CA3 (70.8±7.9 versus 47.4±8.2% contralateral hemisphere, respectively; P < 0.05; Figures 6A and 6C) hippocampal neuron survival measured at 21 days. Treatment with GCEE versus vehicle did not influence lesion volume (10.2±0.8 versus 9.11±0.4mm3; P > 0.05; Figures 6A and 6D), but did reduce occult tissue loss (4.0±0.8 versus 8.3±1.4 mm3; P = 0.01; Figures 6A and 6E) measured at 21 days.
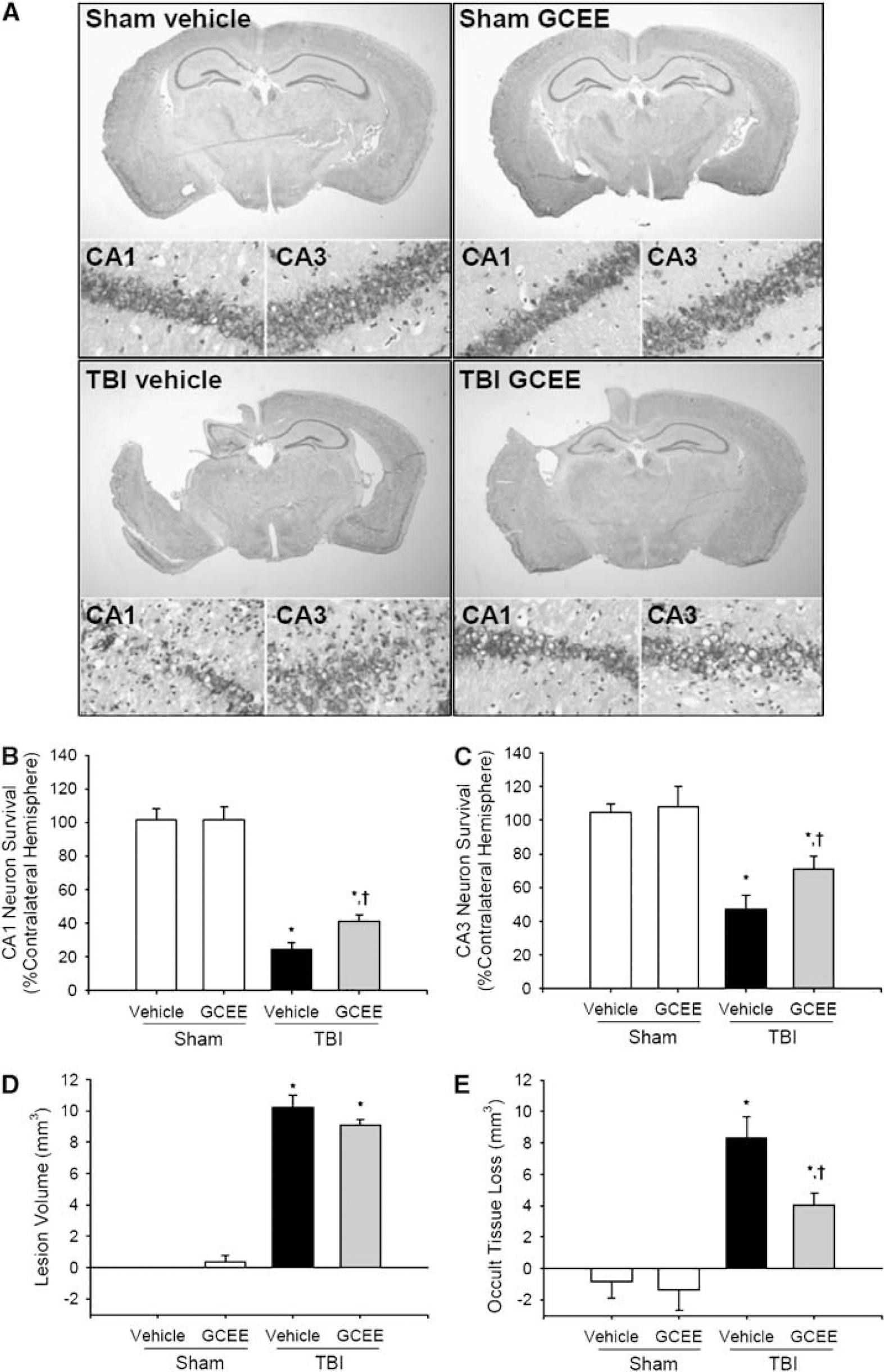
Histologic effects of GCEE after CCI in mice. (
Discussion
The major finding of this study is that autophagy, detected both ultrastructurally and biochemically, is induced in injured brain after TBI in mice. An additional finding is that autophagosomal vacuole formation, as detected by an increase in LC3 II, is partially inhibited by the antioxidant cysteine-donor GCEE. Treatment with GCEE improves Morris-water maze performance and partially reduces histologic damage compared with vehicle treatment after CCI in mice. These data represent the first ultrastructural evidence of autophagosomal vacuole formation and the first to suggest that reducing oxidative stress reduces autophagosomal vacuole formation, after acute brain injury. Taken together, these data suggest that oxidative stress contributes to overall neuropathology after TBI, at least in part by initiating or influencing autophagy. Similar to starvation-induced autophagy, a role for oxidative stress in TBI-induced autophagy is also implicated.
Autophagy is an important and well-defined adaptation to nutrient deprivation-induced stress (Neely and Mortimore, 1974; Seglen and Gordon, 1982), which has recently been discovered to have other, generally considered pathologic, triggers (Chu, 2006; Larsen and Sulzer, 2002). Maladaptive autophagy is implicated in neurodegenerative diseases such as Alzheimer's and Parkinson's disease (Boland and Nixon, 2006). A potential contribution for both microautophagy and macroautophagy in acute cell death after ischemic and TBI has been recently reported. An LC3 shift has been reported in mice of various ages after unilateral carotid artery ligation and hypoxemia (Adhami et al, 2006; Zhu et al, 2003). After closed head injury, upregulation of Beclin-1 has been reported within neurons in injured hippocampus and cortex in mice (Diskin et al, 2005). Our data provide direct ultrastructural evidence of autophagy after acute brain injury (Figures 1 and 2) in addition to biochemical alterations in LC3. Interestingly, autophagosomal vacuoles were detected mainly in cell processes and axons, raising the possibility that autophagy may be involved in dendritic pruning and axonal degeneration, and thus either pathology or plasticity after TBI. Autophagy can contribute to axonal degeneration (Wang et al, 2006), and traumatic axonal damage is a key neuropathologic component of TBI (Povlishock et al, 1992). The majority of the autophagosomal vacuoles contained organelle debris, suggesting that macroautophagy (or ‘mitophagy’) was prominent after TBI. However, multilamellar bodies were also observed, particularly in axons. Multilamellar bodies function in lipid storage and secretion, and are increased under conditions provoking autophagy (Hariri et al, 2000) and in neurons in patients with Alzheimer's disease (Nixon et al, 2005). We also detected biochemical evidence of autophagosomal vacuole formation, a shift from LC3 I to II after TBI. An increase in LC3 II was seen in injured brain at 2, 24, and 48 h, with peak relative abundance at 24 h (Figure 3), although time points after 48 h were not evaluated and as such a later peak in LC3 II after TBI cannot be ruled out.
Oxidative stress is believed to be one mechanism by which nonstarvation-induced autophagy can be initiated, and/or lysosomal clearance impaired, resulting in increased autophagic vacuoles, a term coined ‘autophagic stress' (Chu, 2006). Furthermore, it has recently been shown that oxygen radicals are essential for starvation-induced autophagy in vitro (Scherz-Shouval et al, 2007). For these reasons, we tested the antioxidant GCEE for its capacity to reduce oxidative stress and evidence of autophagosomal vacuole formation. γ-Glutamylcysteinyl ethyl ester crosses the blood—brain barrier and results in maintenance of mitochondrial GSH and function under conditions of oxidative stress induced by buthionine sulfoxamine (Drake et al, 2003). Depletion of GSH and total antioxidant reserves occur after TBI in vivo (Tyurin et al, 2000). Consistent with either upstream initiation or regulation of autophagy by oxidative stress, GCEE treatment resulted in both preservation of antioxidant reserves and a reduction in LC3 II formation (Figure 4). Posttreatment with GCEE not only produced salient biochemical effects, but also prevented behavioral deficits (Figure 5) and reduced histologic damage (Figure 6). γ-Glutamyl-cysteinyl ethyl ester treatment did not affect the primary lesion volume, but it reduced nonlesion (occult) brain tissue loss, perhaps representing brain regions more amenable to therapy. Three important caveats need to be raised. First, GCEE only partially reduced LC3 II formation and partially improved neurologic outcome. Second, it is possible that the beneficial effects of GCEE were unrelated to direct inhibition of autophagy. Third, unbiased stereologic analysis was not used to quantify hippocampal neuron survival. Nonetheless, improvement in multiple outcomes warrants further testing of different dosing strategies of GCEE as a potential neuroprotective agent for clinical use.
Since antioxidant therapies such as GCEE clearly have multiple effects, what remains unclear is whether after TBI oxidative stress provokes autophagy, contributes to secondary damage, or initiates cell death, which provokes autophagy setting the stage for recovery and/or repair. Regardless, it is clear that autophagy and oxidative stress are connected after TBI, similar to autophagy and starvation in vitro, and that one effect of antioxidant therapy is a reduction in macroautophagy. What also remains unclear is whether postinjury alterations in food intake contributed to the observed increase in autophagy. The nutritional status of these mice was not ascertained and mice do tend to lose weight in this TBI model, thus a contribution of nutrient deprivation-induced autophagy, cannot be completely ruled out. Arguing against this, starvation alone up to duration of 48 h does not typically produce autophagy in mouse brain in vivo (Mizushima et al, 2004). However, these study conditions are clinically relevant, as withholding glucose and other nutrients early (first 1 to 2 days) is common clinical practice for patients with moderate-severe TBI (Adelson et al, 2003).
What remains to be determined is whether direct inhibition of autophagy—without inhibition of oxidative stress—is beneficial or detrimental after TBI. For example, transgenic mice with deficiencies in autophagy are known to have abnormal accumulation of proteins associated with neurodegeneration (Hamm et al, 1996). Conceptually, reduced autophagy would be thought to contribute to neuropathology related to amyloidopathies such as Alzheimer's disease. However, clinical studies show marked increases in autophagosomes in human brain with Alzheimer's disease-related pathology compared with those without (Nixon et al, 2005), suggesting that disrupted, rather than reduced, autophagy contributes to neurodegeneration. This could also be the case in humans after TBI. The finding that autophagosomal vacuoles are frequently detected in injured but viable appearing neurons, but rarely detected in dying neurons identified via EM would be consistent with a protective role for autophagy after TBI. However, an alternative possibility is that autophagosomal structures are difficult to detect in the shrunken and hyperchromatic end stage of neuronal death at the EM level (Kalimo et al, 1982). In such neurons, it is difficult to definitively identify many of the subcellular organelles and vacuoles seen at the earlier transition stages of neurodegeneration. For example, after cerebral ischemia, mitochondrial changes (microvacuolation) are classically seen, but these changes are difficult to identify after neuronal death progresses to dense shrunken neurons (Jenkins et al, 1981). Thus, it is possible that autophagosomal vacuoles could be present in electron dense end-stage neurons, but they are distorted and not recognizable.
In conclusion, autophagy is increased after TBI in mice. Inhibition of autophagy may represent a therapeutic target, or alternatively, autophagy may be protective, after TBI. Inhibition of autophagy after TBI may be achieved using antioxidants such as GCEE, but more relatively selective agents, such as the phosphatidylinositol 3-kinase inhibitor 3-methyladenine (Seglen and Gordon, 1982) or extracellular signal-regulated kinase inhibitors such as U0126 (Zhu et al, 2007) warrant future testing.
Footnotes
Acknowledgements
We thank Vincent Vagni, Paula Nathaniel, and Holly Donovan for expert technical assistance with the mouse studies.