
Abstract
Intracranial bleeding is one of the most prominent aspects in the clinical diagnosis and prognosis of traumatic brain injury (TBI). Substantial amounts of blood products, such as heme, are released because of traumatic subarachnoid hemorrhages, intraparenchymal contusions, and hematomas. Despite this, surprisingly few studies have directly addressed the role of blood products, in particular heme, in the setting of TBI. Heme is degraded by heme oxygenase (HO) into three highly bioactive products: iron, bilirubin, and carbon monoxide. The HO isozymes, in particular HO-1 and HO-2, exhibit significantly different expression patterns and appear to have specific roles after injury. Developmentally, differences between the adult and immature brain have implications for endogenous protection from oxidative stress. The aim of this paper is to review recent advances in the understanding of heme regulation and metabolism after brain injury and its specific relevance to the developing brain. These findings suggest novel clinical therapeutic options for further translational study.
Keywords
Introduction
Intracranial bleeding is a critical factor in the clinical diagnosis and prognosis of outcome after traumatic brain injury (TBI) (Marshall et al, 1992; Ribas and Jane, 1992). Heme (or ferroprotoporphyrin IX) is generated as a breakdown product of hemoglobin and other hemoproteins arising from traumatic subarachnoid hemorrhages, intraparenchymal contusions, and hematomas (Figure 1) (Hua et al, 2000; Macdonald and Weir, 1991; Wagner et al, 2003, 1996; Xi et al, 1998). Given the pathogenicity of heme (Huang et al, 2002; Maines and Trakshel, 1993; Regan and Panter, 1996; Ryter and Tyrrell, 2000; Wu et al, 2002), it is surprising that few studies have examined its role in the setting of TBI. In this review, we address recent advances in the understanding of heme regulation in the context of traumatic injury to the adult and developing brain.
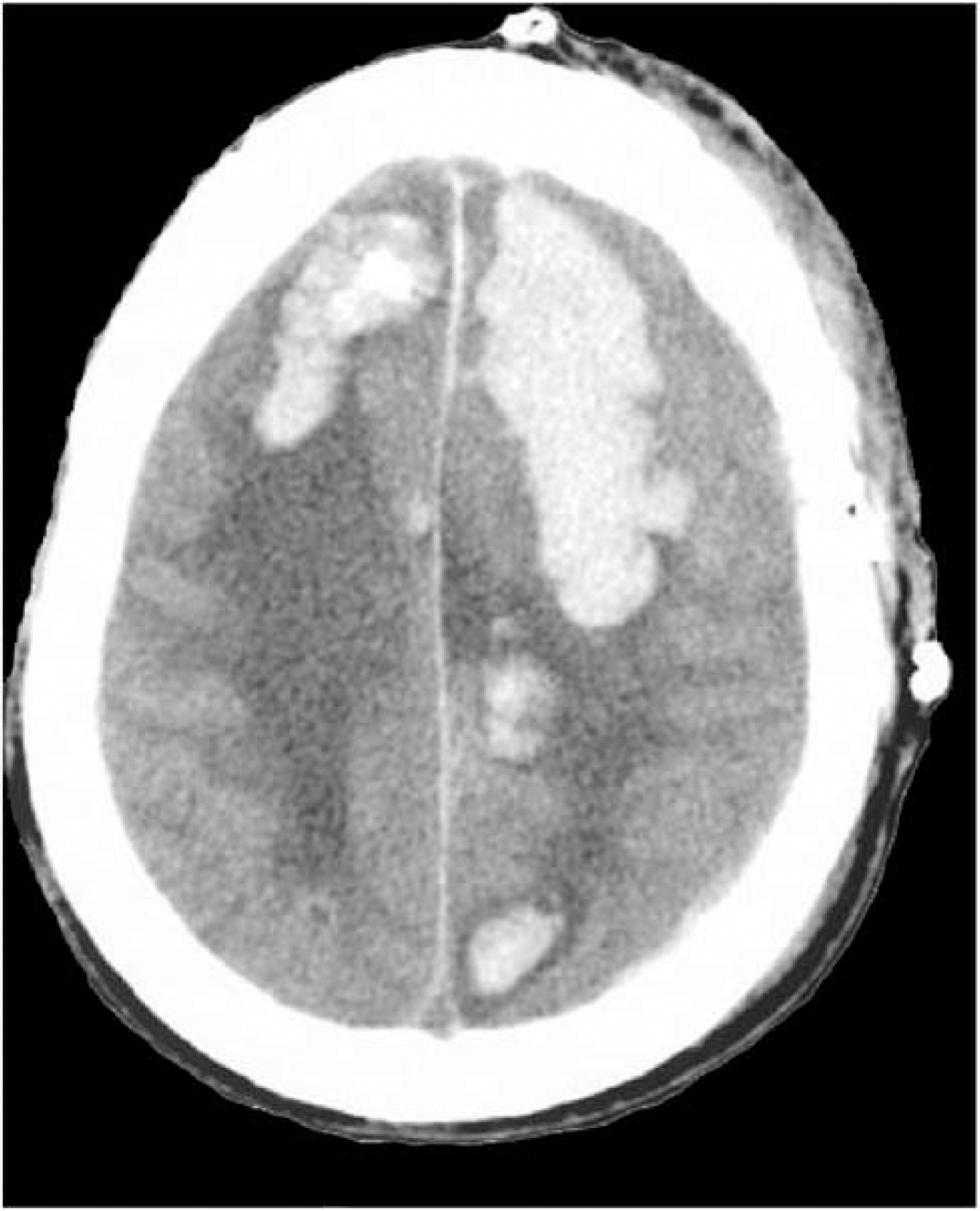
Computed tomographic (CT) scan of human brain after a motor vehicular accident illustrating large traumatic intraparenchymal hematomas. The presence of intracranial bleeding is an important part of the clinical diagnosis and prognosis of outcome after traumatic brain injury. Heme and other blood products are released following trauma resulting in subarachnoid hemorrhages, intraparenchymal contusions, and hematomas, in addition to intrinsic cellular injury.
Heme and Heme Metabolism
Structure/Function
Free heme does not accumulate normally in tissue; it is deposited in tissues only under pathologic conditions (Maines, 1988). The term ‘free heme’ is used in reference to the molecule before its incorporation into hemoproteins. The molecule is integral to life as part of hemoproteins; and as such, carries out oxidative reactions, electron transfer processes, and delivers molecular oxygen (O2) to cells.
The conjugation of the tetrapyrrole macrocycle with a central metal determines the color, planarity, and fluorescence properties characteristic of a given metalloporphyrin (Figure 2). Porphyrin molecules alone do not bind O2 nor participate in enzyme-catalyzed reactions; these properties are conferred on the macrocyclic molecules by chelating a multivalent transition metal, such as iron (Fe), tin, zinc, and attachment of the protein moiety.
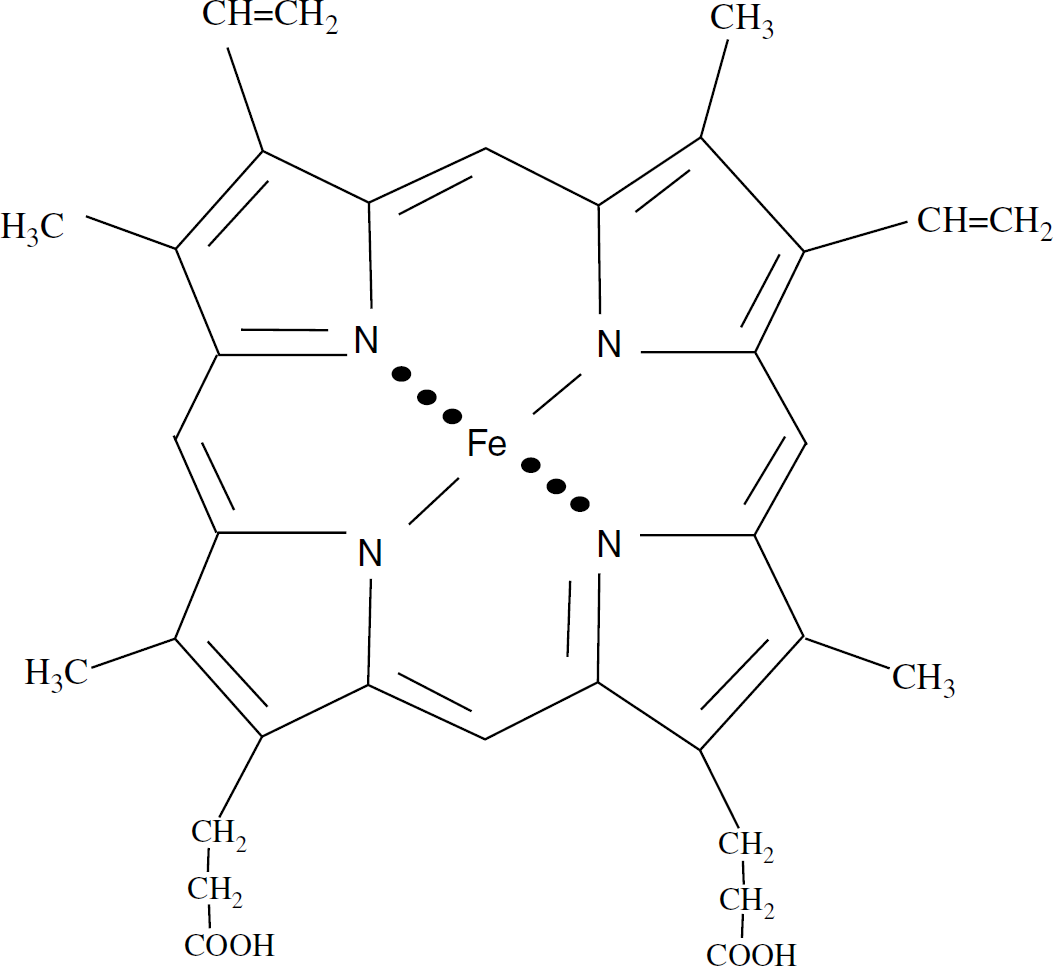
Structure of heme molecule: a square-planar tetrapyrrole with a divalent chelating transition metal, iron (Fe).
The bulk of the heme required for the production of hemoproteins, such as cytochromes, is derived from the de novo synthesis of heme (Maines, 1997; Ponka, 1999). Heme is a major component of hemoproteins such as hemoglobin, myoglobin, cytochromes, guanylate cyclase, and nitric oxide synthase. The heme molecule is synthesized through a series of enzymatic steps that begin with 5-δ-aminolevulinate synthase and end with ferrochelatase.
Heme Catabolism
The heme molecule is degraded by enzymatic and nonenzymatic mechanisms. Both pathways, however, use molecular O2 and require a reducing agent; the latter is needed for activation of O2 and reduction of heme iron from Fe2+ to Fe3+ and/or maintenance of iron in the Fe2+ state for the binding of O2. The breakdown of heme is catalyzed by heme oxygenase (HO), in the presence of NADPH, which serves as the source of the reducing equivalent. The reaction produces equimolar amounts of carbon monoxide (CO), Fe3+, and the α-isomer of biliverdin. Biliverdin is then rapidly converted into bilirubin by biliverdin reductase, which is in excess in all tissues (Figure 3). Nonenzymatic reactions, also called ‘coupled-oxidations’, lead to formation of four isomers of biliverdin from heme.
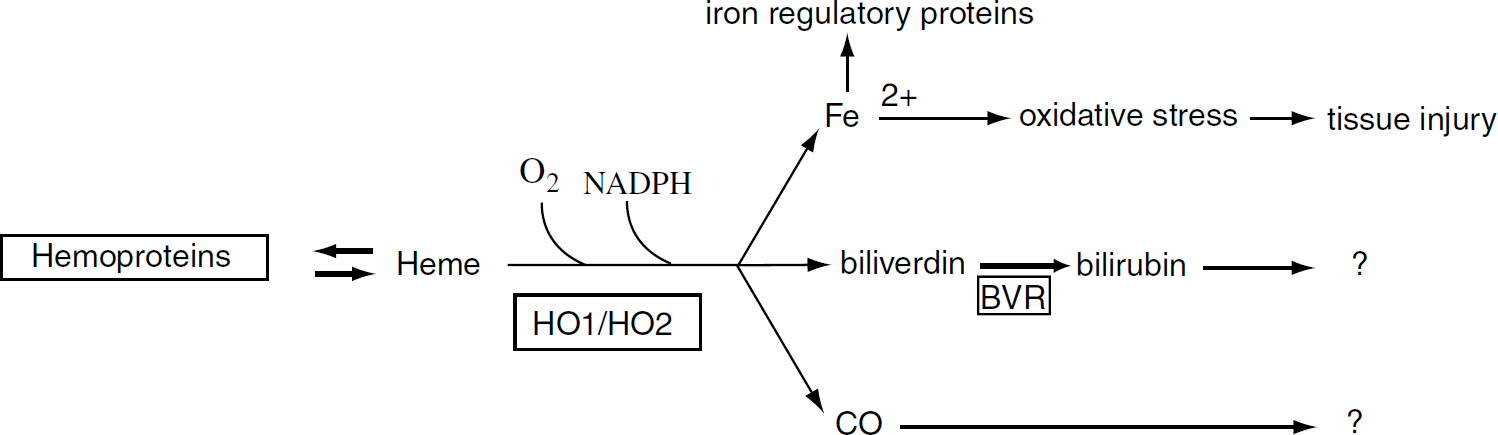
Heme degradation pathways. The products of heme metabolism may have harmful or beneficial effects in the setting of traumatic brain injury or other pathologic conditions, although this has to be further elucidated. Heme is degraded by heme oxygenase (HO) isozymes (HO-1 and HO-2) into iron, one isomer of biliverdin, and carbon monoxide (CO). The enzymatic reaction requires molecular oxygen (O2) and NADPH as a reducing equivalent. Activation of O2 initiates coupled-oxidation: an electron donor starts the oxidative cleavage of methene carbon bridges in heme leading to formation of four isomers of biliverdin. Bilirubin reductase (BVR) converts biliverdin into bilirubin.
Sources of Free Heme
Traumatic brain injury is associated with a host of pathophysiologic conditions, which include mechanical damage, edema, ischemia, and hemorrhage. Each of these processes contributes to the cumulative, and substantial release of free heme from the hydrolysis of hemoproteins. Both intra-and extra-cellular sources of heme likely contribute to its accumulation after injury (Table 1) (Wagner et al, 2003). Cytoplasmic hemoproteins and mitochondrial cytochromes in neurons and glia are intracellular sources of heme. Extracellular sources of heme include hemoproteins that are released from dying cells, and/or derived from hemoglobin that is generated as a consequence of subarachnoid and intraparenchymal bleeding (Sharp et al, 1999). Extravascular erythrocytes lyse in the central nervous system (CNS) via a complement-mediated mechanism (Hua et al, 2000). Furthermore, extracellular hemoglobin is thought to be selectively internalized by microglia throughout the brain after experimental subarachnoid hemorrhage, possibly mediated through the induction of hemopexin (Liem et al, 1975; Matz et al, 1996; Turner et al, 1998). Heme and the products of heme catabolism have all been shown to be distinct bioactive molecules and therefore the subject of considerable interest (Platt and Nath, 1998).
Sources of free heme

Free heme does not accumulate normally in tissues, but rather deposited in the brain under pathologic conditions, such as traumatic brain injury (TBI) and stroke.
Regulation of Heme Oxygenase
In mammals, two principal isozymes of HO have been identified. Heme oxygenase-1 is a heat shock protein (HSP-32) induced under numerous conditions of cellular stress (Ewing and Maines, 1993; Ferris et al, 1999; Fukuda et al, 1996; Geddes et al, 1996; Turner et al, 1998). The constitutively expressed HO-2, in contrast, is the predominant isozyme found in the adult rodent brain and testes and is only regulated by adrenal glucocorticoids (Maines et al, 1996; McCoubrey and Maines, 1994). Two pseudogenes (HO-3a and HO-3b) have also been discovered, but are thought to be nonfunctional derivatives from HO-2 transcripts (Hayashi et al, 2004)
Heme oxygenase-1 and HO-2 are different gene products and share little similarity in transcript number, or size (Cruse and Maines, 1988; Maines, 2000) (Table 2). Heme oxygenase-1 is the smaller of the three with 288 amino acids and has a molecular weight of 30,000 to 33,000 Da (Shibahara et al, 1985); HO-2 is comprised of 316 amino acids and has a molecular weight of 36,000 Da (Rotenberg and Maines, 1990). The enzymes, however, have similar mechanisms of heme catalysis, substrate specificity, and cofactor requirements. Heme oxygenase-1 and HO-2 share similar nucleotide (50% similarity) and amino acid (43% similarity) sequences, and 76% of the amino-acid sequence is conserved in functional domains (Rotenberg and Maines, 1990). Despite major structural differences between HO-1 and HO-2, each isozyme is evolutionarily conserved. For the rat, mouse, and human, the extent of homology for HO-1 is greater than 80%; and for HO-2, similarity among the rat, rabbit, and human cDNA nucleotide sequence is over 90% (Rotenberg and Maines, 1991).
Comparison of heme oxygenase (HO) isozymes, HO-1 and HO-2
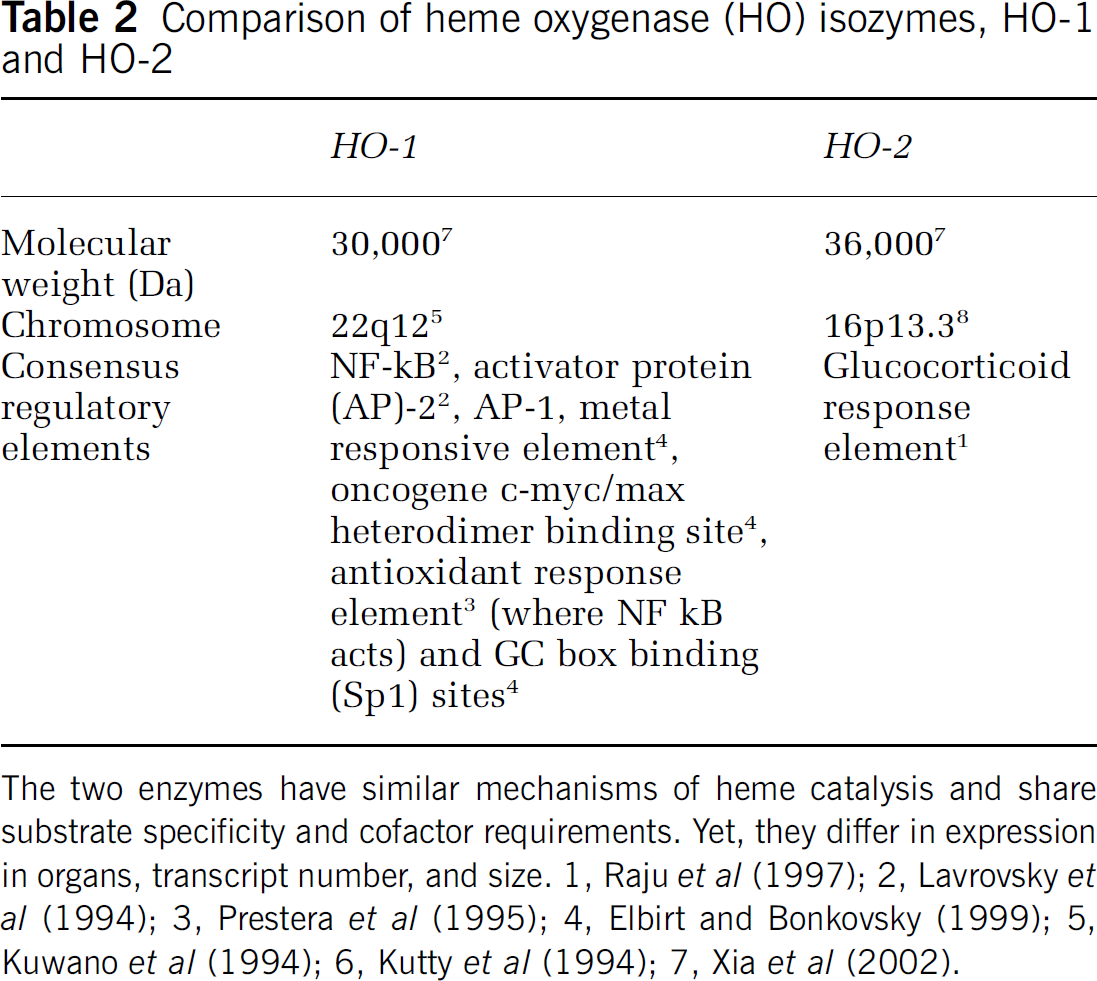
The two enzymes have similar mechanisms of heme catalysis and share substrate specificity and cofactor requirements. Yet, they differ in expression in organs, transcript number, and size. 1, Raju et al (1997); 2, Lavrovsky et al (1994); 3, Prestera et al (1995); 4, Elbirt and Bonkovsky (1999); 5, Kuwano et al (1994); 6, Kutty et al (1994); 7, Xia et al (2002).
Differences between HO-1 and HO-2 expression relate to the presence of regulatory elements in their promoter region. The consensus sequences necessary for binding several regulatory factors—such as heat-shock factor, AP-1, NF-κB, and metal regulatory elements (MREs) (Lavrovsky et al, 1994; Muller et al, 1987; Weber et al, 1994)—are present in the HO-1 gene promoter region; whereas, only a single glucocorticoid response element (GRE) is present and functional in the HO-2 gene promoter region (McCoubrey and Maines, 1994; Raju et al, 1997).
Heme Oxygenase Expression
Under normal conditions, regional and cellular differences exist in the immunoexpression of HO-1 and HO-2 in the adult brain. Although each of these isozymes is expressed in neurons, HO-1 has a very limited distribution—with highest levels of expression within the dentate gyrus of the hippocampus and the ventromedial hypothalamus (Ewing et al, 1992). In contrast, HO-2 is expressed in neurons throughout the forebrain, hippocampus, midbrain, basal ganglia, thalamus, cerebellum, and brain stem (Ewing and Maines, 1997; Maines et al, 1996). Unlike HO-2, which is limited in expression to neurons, HO-1 is also localized in glia (Chang et al, 2003; Fukuda et al, 1996).
After adult TBI, HO-1 and HO-2 isozymes have markedly different cellular and temporal expression patterns (Figure 4). Induction of HO-1 in both astrocytes and microglia/macrophages has been reported in experimental animal models of TBI as well as in human brain injury (Beschorner et al, 2000; Fukuda et al, 1995, 1996). This induction is most prominent in areas of maximal tissue damage and intraparenchymal hemorrhage. Human clinical studies likewise support the induction of HO-1 in microglia/macrophages associated with the hemorrhagic lesion (Beschorner et al, 2000). Heme oxygenases levels, in contrast, are largely unchanged after injury. Although HO-1 is induced after injury and contributes to total HO activity, HO-2 is still the dominant source of HO activity in the injured brain (Chang et al, 2003).
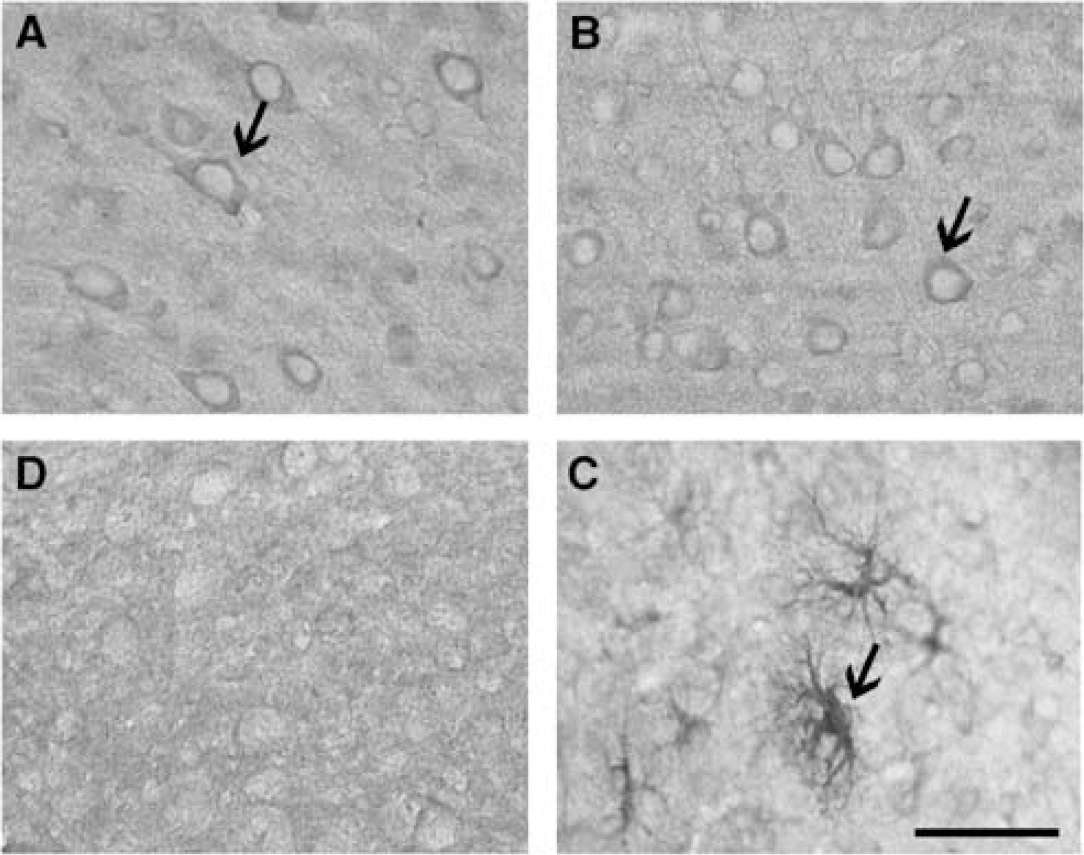
Immunolocalization of heme oxygenase (HO)-2 and HO-1 in the uninjured (
Although both HO-1 and HO-2 catalyze the same biochemical reaction, the fact that they show such different patterns of cellular expression suggests that the two isozymes have individual roles in the setting of brain injury.
Heme Oxygenase as Modulator of Neural Injury
The precise role that each HO isozyme plays in TBI and other pathophysiologic conditions still remains unclear (Platt and Nath, 1998). It is likely that the end-products of heme degradation, namely, CO, bilirubin, and iron, influence outcome. Despite much interest in its putative neuromodulatory functions, CO still has an unclear role in neuroprotection (Baranano and Snyder, 2001; Koehler and Traystman, 2002; Maines, 1997). Some evidence suggests that CO liberated in the course of heme degradation can diminish cerebral vasospasm by promoting relaxation of vascular smooth muscle (Matz et al, 1996; Suzuki et al, 1999; Tanaka et al, 2000).
Although clinically better known for its deleterious effects in kernicterus, bilirubin has showed surprisingly robust antioxidant properties (Dennery, 2000; Dore and Snyder, 1999). Dore et al have reported that these effects occur at the physiologic low nanomolar endogenous levels that occur in the brain, thousands-fold lower than neurotoxic levels of bilirubin that occur during kernicterus (Dore et al, 1999b; Stocker et al, 1987a,b). However, the exact concentration at which bilirubin is indeed protective or toxic is still controversial and currently being studied (Ostrow et al, 2004, 2003; Ostrow and Tiribelli, 2001). Bilirubin is believed to be one of the most potent endogenous antioxidants in mammalian tissue, accounting for the majority of the antioxidant activity of human serum (Gopinathan et al, 1994).
One attractive hypothesis regarding the strong antioxidant abilities of bilirubin involves the rapid regeneration of bilirubin via redox cycling (Baranano et al, 2002). Each molecule of bilirubin that acts as an antioxidant is thereby itself oxidized to biliverdin. The high level of biliverdin reductase immediately reduces biliverdin back to bilirubin. Cell lines designed to express less biliverdin reductase exhibit increased vulnerability to oxidative stress (Baranano et al, 2002). The rapid recycling of bilirubin is therefore a potentially ideal endogenous system that can neutralize the numerous reactive oxygen species generated after TBI. However, this concept needs to be further studied.
Cell culture studies offer insight into the significance of HO-1 induction in microglia. Heme oxygenase-1 induction protects cultured cortical astrocytes, but not neurons, from oxidative stress resulting from exposure to hemoglobin and hydrogen peroxide (H2O2) (Dwyer et al, 1995; Regan et al, 2000). The beneficial effect of HO-1 is in general agreement with many prior observations in neural and nonneural systems (Chen et al, 2003; Clark et al, 2000; Turner et al, 1999).
Heme oxygenase-2, unlike HO-1, is constitutively expressed in neurons throughout the brain. Heme oxygenase-2 protects against cellular injury after TBI, especially in areas of the brain previously shown to be selectively vulnerable to oxidative stress, and is associated with improved behavioral recovery. Heme oxygenase-2 knockout mutants were found to have reduced motor recovery on a rotarod task for generalized motor learning ability and a beam-walking task for sensorimotor processing (Figure 5). Importantly, these experiments found an association between worsened behavioral outcomes and increased cellular injury as a result of the HO-2 gene deletion. Furthermore, HO-2 knockout mice showed greater vulnerability to lipid peroxidation, specifically in the setting of TBI (Chang et al, 2003).
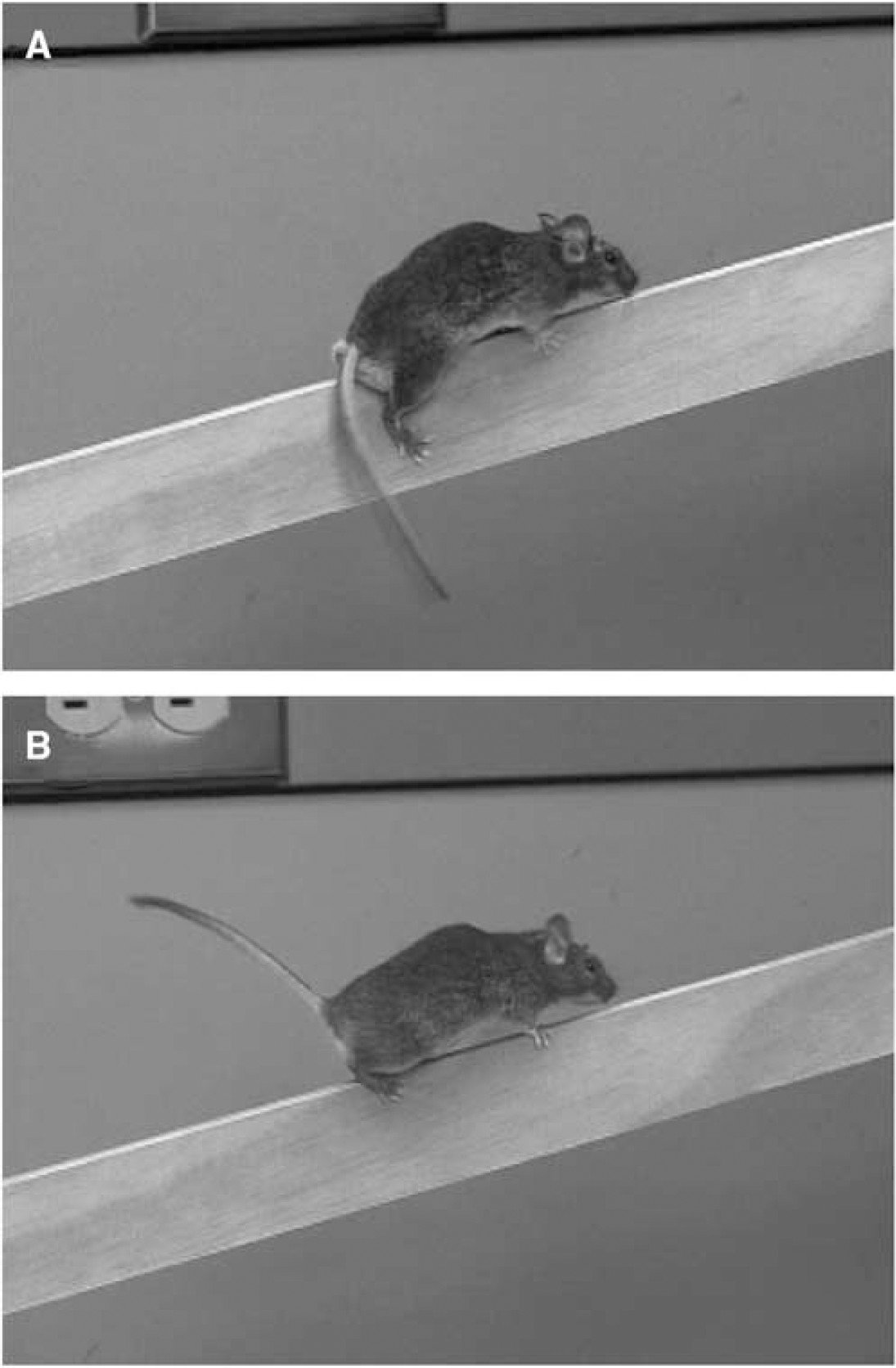
Reduced motor recovery in heme oxygenase (HO)-2 knockout mice after traumatic brain injury. Beam walking depends on the integrity of sensorimotor cortical processing and, as expected, all animals showed a high percentage of contralateral hind-limb foot faults in the first few days after brain injury. Heme oxygenase-2 knockout mice (
The finding that HO-2 confers neuroprotection is consistent with results using other models of injury. Heme oxygenase-2 expression has been shown to be protective against apoptotic cell death in cortical, hippocampal, and cerebellar granule cultures and in an in vivo model of ischemic injury (Dore et al, 2000, 1999a b; Dore and Snyder 1999). Furthermore, others have found a link between HO activity and/or expression and neurodegenerative conditions such as familial Alzheimer's disease or Parkinson's disease (Schipper, 2004). Several single-point mutations in amyloid precursor proteins, which bind HO, have been associated with a significant reduction in HO activity, resulting in decreased bilirubin staining and increased neurotoxicity (Takahashi et al, 2000).
Other genetic and pharmacologic studies, however, have challenged the belief that HO confers protection after brain injury. For example, although HO-1 is inducible, its genetic reduction does not worsen injury in an experimental model of cerebral ischemia (Dore et al, 1999a). Moreover, HO-1 expression is largely limited to reactive glial populations (although that does not necessarily rule out a neuronal protective function) (Ewing et al, 1992; Fukuda et al, 1996; Geddes et al, 1996; Schipper, 1999; Turner et al, 1998). Neuronal and astrocytic cell cultures have recently showed differential effects of HO-2 (Regan et al, 2004). Heme oxygenase-2 gene deletion was actually found to protect murine cortical neurons from hememediated oxidative injury, yet appeared to be more vulnerable to nonheme-mediated oxidative injury (Regan et al, 2004; Rogers et al, 2003). Heme oxygenase-2 deletion interestingly had the opposite effect in astrocytic cell cultures, with 50%–70% increased cell death. Heme oxygenase-2 detection also did not appear to alter cellular expression of HO-1. This increased sensitivity was reversed by increasing HO-1 expression by adenoviral gene transfer (Chen and Regan, 2004).
A central concern is that heme degradation may generate toxic levels of free iron (Huang et al, 2002; Kadoya et al, 1995; Panizzon et al, 1996). Cell injury is speculated to be mediated through the generation of free Fe2+ during heme degradation, which may potentiate free radical-induced damage via the Fenton reaction. Iron, a strong generator of reactive oxygen species, reacts with H2O2 to form the hydroxyl radical, or may decompose membrane lipid peroxides to yield alkoxy and peroxy radicals (Dennery et al, 2003; Rouault, 2001). All three species are capable of initiating lipid peroxidation chain reactions.
Consistent with this view, the administration of HO inhibitors, such as tin protoporphyrin, has been shown to reduce the formation of brain edema, and to reduce injury from ischemic, hemorrhagic, and even traumatic injuries to CA3 hippocampal slices (Huang et al, 2002; Kadoya et al, 1995; Panizzon et al, 1996). Dennery et al have reported that neurons of HO-2 knockout neurons are more vulnerable than wild type to inorganic iron, and concluded that HO-2 expression accelerates hemoglobin neurotoxicity and most likely because of the accumulation of iron. Their findings are consistent with the observations that HO inhibitors attenuate heme-mediated reactive oxygen species formation and cell death (Goldstein et al, 2003; Huang et al, 2002; Wagner et al, 2000).
Red blood cell lysis has been shown to contribute to edema formation after intracerebral hemorrhage at the same time that HO-1 expression and iron accumulation are enhanced, suggesting that heme and its degradation products are associated with breakdown of the blood-brain barrier (Bhasin et al, 2002; Hoff and Xi, 2003; Wu et al, 2003, 2002; Xi et al, 2001). Damage to the vascular endothelium may be because of oxidative stress perpetuated by iron (Ogihara et al, 1999; Wu et al, 2003, 2002) and furthermore the administration of iron chelators attenuates edema after intracerebral hemorrhage (Huang et al, 2002).
It should be noted, however, that tin protoporphyrin and other metalloporphyrins are not only differential inhibitors of HO-1 and HO-2 (Vreman et al, 1998), but have also been reported to inhibit the activity of cytochrome P450s, nitric oxide synthase, soluble guanylate cyclase, and others (Appleton et al, 1999; Grundemar and Ny, 1997; Luo and Vincent, 1994; Meffert et al, 1994; Murray, 1997). They have also been reported to be direct inhibitors of lipid peroxidation (Imai et al, 1990; Wong et al, 2000). Finally, the use of metalloporphyrins in studies regarding the physiologic function of HO-1 or HO-2 also needs to take into account, or at least controlled for, the diminished production of reaction products, CO, bilirubin, and iron as well as the unaltered levels of free heme.
Several reasons might explain why generation of iron is not as harmful as free heme. First, unlike Fe2+, free heme is lipophilic, which enhances lipid peroxidation (O'Brien and Little, 1969; Shaklai et al, 1985). Second, in the adult brain, excess iron can be rapidly sequestered by upregulated iron storage proteins such as ferritin, transferrin, and ceruloplasmin (Patel et al, 2002; Regan et al, 2002; Rouault, 2001). Although iron regulatory capacity may be overwhelmed in the setting of injury, which explains why iron chelators such as deferoxamine have showed neuroprotective benefits (Goldstein et al, 2003; Nakamura et al, 2004). Third, some evidence suggests a role for HO-1 in facilitating the transport of iron out of cells. Transfection of HO-1 into HO-1-deficient mice augments iron efflux and protects against stress-induced apoptotic cell death (Ferris et al, 1999).
Another explanation for the disparity between observed HO effects on brain injury may be that the mechanisms of ischemic, hemorrhagic, and traumatic injuries are different enough across models to generate different outcomes. The loads of heme and degradation products in each context are likely unequal. This may be important given the proven concentration-dependent effects of bilirubin, for example, or the limited capacity of iron regulatory proteins to deal with toxic iron. These arguments raise an important point regarding the complex regulation of heme-derived products in the setting of injury. More studies are needed to fully elucidate these issues.
Vulnerability of the Developing Brain to Injury
Traumatic brain injury is the leading cause of pediatric death and disability in the United States (Adelson and Kochanek, 1998). Children less than 4 years of age exhibit worse motor and cognitive function after TBI than older children (Durkin et al, 1998; Ewing-Cobbs et al, 1989; Koskiniemi et al, 1995; Luerssen et al, 1988). Biologie explanations for the increased vulnerability involve oxidative stress, though the precise mechanisms are unclear (Ruppel et al, 2002). Importantly, there are virtually no pharmacologic treatments that are specifically tailored to the brain-injured child (Bayir et al, 2003a, b).
Both clinical findings and experimental models offer insights into the biologic basis for the vulnerability of the developing brain to traumatic injury. Increased F2-isoprostane and protein sulfhydryl, indicators of oxidative stress, and measures of antioxidant reserves, including ascorbate, glutathione, and free ascorbate radical production, have been reported to be reduced in samples of cerebrospinal fluid collected from brain-injured children (Bayir et al, 2002). These clinical observations suggest that increased oxidative stress in concert with compromised antioxidant reserves may be determinants of vulnerability in the developing brain to injury (Bayir et al, 2002).
Reactive oxygen species and the nonradical oxygen metabolite, H2O2, are integral to the pathophysiology of TBI in the adult brain (DeWitt and Prough, 2003; Shohami et al, 1997). The brain is particularly vulnerable to oxidative damage because of its high rate of oxidative activity and relatively low antioxidant capacity. There are several lines of defense against the generation of reactive oxygen species. These include the antioxidants, superoxide dismutase (SOD)—which exists as Cu, Zn-SOD (SOD1) in the cytoplasm and Mn-SOD (SOD2) in the mitochondria—catalase, and glutathione peroxidase. Superoxide dismutase dismutates superoxide radicals to H2O2 whereas catalase and glutathione peroxidase catalyze the breakdown of H2O2 to water and O2.
Activities and the response of these enzymes to oxidative stress differ between the developing and adult brain. Catalase levels, for example, are low embryonically, triple to reach the highest levels between 10 and 16 days of postnatal life and then decrease dramatically in adulthood (Buard et al, 1992; Lazo et al, 1991). Glutathione peroxidase levels are also low in the embryo and neonate, and then gradually increase to reach their maximum levels during adulthood (Buard et al, 1992; Ceballos-Picot et al, 1992; de Haan et al, 1994). Together, these data suggest that the immature brain lacks antioxidant ‘reserves’ and may help to explain the differential susceptibility of the developing CNS to brain injury.
Experimental studies aimed at reducing oxidative damage have shown encouraging results for the mature nervous system (Adelson and Kochanek, 1998; Mikawa et al, 1996). For example, overexpression of SOD1 results in marked neuroprotection after focal and global ischemic insults and after TBI in the adult rodent brain (Chan et al, 1998; Mikawa et al, 1996). These findings, however, are in contrast to those observed after injury to the immature rodent brain (Ferriero, 2001). SOD1 overexpression in the neonatal rodent exacerbates hypoxic—ischemic brain injury (Ditelberg et al, 1996; Fullerton et al, 1998). This pronounced vulnerability is likely because of the imbalanced overproduction of H2O2 from O2•−, and a lack of compensatory protective mechanisms (Fullerton et al, 1998). This important finding suggests differences in the generation of and response to oxidants between the immature and the mature nervous system.
Glutathione peroxidase activity may be a determinant of vulnerability of the developing brain to injury. This enzyme catalyzes the reduction of mitochondrial and cytosolic H2O2 to H2O (Dringen, 2000; Dringen et al, 2000) (Figure 6). This occurs through the oxidation of glutathione. Glutathione levels are then replenished by glutathione reductase at the expense of NADPH. This glutathione redox cycle is an important endogenous mechanism to detoxify reactive oxygen species.
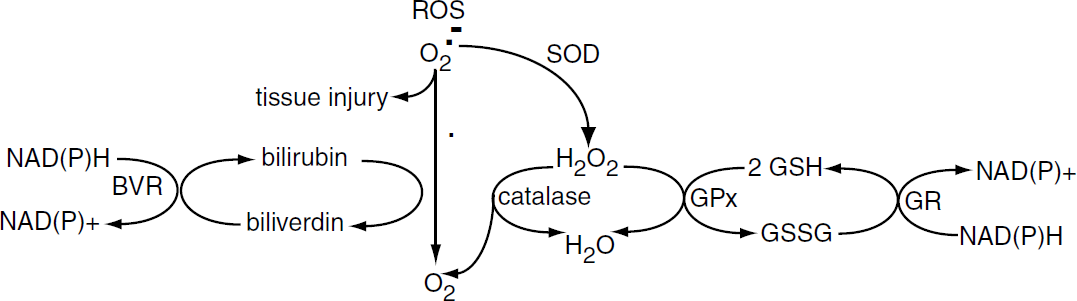
Comparison of bilirubin and glutathione as endogenous brain antioxidants. Reactive oxygen species released after injury are scavenged, protecting cells from oxidative stress, via redox cycling of bilirubin and glutathione-SH (GSH) by biliverdin reductase (BVR) and glutathione reductase (GR), respectively SOD: superoxide dismutase.
There are few studies that have studied glutathione peroxidase in the setting of TBI. Glutathione peroxidase activity is upregulated within the first several days after traumatic injury to the adult rat brain (Goss et al, 1997). Recent studies from our group show a distinct difference in how the immature brain responds to injury. We have compared glutathione peroxidase activity in both the adult and developing murine brain (postnatal day 21) after TBI (Fan et al, 2003). Activity is similar in the uninjured adult and developing brain and significantly increases within the adult brain by 24 h after injury, a finding consistent with that observed in the traumatized rat brain (Goss et al, 1997). In contrast, there is no increase in activity in the traumatized, immature brain. These findings have led us to postulate that the immature brain has a limited compensatory response to injury.
Heme Oxygenase Expression in the Developing Brain
Little attention has been directed at the role of heme metabolism during development. There is clear evidence for both a uterine and placental gestational pattern of expression of HO, which parallels the pattern of expression of growth factors, including vascular endothelial growth factor in the rodent. Such a relationship suggests a role for HO in intrauterine growth (Kreiser et al, 2003).
The developing brain has different expression patterns of HO-1 and HO-2. Baseline levels of HO-1 transcript are very low, and continue to undergo age-dependent decrease (Bergeron et al, 1998; Sun et al, 1990). It is highest in the rat brain at postnatal day 7, decreases 2- to 3-fold by postnatal day 21 and reaches its lowest level by adulthood. Heme oxygenase-1 exhibits a more widespread, regional distribution in the developing brain as compared with the adult (Table 3). Expression is in similar cell types, including neurons and glia in the developing and adult brains, with one exception where vascular expression is limited to the developing brain (Bergeron et al, 1998). Using a transgenic (HO-1-luc) murine model, where the transgene is comprised of the full-length HO-1 promoter driving expression of the reporter gene luciferase (luc) (Zhang et al, 2001), brain HO-1 transcriptional activity, as assessed by in vivo bioluminescence imaging, has been shown to follow a similar developmental expression pattern (Bergeron et al, 1998; Wong et al, 2002). Therefore, it appears that HO-1 expression in the brain is developmentally regulated, but the exact mechanism of this regulation needs to be further studied.
Regional expression of HO-1 during development in gray and white matter


Whereas HO-1 transcription and protein levels decrease with age, HO-2 transcripts and protein gradually increase from postnatal day 1 to adulthood, when they remain stable (Figure 7) (Sun et al, 1990). It is hypothesized that these HO expression patterns arise from age-specific regulation of tissue differentiation, cell population changes, loss of specific HO-1 inducers, or gene silencing (Wong et al, 2002). Heme oxygenase-2 appears to be restricted in expression to neurons in both the developing and adult brain (Ewing and Maines, 1997).
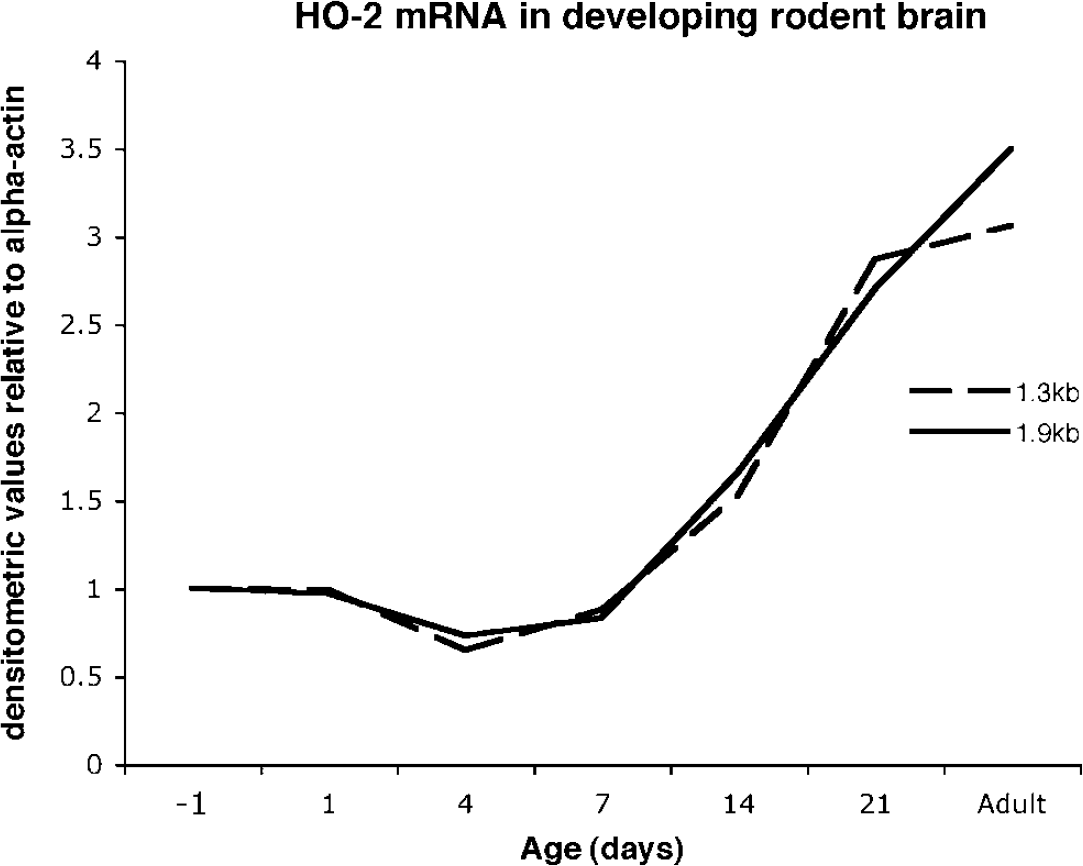
Heme oxygenase (HO)-2 mRNA levels in developing rat brain. Production of HO-2 mRNA is subject to developmental regulation and is present at relatively low levels at birth compared with adulthood (two HO-2 homologous transcripts, dashed line= 1.3 kilobases (kb) transcript, solid line = 1.9 kb). Adapted from Sun el al (1990).
Heme oxygenase-2 activity during development is regulated by adrenal glucocorticoids. Adrenal glucocorticoids follow a similar developmental expression pattern to HO-2, which may be because of the GRE that has been characterized on the promotor region of the HO-2 gene (McCoubrey and Maines, 1994). Administration of corticosterone increases HO-2 mRNA and protein levels in the maturing brain, indicating that the GRE is functional and has the potential to positively regulate HO-2 expression during development (Ewing and Maines, 1997; Maines et al, 1996; Raju et al, 1997). This relationship is important to consider in the highly controversial context of steroid treatment after TBI.
The formation of synapses, myelination, dendritic growth, and vascular proliferation are all crucial aspects of normal maturation of the brain (Dobbing and Sands, 1979; McDonald and Johnston, 1990; Paxinos, 1995; Robertson et al, 1985). How HO interacts with these developmental events is not entirely clear. Studies of HO-1 and HO-2 mutant mice have begun to reveal the respective roles of HO-1 and HO-2 in development. Heme oxygenase-2 mutant mice are fertile, generate expected Mendelian ratios, and are physically indistinguishable from their wild-type littermates. They appear to develop normally, exhibit no overt abnormalities in brain structure and function, and have a normal life expectancy (Poss et al, 1995).
Heme oxygenase-1 mutant mice, however, do not yield the expected Mendelian ratios. Only 20% of homozygous mutant litters survive, thus demonstrating tremendous fragility. Those animals reaching adulthood are anemic. There is a notable reduction in erythrocyte number and size and by 20 weeks of age serum iron values are severely reduced. Mutant animals likewise show accumulation of iron in the kidney and liver and chronic inflammation (Poss and Tonegawa, 1997).
In 1999, a 6-year-old boy was the first human diagnosed with HO-1 deficiency. This individual presented with severe growth retardation, persistent hemolytic anemia characterized by marked erythrocyte fragmentation and intravascular hemolysis, leukocytosis, thrombocytosis, coagulation abnormality, elevated serum levels of haptoglobin, ferritin, and heme, a low serum bilirubin concentration, and hyperlipidemia (Kawashima et al, 2002; Yachie et al, 1999). Structural evidence of genomic exon-deletion mediated by Alu—Alu recombination in a human case with HO-1 deficiency (Saikawa et al, 2000). The observed growth retardation, anemia, and abnormal iron deposition are all characteristics seen in HO-1-deficient mice. However, in the human there was a more severe involvement of endothelial cells and the reticuloendothelial system, resulting in intravascular hemolysis, disseminated intravascular coagulation, and amyloidosis. This is in contrast to the predominant iron metabolic disorders observed in HO-1-deficient mice.
The Developing Brain: Iron and Iron-Management Proteins
The functional significance of HO during maturation of the brain should be considered in the context of the developmental regulation of the end-products of heme metabolism, including iron and bilirubin as well as the iron-management proteins ferritin and transferrin. Each is critical to development and most likely influences the overall vulnerability of the developing brain to injury.
Iron
Iron, a universal constituent of molecules generating mitochondrial energy, is important for growth and cellular differentiation (Connor et al, 2001; Roskams and Connor, 1994). It is heterogeneously distributed in the adult brain with highest concentrations in white matter relative to gray matter and in subcortical regions, including the globus pallidus, substantia nigra, and dentate nucleus, relative to the cortex (Connor et al, 1995a). Importantly, this distribution is not apparent until the lattermost stages of brain development. During early postnatal development, iron is used for mitochondriogenesis, neuronal maturation, synthesis of neurotransmitter, and myelination (Taylor and Morgan, 1990). Iron first appears in microglia within myelinogenic foci in white matter (Connor et al, 1995a). With maturation, this localization shifts from microglia to oligodendrocytes. Such a shift coincides with the role of iron in the production of myelin and the maturation of myelin-producing oligodendrocytes (Connor and Menzies, 1996; Hulet et al, 1999). Myelination in the rodent peaks between postnatal day 8 and 14, a period when the transport of iron across the blood—brain barrier is also maximal (Connor and Menzies, 1996; Crowe and Morgan, 1992; Moos and Morgan, 2001; Taylor et al, 1991; Taylor and Morgan, 1990). It is thought that oligodendrocytes, in close proximity to the micro-vasculature, may accumulate iron that has been transported across the barrier (Connor et al, 2001).
Transferrin
Regulation of iron in the brain is in part governed by transferrin, an iron mobilization protein, the transferrin receptor, and ferritin, the iron storage protein (Banks et al, 1988; Burdo et al, 1999; Dwork, 1995; Jefferies et al, 1984). Iron that is not in use by the cell is bound to intracellular ferritin, for reutilization or long-term storage (Connor et al, 1994; Drysdale, 1988; Seligman and Huebers, 1987).
The developmental protein expression of transferrin parallels that of iron. Transferrin is highest at birth in the cerebral cortex, midbrain, cerebellum, and pons of the rodent, decreases to a minimum between postnatal day 12 and 24, and then increases until adulthood (Roskams and Connor, 1994) (Figure 8). The decrease in transferrin during development of the brain coincides with maturation of the blood—brain barrier. This temporal relationship implicates plasma as the vehicle for transferrin during early postnatal development. The subsequent increase in transferrin after establishment of the blood—brain barrier is attributed to its local production. Transferrin mRNA and protein are localized to oligodendrocytes and epithelium of the choroid plexus (Connor et al, 2001). Although the function of transferrin in oligodendrocytes has not been fully elucidated, its expression corresponds with the developmental pattern of myelination (Connor et al, 2001).
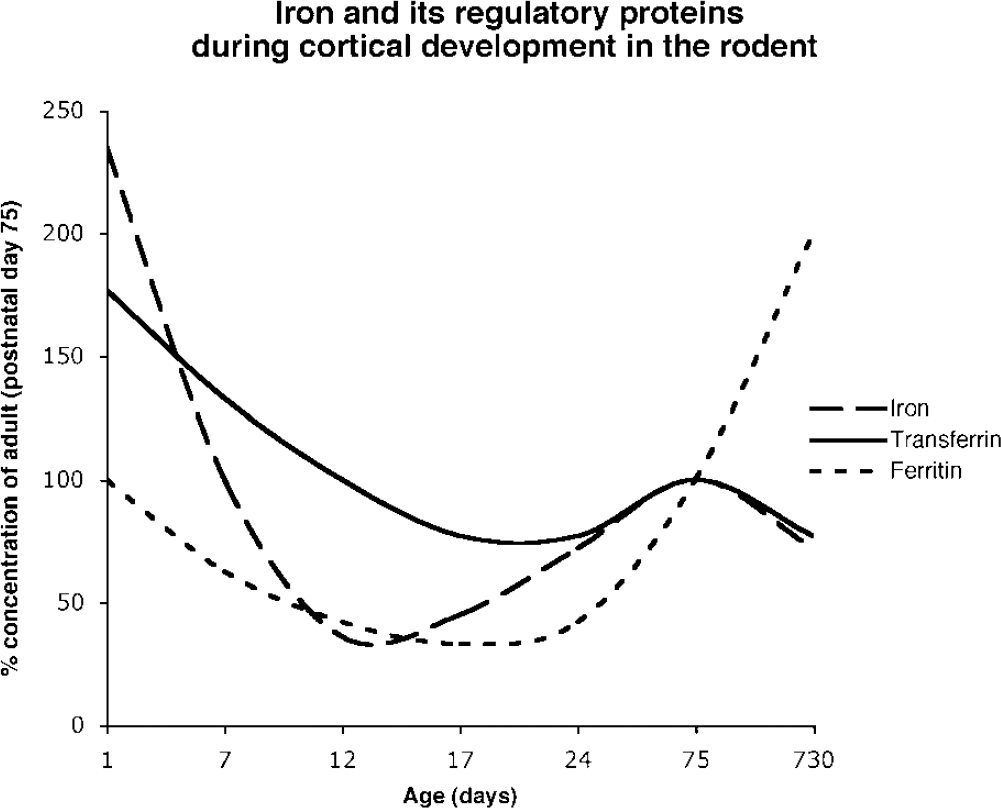
Relative concentrations of iron and iron-regulatory proteins (transferrin and ferritin) during development in the rodent cortex (compared with adult). Ferritin, the principal binding protein for iron, is at its lowest concentration in the brain during the third postnatal week. Adapted from Roskams and Connor (1994).
The transferrin receptor is developmentally regulated in the endothelium, epithelium, and neurons (Moos et al, 1998). Transferrin receptor immunor-eactivity in the brain capillary endothelial cells peaks at postnatal day 21; whereas, a constant level of expression is found in the epithelium of the choroid plexus throughout development. The receptor is primarily localized to gray matter rather than white matter. Immunoreactivity in neurons is apparent as early as birth and reaches maximal levels at postnatal day 21 (Moos et al, 1998). Oligodendrocytes, which express the highest levels of iron in the brain, may not play a significant role in iron delivery given the preferential distribution of the transferrin receptor in the gray matter (Moos et al, 1998).
Ferritin
The concentration of ferritin in the brain at birth approximates that of the adult. There is a sharp decline during development, reaching the lowest values between postnatal days 17 to 24. Thereafter, protein levels increase to adult levels (Moos et al, 1998; Percy et al, 1998; Roskams and Connor, 1994). This age-dependent increase in ferritin occurs in both the rodent and human brain (Connor et al, 1995b; Focht et al, 1997).
Assembled in the cell cytosol, ferritin has two subunits (L-ferritin and H-ferritin) with distinct functions and cellular distributions. Neurons contain abundant H-ferritin, which is important for rapid uptake and reutilization of iron. In contrast, L-ferritin, involved in long-term iron storage, is the dominant form in microglia (Levi et al, 1992). Both L- and H-ferritin are expressed in oligodendrocytes. During early brain development, ferritin is initially expressed in microglia. The onset of myelination coincides with a switch in localization from microglia to oligodendrocytes (Cheepsunthorn et al, 1998).
Bilirubin
In human newborns, approximately 80% to 90% of bilirubin originates from the degradation of heme from the hemoglobin of senescing red blood cells and the remainder is attributed to the catabolism of hemoproteins (Berk et al, 1974; Mancuso, 2004). Bilirubin is typically bound to plasma albumin in the blood stream during the immediate postnatal period. In utero, unconjugated bilirubin is actively transported across the placenta to the maternal circulation where it is conjugated and excreted. Directly after birth, the newborn experiences a large increase in the load of unconjugated bilirubin because of the reduced lifespan of fetal erythrocytes (Landaw, 1988). Unconjugated or free bilirubin in the absence of insufficient albumin is lipid soluble and can be neurotoxic (Shapiro, 2003). Physiologic jaundice in human infants is a consequence of immaturity of the hepatic enzyme uridine diphosphoglucuronate glucuronosyltransferase in concert with an increased hemoglobin load (Shapiro, 2003). By 2 weeks, bilirubin conjugation and subsequent hepatic glucuronide excretion and subsequent enteral hydrolysis is comparable to the adult where the plasma concentration of unconjugated bilirubin is stabilized below 20 μmol/L (Ostrow et al, 2002).
Heme Oxygenase and Neonatal Traumatic Brain Injury
There are few studies that have examined HO expression after injury to the immature brain (Acarin et al, 2002; Bergeron et al, 1997). Heme oxygenase-1 is induced in macrophages and glia in the ipsilateral cortex, hippocampus, and striatum within the first week after neonatal hypoxia/ischemia (Bergeron et al, 1997). A similar cellular pattern of HO-1 induction has been showed after excitotoxic injury in the immature rodent brain (Acarin et al, 2002). Importantly, induction of HO-1 does not coincide with a change in HO activity (Bergeron et al, 1997). Rather, HO enzymatic activity remains unchanged in the ipsilateral cortex and subcortical regions compared with that of the contralateral hemisphere or uninjured, control brains. This increase in HO-1 protein in the absence of a change in enzymatic activity has several possible interpretations. Because both HO-1 and HO-2 contribute to HO activity, it is possible that after injury neuronal loss leads to a reduction in HO-2 activity. Such a reduction may not be reflected in a change in overall HO activity, as increased activity of HO-1 may provide adequate compensation and offset this reduction. Alternatively, it is also possible that HO-2, which is the primary source of HO activity in the uninjured brain (Chang et al, 2003), remains the dominant source of HO activity in the injured brain. In this context, any increases in HO-1 activity, which are expected to be relatively small, may be masked by that contributed by HO-2.
In the developing brain, TBI generates pronounced intraparenchymal bleeding and overt tissue destruction similar to that observed in the adult brain. Collectively, these events likely contribute to the accumulation of heme. Generated iron may then react with H2O2 to form the hydroxyl radical and/or with lipids to generate alkoxy and peroxy radicals. Free radical generation can be particularly damaging to the immature brain because of a high concentration of lipids, high rate of oxygen consumption, and decreased antioxidant defense, which all can lead to damage to immature white matter (Ames et al, 1993; Halliwell 1992; Watson, 1993).
Anatomical studies show iron accumulation in the injured, developing brain. There is increased iron deposition in the basal ganglia, thalami, and white matter in children with severe ischemia/anoxia (Dietrich and Bradley, 1988). Similar findings of excess iron deposition have been observed in brain regions that are immediately adjacent to the site of maximal tissue injury after experimental hypoxia/ischemia at postnatal day 7 (Cheepsunthorn et al, 2001; Palmer et al, 1999). Moreover, there is a rapid appearance of iron staining (as early as 1 h recovery) in the developing brain, exposed to hypoxia/ischemia compared with a more delayed onset in the adult (Kondo et al, 1995; Palmer et al, 1999). Although the biologic basis for this difference is unclear, it is nevertheless intriguing as it may offer an explanation for the heightened vulnerability of the immature brain to injury.
There are few studies that have examined the pathogenicity of iron after TBI. Pharmacologic strategies to chelate iron have offered some insight. Deferoxamine, which scavenges ferric iron as well as the hydroxyl radical and the peroxynitrite anion, improves spatial memory in the adult rodent after TBI (Long et al, 1996) and improves forelimb grip (Panter et al, 1992). Oxidative stress is reduced in cultures of neurons and astrocytes that have been treated with deferoxamine. (Regan and Rogers, 2003). In the developing brain, there is histopathologic evidence for neuroprotection in rodents that have been treated with deferoxamine (Palmer et al, 1999, 1994; Sarco et al, 2000).
Experimental exposure to iron in the neonate results in abnormal motor development and cognitive deficits that are apparent later in adult life (Fredriksson et al, 1999). There are a number of studies that have addressed how iron may influence brain development. After a hypoxic/ischemic insult, instead of the normal switch of iron staining from microglia to oligodendrocytes, iron staining remains in microglia. It is thought that impaired myelination results from the sequestration of iron by microglia, thus limiting availability to oligodendrocytes (Cheepsunthorn et al, 2001; Connor and Menzies, 1996; Hulet et al, 1999).
The cytotoxic effects of iron are typically limited by iron-management proteins, including transferrin and ferritin. Their effectiveness may be determined by their relative activities and cellular distribution. At postnatal day 21, neither transferrin nor ferritin have reached adult concentrations (Roskams and Connor, 1994). Whether or not this relatively lower level of expression confers vulnerability is not clear. Ferritin expression is prolonged in microglia; whereas, its expression is delayed in oligodendrocytes after neonatal hypoxia/ischemia (Cheepsunthorn et al, 2001; Connor et al, 1995b). This most likely accounts for the shift in iron staining from oligodendrocytes to microglia in the injured immature brain. The preferential expression of ferritin in microglia also coincides with the induction of HO-1 in microglia in neonatal hypoxia/ischemia (Bergeron et al, 1997). The presence of both ferritin and HO-1 in microglia raises some uncertainty as to how microglia may function in the injured brain. One scenario may be that microglia serve to sequester iron and thus reduce iron-related toxicity. Such a protective response, however, may be dependent on the relative level of HO-1 activity, where high levels of HO-1 may actually reverse cytoprotection. This has been shown to be the case in an experimental model of hyperoxia-induced oxidant stress where high HO-1 expression is associated with the accumulation of reactive iron (Suttner and Dennery, 1999).
The importance of antioxidants may vary according to the maturational state of the brain. It has been suggested that bilirubin, which is lipophilic, may protect membranes from lipid peroxidation (Baranano et al, 2002). Thus, there may be greater dependence on bilirubin to protect a ‘myelinating’ brain from reactive oxygen species. Inadequate levels of bilirubin in the injured brain during this critical period of development could alter ongoing myelination and/or promote demyelination. In vitro studies support this possibility. Bilirubin, at relatively low concentrations, significantly reduces H2O2-mediated toxicity in cultured oligodendrocytes as compared with cultures treated with either vehicle or the antioxidant α-tocopherol (Liu et al, 2003). However, the protective potential of bilirubin is limited. At high concentrations, free bilirubin can act as a potent neurotoxin (McDonald et al, 1998; Shapiro, 2003, 2005).
Heme Oxygenase as a Therapeutic Target After Injury to the Adult and Developing Brain
Research on heme regulation by HO isozymes has elucidated new aspects of oxidative stress in the pathophysiology of TBI. Whereas many studies suggest a protective function for HO against cellular loss, further studies are required to evaluate complex functional outcomes such as behavioral recovery. Future studies will need to characterize in more detail the effects of HO compartmentalization in the brain. Moreover, significant research still needs to be performed in understanding the implications of developmental differences for HO-mediated effects. Finally, future research will need to concentrate on the most important components of the heme regulation pathway for targeted neuroprotection therapy.