
Abstract
Heme and iron metabolism are of considerable interest and importance in normal brain function as well as in neurodegeneration and neuropathologically following traumatic injury and hemorrhagic stroke. After a cerebral hemorrhage, large numbers of hemoglobin-containing red blood cells are released into the brain's parenchyma and/or subarachnoid space. After hemolysis and the subsequent release of heme from hemoglobin, several pathways are employed to transport and metabolize this heme and its iron moiety to protect the brain from potential oxidative stress. Required for these processes are various extracellular and intracellular transporters and storage proteins, the heme oxygenase isozymes and metabolic proteins with differing localizations in the various braincell types. In the past several years, additional new genes and proteins have been discovered that are involved in the transport and metabolism of heme and iron in brain and other tissues. These discoveries may provide new insights into neurodegenerative diseases like Alzheimer's, Parkinson's, and Friedrich's ataxia that are associated with accumulation of iron in specific brain regions or in specific organelles. The present review will examine the uptake and metabolism of heme and iron in the brain and will relate these processes to blood removal and to the potential mechanisms underlying brain injury following cerebral hemorrhage.
The role of iron and heme in the brain has been reviewed several times in recent years (Connor and Menzies, 1995, 1996; Connor et al., 2001). However, the fast pace of gene discovery has revealed additional genes related to the transport and metabolism of heme and iron in the brain and other tissues. Examples of such genes include the divalent cation transporter (DCT-1; also known as DMT-1), stimulator of iron transport (SFT), paraferritin (a complex of an integrin, mobilferrin, and flavin monooxygenase), lactoferrin and its receptor, melanotransferrin (MTf), ceruloplasmin (CP), the hemachromatosis gene, the heme oxygenase (HO)-3 gene, and hephaestin (Conrad and Umbreit, 2000; Feder, 1999; Maines, 1997; McKie et al., 2000; Ponka, 1999, 2002; Qian and Shen, 2001; Rolfs and Hediger, 1999; Wessling-Resnick, 1999). Furthermore, in the last few years, several new heme and iron transport proteins were identified: ferroportin-1, IREG-1, and MTP-1 (Abboud and Haile, 2000; Donovan et al., 2000; McKie et al., 2000). CD163, a haptoglobin-heme receptor (Kristiansen et al., 2001), and an adenosine triphosphate (ATP)-requiring iron transporter that participates with HO to export intracellular iron (Baranano et al., 2000). Neuroglobin, a new oxygen carrying hemoglobin that is localized to the central nervous system (CNS), has been described (Burmester et al., 2000).
Moreover, the role for HO in brain heme and iron metabolism is being elucidated. HO is the enzyme that metabolizes heme to biliverdin, iron, and carbon monoxide (Maines, 1997). This enzyme function is carried out by three separate genes that code for three separate proteins, HO-1, HO-2, and HO-3 (Maines, 1997). The parallel between the nitric oxide (NO) producing enzyme nitric oxide synthase (NOS) and the carbon monoxide (CO) producing enzyme HO has been noted by several investigators (Maines, 1997; Snyder et al., 1998). Also, HO-1 appears to be an important protein in iron efflux from cells (Ferris et al., 1999).
The metabolism of iron and heme is of special interest for the brain. Some strokes are associated with intracerebral (ICH) or subarachnoid (SAH) hemorrhage and the release of large amounts of hemoglobin into the extracellular spaces. Removal requires the transport and metabolism of heme and the metabolism of the iron released during heme metabolism. In addition, some degenerative diseases like Alzheimer's and Parkinson's diseases and Friedrich's ataxia are associated with accumulation of iron in specific brain regions or in specific organelles. Thus, this review will examine the uptake and metabolism of heme and iron in the brain and will also relate these processes to the mechanisms of blood removal following hemorrhagic stroke. We will review the extracellular and intracellular sources of both iron and heme and their transporters at the cell and mitochondrial membranes. There are different HO isozymes and different iron metabolic proteins in the various cell types in the brain. Unfortunately, there are many gaps in our knowledge of brain iron metabolism that can be filled only by currently available information from other organs. Lastly, because of space limitations, we have frequently cited review articles rather than the original publications.
HEME METABOLISM
Hemopexin/hemopexin receptor: Extracellular heme transporter
Hemopexin is the intravascular protein that binds free heme (Tolosano and Altruda, 2002). Hemopexin protects cells lacking hemopexin receptors by tightly binding heme and abrogating its deleterious effects and preventing nonspecific heme uptake. Another heme binding protein in blood is albumin, but it has a lower affinity for binding heme. Thus, albumin in the extracellular space could also bind free heme from hemorrhage or heme released from dying cells (Grinberg et al., 1999).
The hemopexin/heme complex binds to specific hemopexin receptors, which shuttle the heme into cells (Tolosano and Altruda, 2002). This shuttle process involves the stimulation of endocytosis by the heme/hemopexin complex after receptor binding with release of heme within the cell (Smith and Hunt, 1990). Hemopexin is retained within the endocytotic vesicle and returned to the cell membrane where it is released outside the cell. Intracellular heme induces HO-1 and down regulates the transferrin receptor (TfR), the latter indirectly through heme degradation and iron responsive proteins (IRPs) (Alam and Smith, 1989) (Fig. 1). Biochemical responses that occur in cells with hemopexin receptors upon encountering heme-hemopexin were recently described (Eskew et al., 1999).
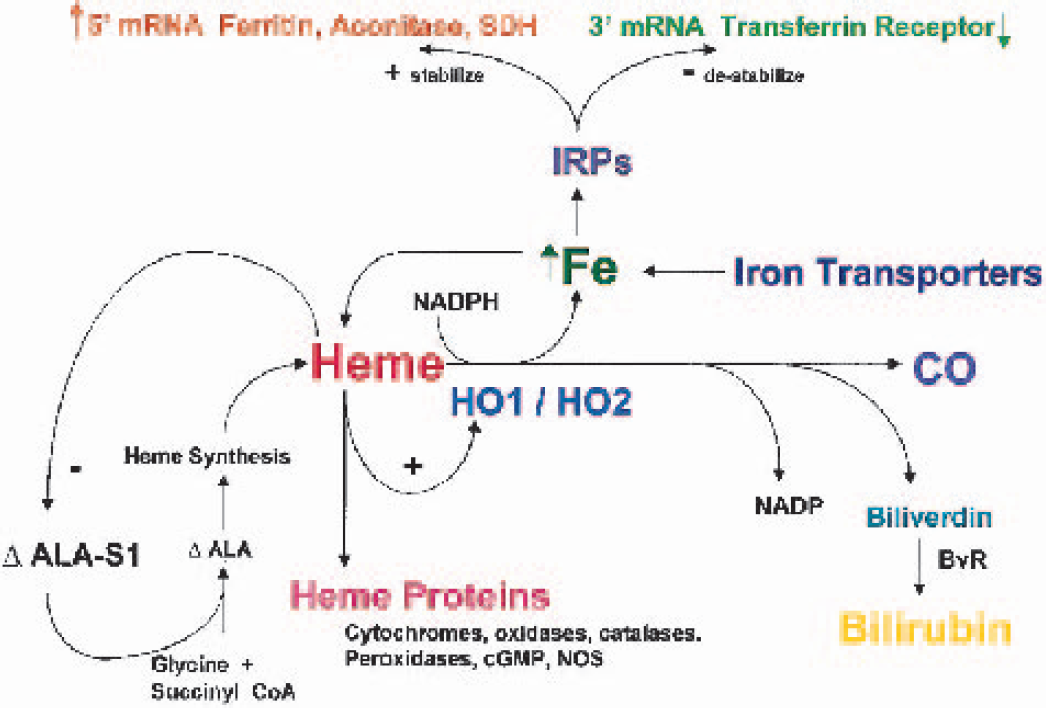
Increased intracellular iron levels act at the posttranscriptional level through iron response proteins (IRPs) to stabilize ferritin mRNA, thereby promoting iron storage and simultaneously destabilizing transferrin receptor mRNA to decrease iron (FE) uptake. Diagram also depicts heme synthesis regulated through 5-aminolevulinic acid synthase. Heme degradation occurs by constitutive and inducible heme oxygenase (HO) isoforms, HO-2 and HO-1, respectively. HO-2 and HO-1 degradation of heme generates biologically active products including iron, carbon monoxide (CO), and bilirubin from biliverdin by biliverdin reductase (BvR). Heme acts directly on delta-5-aminolevulinic acid synthase (ΔALA-S)1 to reduce synthesis and on heme oxygenase to stimulate its degradation. Heme acts indirectly through iron release from HO and IRPs to affect ferritin and transferrin receptor synthesis. SDH, succinate dehydrogenase; ΔALA = delta amino levulinic acid; NADP, nicotinamide adenine dinucleotide phosphate, CoA, coenzyme A; cGMP, cyclic guanosine monophosphate; NOS, nitric oxide synthase.
Hemopexin and heme transport into the brain
Heme enters the extracellular space in brain when cells die, be they neurons or glia. In addition, when hemorrhages occur in the parenchyma or in the SAH space, the hemoglobin released from red cells forms a massive load of extracellular heme. These heme molecules must be bound extracellularly, transported into cells by way of transporters, and metabolized within the cell by HO (Sung et al., 2000) (Figs. 2 to 4).
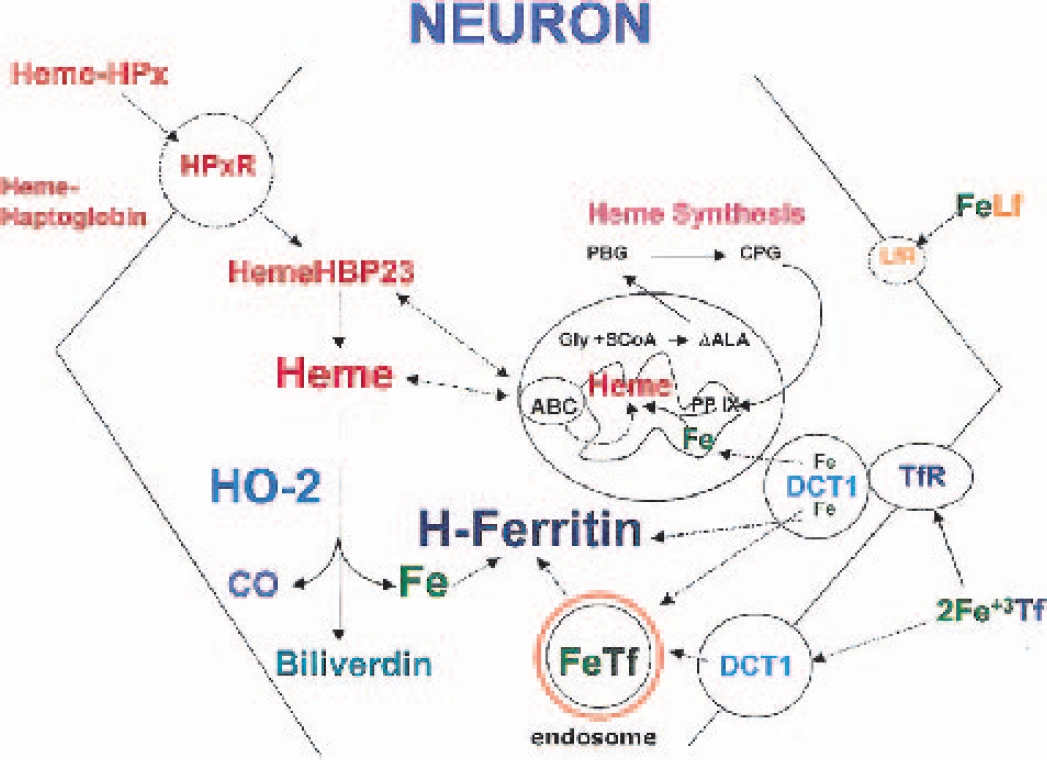
Proposed scheme for iron (FE) and heme metabolism in neurons. Iron bound to extracellular transferrin can be transported into neurons through transferrin receptors (TfR) coupled to the divalent cation transporter (DCT)-1. Intracellularly, iron bound to transferrin/TfR is located in endosomes. Lactoferrin receptors (LfR) on neurons serve in the transferrin-independent pathway to transport iron from iron containing lactoferrin across neurons membranes. Intracellular iron can also be generated after heme degradation by constitutive heme oxygenase (HO)-2 present in neurons. This iron is likely to be immediately bound to H-ferritin, the predominant isoform in neurons. Extracellular heme can be transported into neurons either bound to hemopexin (HemeHPx) or haptoglobin and transported to the cytoplasm by way of the hemopexin receptor (HPxR). Intracellular heme bound to heme binding proteins (HBP)23 can be transported into the mitochondria for cytochrome synthesis by protein chaperones (ABC iron transporters). Heme can also be synthesized by a pathway that includes both cytoplasmic and mitochondrial enzymatic steps. ΔALA, delta amino levulinic acid; PBG, porphobilinogen; CPG, coproporphyrinogen; PPIX, protoporphyrin IX; CO, carbon monoxide; FeTf, iron transferrin; Gly, glycine.
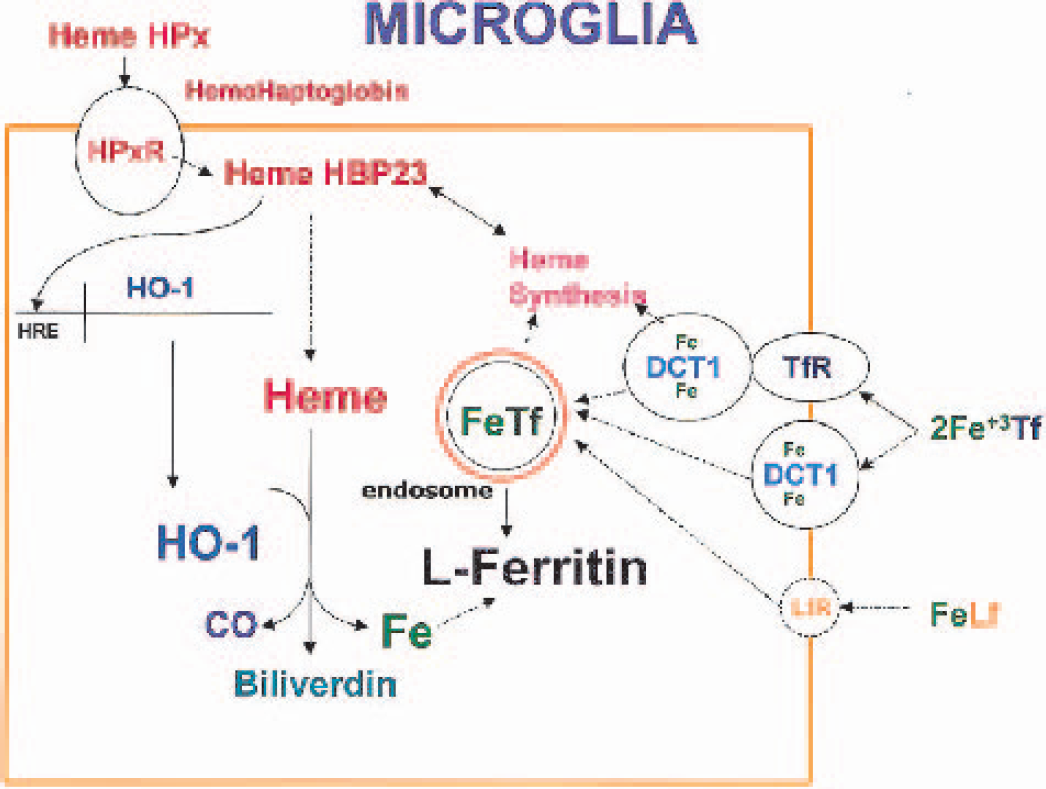
Proposed scheme for iron (FE) and heme metabolism in microglia. Extracellular heme from red-cell breakdown following hemorrhagic stroke can be transported into microglia and then bound by intracellular heme binding proteins. Increased intracellular heme levels can lead to heme oxygenase (HO)-1 induction through a heme response element (HRE) in the HO-1 promotor. In microglia L-ferritin is the predominant isoform for intracellular iron binding. Microglia also can transport iron intracellularly by transferrin-dependent and independent pathways. HPx, hemopexin; HPxR, hemopexin receptor; CO, carbon monoxide; FeTf, iron transferrin; DCT, divalent cation transporter; TfR, transferrin receptors; LfR, lactoferrin receptor; HBP, heme binding protein; FeLf, iron lactoferrin.
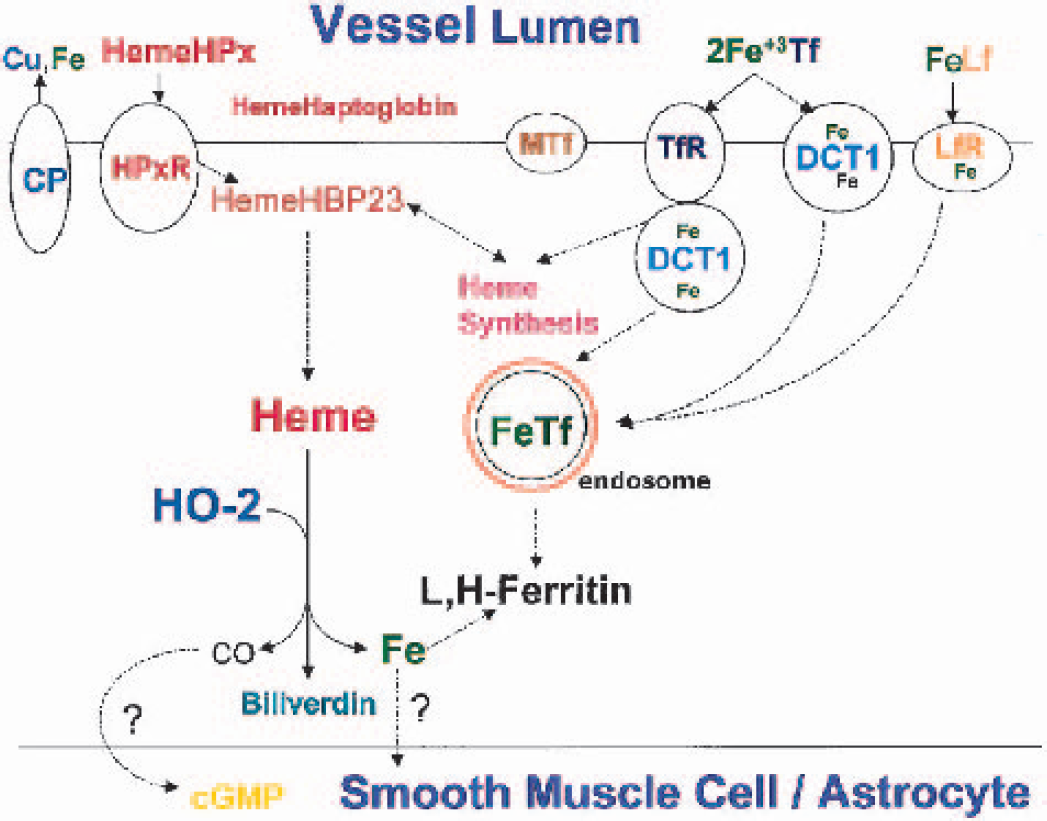
Proposed scheme for iron (Fe) and heme metabolism in endothelial cells. Cerebral vessel endothelial cells contain many of the same iron- and heme-related proteins as neurons and microglia. Ceruloplasmin (CP) is the copper-containing oxidase that transports copper and is also crucially involved in facilitating iron egress from brain. Melanotransferrin (MTf) is another iron-binding protein in transferrin-independent pathway for iron uptake. Recent findings demonstrate that mutations in this gene result in progressive degeneration of the basal ganglia and retina in association with iron deposition in these regions. Carbon monoxide (CO) generated from heme oxygenase (HO)-2 activity can activate guanylate cyclase leading to cyclic guanosine monophosphate (cGMP) generation. cGMP regulates cerebral vessel diameter through its effects on vascular smooth muscle cells. Cu, copper; HPx, hemopexin; HPxR, hemopexin receptor; HBP, heme binding protein; TfR, transferrin receptors; DCT, divalent cation transporter; FeLf, iron lactoferrin; LfR, lactoferrin receptor; FeTf, iron transferrin; MTf, melanotransferrin.
Hemopexin, which is found in vessels throughout the body (Balla et al., 1993), is reported to be present in brain vessels but not in the brain parenchyma (Camborieux et al., 1998). The mRNA for hemopexin is detected in the neural retina (Hunt et al., 1996) and cultured photoreceptors but not in pigment epithelial cells (Chen et al., 1998). Hemopexin mRNA is expressed by most cells of the neural retina including the photoreceptors and, notably, the ganglion neuronal cells (Chen et al., 1998). In the retina, hemopexin protects against heme-mediated toxicity (Hunt et al., 1996). The blood-retinal barrier, consisting of retinal pigment epithelial cells and retinal endothelial cells, prevents hemopexin and haptoglobin, anti-oxidant protective plasma proteins normally synthesized by the liver, from entering the neural retina. If present, these proteins must, therefore, be made locally.
It is possible that hemopexin mRNA might be induced in the injured brain in microglia and not be detectable in the normal brain, much like HO-1. The hemopexin promoter drives reporter-gene expression in the brain, suggesting that hemopexin is found in the brain (Tolosano and Altruda, 2002). Heme and hypoxia do not induce hemopexin (Wenger, 2002). One study did find that neurons and scattered glial cells in the human brain immunostained for hemopexin, whereas oligodendrocytes and choroid plexus did not (Morris et al., 1993). In injured peripheral nerves, hemopexin is upregulated in the Schwann cells, fibroblasts, and macrophages (Madore et al., 1999). Hemopexin synthesis is controlled by interleukin (IL)-6 that is produced in various cell types that also produce hemopexin. IL-6 control is through both paracrine and autocrine mechanisms (Camborieux et al., 2000).
Uptake of heme/hemopexin complexes into macrophages (Liem et al., 1975) suggests that the brain macrophage, the microglial cell, may also take up heme/hemopexin complexes (Fig. 3). Heme is probably transported in microglia in the brain. The presence of hemopexin in neurons in one study suggests that neurons can also transport heme proteins in and out of the cell. Sharp and colleagues have shown that biotinylated hemoglobin is taken up into both neurons and microglia throughout the entire brain (Turner et al., 1998). These results suggest that hemoglobin is transported into these cells, and if so, it is likely that hemopexin mediates this heme transport. Our data suggest that hemopexin receptors should be sought on capillary endothelial cells, microglia, and possibly on neurons. A unique polymorphism has been found in hemopexin in blacks (Kamboh et al., 1993) that might be of importance for ICH and SAH hemorrhage.
The heme/hemopexin complex regulates metallothionein gene expression found in brain blood vessels. Heme-hemopexin or cobalt protoporphyrin-hemopexin (a model ligand for hemopexin receptor occupancy) increases transcription of the metallothionein (MT)-1 gene. A small region of the murine MT-1 promoter is sufficient to increase transcription in response to heme-hemopexin. Protein kinase C but not protein kinase A inhibitors block this MT-1 transcription as do N-acetylcysteine, glutathione, superoxide dismutase, and catalase. These and additional studies suggest that heme-hemopexin induces MT-1 by way of antioxidant response elements and metal/heme response elements in the MT-1 promoter (Ren and Smith, 1995).
Haptoglobin
The mRNAs for haptoglobin are also detected in the neural retina and cultured photoreceptors but not in pigment epithelial cells (Chen et al., 1998). In situ hybridization showed that haptoglobin mRNA was located principally in the photoreceptor cells, cells of the inner nuclear layer, and some cells of the ganglion cell layer. The role of haptoglobin in the brain for binding extracellular heme proteins is uncertain, but it is present and does bind free heme proteins in other tissues (Balla et al., 1992).
Recently, a macrophage hemoglobin scavenger receptor (CD163) that binds complexes of haptoglobin and hemoglobin was reported (Kristiansen et al., 2001). This receptor is an acute phase-regulated and signal-inducing protein and was present after intravascular hemolysis. After receptor binding and endocytosis, the complex is digested and releases heme that is metabolized by HO. The activation of this system may also trigger an antiinflammatory cytokine response.
Heme binding proteins: Intracellular
Heme inside the hemopexin/hemopexin receptor/endosome complex is released into the cytoplasm where it can be bound directly by HO or possibly by a heme-binding protein (HBP) like HBP23 (Ponka, 1999) (Fig. 2). Heme transported into macrophages may be bound by HBP23. Lipopolysaccharide stimulates induction of HBP23 in liver Kupffer cells by way of an NO-mediated mechanism (Ponka, 1999). Pre-induction of HBP23 protects Kupffer cells against hydrogen peroxide (Immenschuh et al., 1999). It will be important to determine whether HBP23 is expressed in microglia, particularly after injury. HBP23 is induced by chromium mesoporphyrins, other metal protoporphyrins, as well as protoporphyrin IX (Ponka, 1999).
There are other known HBPs found in a variety of different cells (Ponka, 1999). It is possible that different HBPs might be present in the cytoplasm of different cells in the brain and therefore be quite specialized like the iron-transport proteins and heme-metabolizing proteins. For example, HO-3 is homologous to HO-1 and HO-2 but does not appear to have HO enzymatic activity (Maines, 1997). It is possible that HO-3 could serve as an intracellular transport and chaperone protein, chaperoning heme either to HO-1 and HO-2 or to the mitochondria for import into mitochondria.
Recently, a novel gene named ckSoul that is strongly expressed in the retina and pineal gland of chicken was discovered using two-tissue suppression subtractive hybridization. The protein product of ckSoul is similar to a novel HBP (p22 HBP) and to an uncharacterized mammalian gene in the expressed sequence tag database. The mouse transcript of this new gene is expressed in the retina and may represent the mammalian ortholog of ckSoul (Zylka and Reppert, 1999).
HasA is a recently characterized bacterial protein involved in heme acquisition and iron metabolism that can bind free heme as well as capture it from hemoglobin (Izadi et al., 1997). HasAh, an HasA homologue or a protein that shares a common epitope with HasA, is reported to be abnormally localized to the neurofibrillary pathology in Alzheimer's disease (AD) but not in normal-appearing neurons in AD brains or in age-matched controls (Castellani et al., 2000). The authors hypothesize that this novel HBP may contribute to oxidative stress through its ability to bind heme and render iron available for free radical generation through the Fenton reaction.
Mitochondrial heme transporters
In bacteria, there are a series of protein chaperones (ABC proteins) that bind heme and transport it into mitochondria for participation in cytochrome c maturation (Goldman and Kranz, 2001). In addition, a heme transporter is necessary for the synthesis of cytochrome c. Cytochrome c maturation in Escherichia coli requires the “ccm operon,” which encodes eight membrane proteins (CcmABCDEFGH). Some of the mammalian ABC mitochondrial heme transport protein homologues have been cloned. Mutation of the ABC mitochondrial heme transporter results in marked iron accumulation in the mitochondria and oxidative damage. This scheme of heme trafficking in bacteria provides the framework for a series of mitochondrial chaperones that must also exist in mammalian mitochondria that shuttle heme in and out of mitochondria during synthesis and degradation of mitochondrial heme/cytochrome proteins (Goldman and Kranz, 2001) (Fig. 2).
Heme synthesisz
The bulk of the heme required for heme proteins like the cytochromes probably comes from de novo synthesis of heme. Heme is a complex of iron with protoporphyrin IX. Heme is a major component of heme proteins such as hemoglobin, myoglobin, cytochromes, gyanylate cyclase, and NOS. There are great differences in rates of heme synthesis between different organs, with erythroid cells having the highest rates, with the liver being second. There are two genes for 5-aminolevulinic acid synthase, the first enzyme in heme synthesis (Ponka, 1999). The S1 gene is ubiquitous and in the brain, whereas the S2 gene is found only in red cells. Iron controls heme synthesis in red cells because of IRP binding to an iron-responsive element (IRE) in the 5′ untranslated region (UTR) of the 5-aminolevulinic acid synthase-2 mRNA. Iron does not appear to regulate heme synthesis in nonerythroid cells. Instead, heme feeds back on 5-aminolevulinic acid synthase-1 to inhibit the regulatory enzyme (Ponka, 1999). In the brain, most heme synthesis is accounted for by mitochondrial cytochromes and iron-binding proteins in neurons and glia (Figs. 1 and 2).
Eight enzymes are required to synthesize heme, four in the mitochondria and four in the cytoplasm (Ponka, 1999). The first enzyme, delta amino levulinic acid synthase, is found in the mitochondria, forming delta amino levulinic acid (ΔALA). Porphyrobilinogen is then formed in the cytoplasm from ΔALA. Two more enzymes are required to synthesize coproporphyrinogen, which is then transported back into the mitochondria. Ferrochelatase adds iron to protoporphyrin IX to form heme (Ponka, 1999). Why some steps of heme synthesis are in the mitochondria and some are in the cytoplasm is unknown. This series of complex steps suggest that there are heme/porphyrin and iron transporters in mitochondrial membranes as noted above and below.
Heme metabolism
Heme inside any given cell could be derived from either endogenous or exogenous sources. Endogenous sources of heme in neurons and glia would be derived mainly from cytoplasmic heme proteins and mitochondrial cytochromes and would be involved in the normal turnover of the heme-containing proteins. Exogenous sources of heme could be derived from the death of neighboring cells that would release their heme proteins or from heme derived from hemoglobin that occurs following ICH or SAH in brain. The heme would then be metabolized by HO in equimolar amounts to biliverdin, carbon monoxide, and iron (ferrous) (Maines, 1997). The CO produced could act on cyclic guanosine monophosphate within the cell or immediately adjacent cells (Durante and Schafer, 1998) (Fig. 4). The iron released following the HO reaction becomes associated with mobilferrin and paraferritin in gastrointestinal cells, which act as ferrireductases to make iron available for production of iron-containing end products such as heme proteins (Umbreit et al., 1998; Uzel and Conrad, 1998). Though it is unknown what protein(s) bind the iron released by HO metabolism in the brain, it is likely that the iron is bound by H/L ferritin as discussed below (Figs. 2 to 4). The biliverdin formed is metabolized to bilirubin, which serves as an antioxidant but can be toxic to the newborn brain.
HO-1 and heme metabolism
Though HO-1 is a HO enzyme, it is also known to be a heat shock protein, HSP32 (Dwyer et al., 1992; Ewing and Maines, 1991; Maines, 1997). The HO-1 promoter contains two heat shock elements, at least one of which binds heat shock factor and makes the gene responsive to heat shock and other stresses that denature proteins within cells. HO-1 mRNA and activity can be increased several fold by heme (Tyrrell, 1999), other metalloporphyrins, and transition metals (Elbirt and Bonkovsky, 1999). The mouse HO-1 gene has five exons and four introns. A single major transcription initiation site is used for its constitutive and heme or metal induced expression. Basal promoter activity is localized within 149 bp of the coding sequence. There are many consensus regulatory elements in the 5′ UTR of HO-1, including activator protein (AP)-1, metal responsive element, oncogene c-myc/max heterodimer binding site, antioxidant response element where NFκB acts (Prestera et al., 1995), and GC box binding (Sp1) sites (Elbirt and Bonkovsky, 1999). The activator protein-1 site in HO-1 could mediate Jun induction of HO-1 in dying cells (Matsuoka et al., 1999). Nrf2, a cap ‘n’ collar leucine zipper transcription factor, also regulates induction of the HO-1 gene (Alam et al., 1999). NO and NO donors are capable are inducing HO-1 protein expression (Hanson and Leibold, 1999) possibly by way of NFκB (Foresti and Motterlini, 1999).
Mild hypoxia does not induce HO-1 in the brain (Bergeron et al., 1997), although hypoxia can induce HO-1 in the lung by way of a hypoxia-inducible factor (HIF)-dependent and independent mechanism (Hartsfield et al., 1999). HO-1 is induced by hypoxia by way of a HIF-independent mechanism in other cells as well (Wood et al., 1998).
Inhibition of mitochondrial electron transport induces HO-1 (Elbirt and Bonkovsky, 1999). Using deletionreporter gene constructs, arsenite-dependent induction of HO-1 was mediated by extracellular signal-regulated kinase and p38 (a homologue of the yeast HOG-1 kinase) but not c-jun N-terminal kinase, mitogen-activated protein kinase pathways (Elbirt and Bonkovsky, 1999). Inhibiting the endoplasmic reticulum calcium ATPase with thapsagargin markedly induces HO-1 (Gissel et al., 1997). This has important implications for ischemia where calcium accumulation in the endoplasmic reticulum could signal many stress responses (Gissel et al., 1997).
HO-1 in vessels and endothelium
The induction of HO-1 in vessels is of interest because HO enzymes release CO (Fig. 4). In addition, HO-1 induction is regulated in part by the vascular transcription factor Ets family of proteins (Deramaudt et al., 1999). TNF-α and IL-1β induce HO-1 in endothelial cells by way of protein kinase C, calcium, and phospholipase A2 (Terry et al., 1999). Iron induces HO-1 in pulmonary endothelium (Fogg et al., 1999) as does peroxynitrite (Foresti and Motterlini, 1999). Pre-induction of HO-1 decreased apoptosis in endothelial cells produced by peroxynitrite (Foresti and Motterlini, 1999). Induction of HO-1 in aorta with free hemoglobin decreases vasoreactivity (Gaine et al., 1999). HO-1 knockout mice develop heart failure during chronic hypoxia, suggesting that HO-1 is important for cardiovascular compensation to chronic hypoxia (Yet et al., 1999). Transfection of HO-1 into endothelial cells using viral vectors protects endothelial cells against oxidant-mediated injury (Yang et al., 1999).
Mildly oxidized LDL from phospholipids also induces HO-1 in smooth muscle cells of vessels (Ishikawa et al., 1997). Shear stress can also induce HO-1, providing a possible mechanism where increased shear stress could lead to local vasodilation by way of induction of HO-1 and increased production of CO. Thiol compounds interact with NO to induce HO-1 (Foresti and Motterlini, 1999), and the interactions of eNOS and HO-1 suggest that both could regulate endothelial responses (Seki et al., 1997).
HO-2 (and HO-1) in the brain blood vessels may play a role in vascular tone in resistance vessels (Kozma et al., 1999). HO-2 has been reported to control vasoactive responses by the release of CO in the carotid of the developing brain (Leffler et al., 1999). In addition, HO might play a role in coupling either cell activation or cell injury to increases of blood flow because inhibiting CO production decreased blood flow increases produced by kainate-induced seizures (Montecot et al., 1998). The nonspecific effects of metalloprotoporphyrins, however, has made it difficult to interpret in vivo studies of CO production from vessels and other tissues (Leffler et al., 1999).
HO-1 induction by cerebral ischemia
Heat shock induces HO-1 in the brain and other tissues (Dwyer et al., 1992; Ewing and Maines, 1991). Focal ischemia and global ischemia induce HO-1 in the brain (Geddes et al., 1996; Koistinaho et al., 1996; Matsuoka et al., 1998b; Nimura et al., 1996; Paschen et al., 1994; Takeda et al., 1994, 1996). After focal ischemia, the HO-1 protein is induced in vessels in the core of the infarct (Nimura et al., 1996). HO-1 protein is also induced in neurons, astrocytes, and microglia at the margins of the infarct in regions outside the infarction (Geddes et al., 1996; Nimura et al., 1996). In addition, HO-1 is induced in the cortex at some distance from any infarction including the entorhinal cortex, cingulate cortex, and occipital cortex. This diffuse cortical induction occurs mainly in microglia (Nimura et al., 1996). This diffuse induction is likely caused by spreading depression induction of HO-1 that can be modulated by serum glucose (Koistinaho et al., 1999). The mechanism of HO-1 induction by spreading depression could be by way of c-fos/jun induction by spreading depression and its actions on activator protein-1 sites in the HO-1 promoter (Elbirt and Bonkovsky, 1999).
After global ischemia, HO-1 is transiently expressed in pyramidal neurons in the cortex and hippocampus (Matsuoka et al., 1998b; Takeda et al., 1996). However, it is expressed mainly in microglia at later times, and mainly in microglia that express major histocompatibility complex class II antigens (Matsuoka et al., 1998b).
The role of HO-1 in ischemic injury is unclear. In the HO-1 transgenic mouse stroke, sizes are smaller compared with wild-type controls (Panahian et al., 1999), suggesting HO-1 induced protection. However, this was an unusual mouse because the HO-1 gene was under the control of a neuron-specific enolase promoter (Panahian et al., 1999). This would have targeted the HO-1 to neurons that already have HO-2 present in the cell. Therefore, increasing HO activity in neurons can apparently improve stroke. However, in mice with knockouts of the HO-1 gene, the HO-1 knockout mice have similar infarct volumes to wild-type mice (Dore et al., 1999a). The failure to detect an effect in the HO-1 knockout mouse could be due in part to the fact that HO-1 is expressed at barely detectable levels in the normal brain and hence might not have an effect related to acute stroke because it is induced over many hours after the stroke. Induction of HO-1 with hemin protects the cortex and striatum but not the hippocampus from injury produced by global ischemia (Takizawa et al., 1998). Increasing HO-1 in cultured neurons can protect them against Abeta peptide and against hydrogen peroxide (Le et al., 1999). However, although HO-1 generally protects cells, marked overexpression of HO-1 in cells can lead to iron accumulation and their death (Suttner and Dennery, 1999). This may help explain why inhibiting HO-1 with protoporphyrins in some cells protects against injury (Dwyer et al., 1998). This could relate to the levels of iron-binding and ironstorage proteins because cells with high ferritin levels would tend to be protected against high HO-1, whereas those with lower levels of iron-binding proteins might be more vulnerable (Jelinski et al., 1999).
HO-1 induction by oxidative stress
HO-1 expression appears to be an excellent marker of oxidative stress related to cell injury in the brain (Sharp, 1995). Glutathione depletion induces HO-1 in the brain (Ewing and Maines, 1993), possibly being mediated by way of an NF-κB site in the HO-1 gene (Lavrovsky et al., 1993). HO-1 is induced following a variety of injuries that increase oxidative stress, including focal ischemia, global ischemia, kainate induced injury, traumatic injury (Dwyer et al., 1996; Fukuda et al., 1996; Matsuoka et al., 1998a) and cell injury, and death related to N-methyl-D-aspartate receptor antagonists (Rajdev et al., 1998). In addition, antioxidants decrease HO-1 expression caused by SAH-induced ischemic injury (Turner et al., 1999), and antioxidants decrease HO-1 induction produced during cell injury produced by MK-801 and phencyclidine (Rajdev et al., 1998).
HO-1 is also induced in the brain in a number of disease states that are associated with increased oxidative stress. HO-1 is induced in the midbrain in Parkinson's disease (Schipper, 2000) and areas of amyloid deposition and plaque formation in AD (Markesbery, 1997; Schipper 2000).
Heme metabolism by HO-2 in the brain
HO-2 is present chiefly in the brain and testes and, unlike HO-1, appears to be induced by only a single class of molecules, the adrenal glucocorticoids (Maines, 1997). The HO-2 transcript found in testis is unique to this organ (Liu et al., 2000). HO-2 is constitutively expressed in most neurons in the brain (Verma et al., 1993; Vincent et al., 1994) and therefore is always present to metabolize heme molecules derived from endogenous cytochromes and other heme proteins as well as from any exogenous source. Neuronal HO-2 is inducible by glucocorticoids by way of a glucocorticoid response element in its promoter, particularly in neonatal rats (Maines 1997). Developmentally linked induction of HO-2 expression by these steroids in neurons of the motor and cognition systems may affect brain growth and differentiation (Maines, 1997).
The rat-brain HO-2 gene has five exons and four introns. The promoter contains a glucocorticoid response element but no heat shock elements, accounting for its failure to respond to heat shock (Ewing and Maines, 1991, 1995). HO-2 is encoded by two mRNA transcripts, 1.3 and 1.9 kb, and the transcripts are the products of a single gene and differ in the use of the polyadenylation signal (Maines, 1997). A 24 amino acid region is found in HO-2 that exhibits nearly complete similarity in predicted secondary structure and is found in the rabbit, rat, mouse, and human HO-1 and HO-2 genes. It is believed that this peptide mediates the heme-binding and isomerspecific tetrapyrrole cleavage activities of the HO enzymes (Maines, 1997).
HO-2 appears to play a protective role during ischemia because mice with knockouts of the HO-2 gene (Poss et al., 1995) have larger infarction volumes than wild-type controls (Dore et al., 1999a). The protective effects of HO-2 could be due in part to the antioxidant properties of biliverdin/bilirubin produced by HO-2 because bilirubin can protect neurons against oxidative stress (Dore et al., 1999b). If true, it is difficult to understand how inhibiting HO activity with protoporphyrins protected the brain against stroke in at least one study (Kadoya et al., 1995). This could be because of nonspecific effects of these compounds on NOS and other molecules.
The HO-3 isoform
HO-3 is the third HO gene to be identified (Maines, 1997). It appears that this gene does bind heme, but that it does not have active HO activity. The role of HO-3 in the brain and other tissues remains to be clarified. It could serve as an HO under particular circumstances or in some cells, or it might serve as an intracellular heme chaperone binding heme delivered by the hemopexin receptor to mitochondria, to HO-1 or HO-2, or to the nucleus where heme would induce HO-1.
Heme and iron regulation of HO-1 and ferritin
The inhibitor of heme degradation, tin mesoporphyrin IX, reduces the ability of exogenous hemin to induce ferritin synthesis but enhances HO synthesis (Eisenstein and Blemings, 1998). The iron chelator Desferal suppresses the ability of hemin to induce synthesis of ferritin but not of HO. The heme synthesis inhibitor succinylacetone does not block iron induction of ferritin synthesis. Inorganic iron significantly induces the synthesis of ferritin but not of HO. Increasing delta-aminolevulinic acid to stimulate heme synthesis represses the ability of inorganic iron to induce ferritin synthesis while activating HO synthesis. The data suggest that release of iron by HO plays an essential role in the induction of ferritin synthesis by heme, and chelatable iron (non-heme–derived iron) can regulate ferritin synthesis independently of heme formation (Eisenstein and Blemings, 1998).
Neuroglobin
Recently, a third type of oxygen-carrying globin protein termed “neuroglobin” is reported to be expressed at small but differential levels throughout the brain in mice and humans (Burmester et al., 2000). Expression in the cortex is four times higher than in the hippocampus. Neuroglobin is the first hexacoordinate hemoglobin to be described in vertebrates. This structural similarity is present in plant and bacterial hemoglobins (Trent et al., 2001). The primary amino acid sequence of neuroglobin is only about 25% identical to hemoglobin and myoglobin, but the physical structure of the protein and its heme pocket predict that in sufficiently high local concentration in neurons or surrounding cells neuroglobin could act as a reservoir to augment oxygen supply to meet the increased demand for oxygen during neural activity (Trent et al., 2001). Interestingly, neuronal globins are also present in various invertebrates and in the mollusk Tellina alternata; nerve excitability is dependent on the oxygen stored in neuronal globin.
Neuroglobin expression is increased by neuronal hypoxia in vitro and focal cerebral ischemia in vivo and helps to promote neuronal survival (Sun et al., 2001). In addition, these authors showed that neuronal survival after hypoxia was reduced by inhibiting neuroglobin expression with an antisense oligodeoxynucleotide and enhanced by its overexpression. Neuroglobin's protective effect appears to be specific for hypoxia versus other stressors, e.g., binding NO.
IRON METABOLISM
The regions of the brain with the highest levels of iron include the basal ganglia, substantia nigra, and deep cerebellar nuclei (Connor and Menzies, 1995; Koeppen, 1995). Oligodendrocytes have the highest levels of iron of any cell type in the brain (Connor and Menzies, 1996). This may help explain why these cells are particularly sensitive to oxidative stress (Smith et al., 1999). Exposure of cultures to xanthine/xanthine oxidase systems leads to selective degeneration of oligodendrocytes, presumably by way of the peroxide radical. The oligodendrocyte xanthine/xanthine oxidase and glucose/glucose oxidase induced cell death can be prevented by catalase, suggesting that hydrogen peroxide production is particularly damaging to oligodendrocytes because superoxide dismutase (SOD) and other radical scavengers do not protect. Oligodendrocytes are also sensitive to glutamate, an injury also mediated by free radicals (Oka et al., 1993).
Concentrations of brain iron are highest at birth (Connor et al., 1994), decrease during the first 2 weeks of life, and then steadily increase throughout life. This steady increase of iron suggests that the brain does not have ready means of clearing iron out of cells once it accumulates and also suggests that the brain may be subjected to greater oxidative stress with age.
Iron responsive proteins and iron response elements
Cellular iron uptake and storage are regulated by cytoplasmic proteins called iron regulatory proteins-1 (IRP-1) and −2 (IRP-2) that act at the posttranscriptional level (Aisen et al., 1999; Eisenstein and Blemings, 1998; Haile, 1999) (Fig. 1). IRP-1 and IRP-2 detect levels of iron, and when iron is scarce, bind to stem-loop structures in target mRNAs known as IRE on the 5′ UTR of ferritin and related mRNAs and the 3′ UTR of the TfR and related mRNAs. Such binding inhibits translation of the ferritin mRNA and stabilizes the TfR mRNA (Ponka et al., 1998). This increases iron uptake and decreases iron storage. The opposite situation occurs when iron is in excess (Strahan et al., 1992).
Multiple genes are regulated by the IRPs. Iron scarcity induces binding of IRPs to the single IRE in ferritin, erythroid 5-aminolevulinic acid synthase, aconitase, and succinate dehydrogenase mRNAS to suppress translation initiation (Aisen et al., 1999; Eisenstein and Blemings, 1998; Haile, 1999). Simultaneous interaction of IRPs with multiple IREs in the 3′ UTR of the TfR mRNA selectively causes its stabilization. The DCT-1, described below, also has an IRE in its 3′ UTR and is likely upregulated by binding of IRPs (Aisen et al., 1999; Rolfs and Hediger, 1999). Iron can also modulate the expression of amyloid precursor protein possibly by way of an unconfirmed IRE in the amyloid precursor protein mRNA (Tanzi and Hyman, 1991). The iron responsiveness of TfR has led to the routine measurement of the TfR in anemias because serum TfR is elevated when whole-body iron decreases (Cook, 1999).
The iso-IRPs have sequence homology to the aconitases, and at least one iso-IRP can be converted to an aconitase (Beinert and Kennedy, 1993). Factors that target iso-IRE/iso-IRP interactions in mRNA include environmental iron, oxygen and reoxygenation, NO, hydrogen peroxide, ascorbate, growth factors, and protein kinase C-dependent phosphorylation of IRPs (Theil, 2000). IRP-1, which can function as an aconitase, is regulated by iron sulfur clusters, with iron stimulating disassembly of the iron-sulfur cluster of IRP-1 (Gruer et al., 1997). IRP-2 is regulated directly by iron, being rapidly degraded in the presence of iron (Rouault and Klausner, 1997; Theil, 2000). IRP-1 is the same molecule as mitochondrial aconitase, except that IRP-1 is an alternative cytosolic form of (not mitochondrial) aconitase that is devoid of its cubane Fe-S cluster (Gruer et al., 1997; Rouault and Klausner, 1997; Theil, 2000). In most cell lines tested, levels of IRP-2 are inversely regulated by iron levels because of iron-dependent regulation of the half-life of the protein. In addition to changes in total amounts of IRP-2, binding to IREs by IRP-2 can also vary up to fourfold in the absence of any change in IRP-2 protein levels. Lastly, several studies show that heme can regulate the levels of IRP-1 and IRP-2, providing one of the most direct links between heme and iron metabolism (Fig. 1).
Recently, LaVaute et al. (2001) reported that mice with a targeted disruption of the gene encoding IRP-2 (Ireb-2) misregulate iron metabolism in the intestinal mucosa and the CNS. In adulthood, Ireb-2(−/−) mice develop a movement disorder characterized by ataxia, bradykinesia, and tremor. Iron accumulates in white matter tracts and nuclei throughout the brain several months before the onset of neurodegeneration and movement disorder symptoms. Ferric iron accumulates in neuronal cytosol and oligodendrocytes in distinctive brain regions. Abnormal accumulations of ferritin colocalize with iron accumulations in populations of neurons that degenerate, and iron-laden oligodendrocytes accumulate ubiquitinpositive inclusions. Thus, misregulation of iron metabolism leads to neurodegenerative disease in Ireb-2(−/−) mice and may contribute to the pathogenesis of comparable human neurodegenerative diseases.
IRPs, NO, HO, and oxygen
The iron regulatory proteins (IRP-1 and IRP-2) are sensitive to NO synthesis and oxidative stress. IRP-1 possesses a redox-active Fe-S cluster and exhibits aconitase activity (Bouton, 1999). IRP-2 has redox-sensitive cysteine residues. Under proper redox conditions, both IRPs bind to IREs in iron-responsive target genes (Bouton, 1999). Therefore, peroxynitrite and reactive oxygen species may stimulate the translation of iron-binding proteins and iron-transport proteins directly (Bouton, 1999; Hanson and Leibold, 1999).
In addition, molecular oxygen may also directly activate IRPs (Hanson and Leibold, 1999). This raises the possibility that one of the IRPs could serve as an oxygen sensor and could regulate HIF or endothelial Per/Arnt/Sim (PAS) domain protein mRNA stability or translation. Activation of N-methyl-D-aspartate receptors stimulates IRP binding to mRNA by way of actions of NO on the Fe-S cluster (Bouton, 1999; Hanson and Leibold, 1999).
IRP-1 and IRP-2 are found in the brain (Hu and Connor, 1996). IRP-2 but not IRP-1 is upregulated in Alzheimer brain tissue in regions of neuronal inclusions and other areas of redox sensitive iron. IRP-1 appears to be the predominant species in the human brain and can bind to brain ferritin mRNA (Hu and Connor, 1996).
NO and iron
NO interactions with iron are some of the most important biological reactions in which NO participates (Cooper, 1999). Reversible binding to ferrous haem iron is responsible for the observed activation of guanylate cyclase and inhibition of cytochrome oxidase. Unlike CO or oxygen, NO can also bind reversibly to ferric iron. The latter reaction is responsible for the inhibition of catalase by NO. NO reacts with the oxygen adduct of ferrous haem proteins (e.g., oxyhemoglobin) to generate nitrate and ferric heme. This reaction is responsible for the majority of NO metabolism in the vasculature. NO can also interact with iron-sulphur enzymes (e.g., aconitase, nicotinamide adenine dinucleotide dehydrogenase). It is likely that NO metabolites, including peroxynitrite and nitroxyl anion, also have important interactions with iron (Cooper, 1999). The NO-generating agent sodium nitroprusside releases iron from the iron storage protein ferritin. This can be prevented by the NO scavenger hemoglobin. Therefore, NO generation in vivo could lead to the mobilization of iron from ferritin, disrupting intracellular iron homeostasis and increasing the level of reactive oxygen species (Cooper, 1999).
Iron transport
Transferrin
The production of transferrin by the choroid plexus would ensure that any iron released during secretion of CSF would be rapidly bound by transferrin. The transferrin in CSF is saturated with iron (Bradbury, 1997). Transferrin is synthesized by cells in the choroid plexus that secrete transferrin into the CSF (Connor and Menzies, 1995). The synthesis of transferrin by the choroid plexus is regulated by serotonin and likely other neurotransmitters by way of cyclic adenosine monophosphate. Transferrin is necessary for the transport of iron but not manganese across the blood-brain barrier, and there is a transferrin-independent uptake mechanism for iron in the choroid plexus. In addition, these data suggest that endogenous synthesis of transferrin is necessary for iron transport from the choroid plexus (Connor and Menzies, 1995, 1996).
The role of transferrin in transporting iron into the brain has been investigated in hypotransferrinemia, a genetic defect in mice resulting less than 1% of normal plasma transferrin concentrations. With dietary iron deficiency, tissue iron concentrations decreased, whereas brain concentrations remained the same or increased. These results imply that there is an alternative iron delivery system to the brain (Connor and Menzies, 1995) as described below. There were no differences in tissue distribution of 54Mn despite the differences in circulating Tf concentrations and body-iron stores, suggesting that non–Tf- dependent mechanisms for Mn transport. Iron deficiency upregulates ferritin protein, possibly by way of an IRP-related mechanism (Moos et al., 1999).
Hypotransferremic mice are hypomyelinated, suggesting that transferrin and iron may be essential for the formation of normal myelin (Connor and Menzies, 1996). The brain is the only organ where a postnatal increase of transferrin mRNA occurs, and this occurs mainly in oligodendrocytes (Connor and Menzies, 1996). Any role for transferrin in the intracellular trafficking of iron is unknown.
Transferrin can also be synthesized throughout all brain regions following injury (Kondo et al., 1995). Transferrin protein, which migrates as a 80 kDa band on Western blots, increases in rat brain following occlusion of the middle cerebral artery (Lu et al., 1999). Immunohistochemically, transferrin protein is found in the periinfarct region and in hippocampal CA1 and dentate gyrus ipsilateral to the middle cerebral artery occlusion (Lu et al., 1999). Neurons and astrocytes can also express transferrin protein in vivo (Dwork et al., 1988). Neurons, astrocytes, and oligodendrocytes express transferrin mRNA in vitro. Transferrin is markedly induced in microglia following a variety of brain injuries including ischemia (Kondo et al., 1995).
Transferrin is upregulated in microglia as well as astrocytes and oligodendrocytes following motor neuron injury (Tornquist et al., 1997). Cytokines, including TNF-α, IL-1, and gamma interferon, induce transferrin in macrophages (Djeha et al., 1995), suggesting they might do the same in microglia and suggesting that transferrin might be an NF-κB target gene.
Transferrin receptors
Within the brain, parenchyma TfRs are highest on neurons using both immunocytochemical and autoradiographic techniques (Connor and Menzies, 1995, 1996). Of interest is the finding that the density of TfRs correlates with the density of cytochrome staining in the brain (Morris et al., 1994a, 1994b), a finding that is consistent with the proposition that most of the iron used in neurons would be mostly bound in cytochromes in mitochondria (Morris et al., 1994a, 1994b). TfRs are upregulated on regenerating motor neurons following axotomy. Iron uptake increases in these motor neurons as well. Loss of dopamine neurons following treatment of mice with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) results in loss of TfRs in the midbrain, the loss presumably occurring because of the neuronal loss. Patients with Parkinson's disease and AD have loss of TfRs on neurons (Morris et al., 1994a, 1994b).
TfRs are often not detectable on astrocytes, microglia, and oligodendrocytes using immunocytochemical methods in the normal brain (Moos, 1996). Cultured astrocytes, however, have TfRs on their membrane surface (Qian and Shen, 2001). Normal oligodendrocytes have been shown to express low levels of TfRs using sensitive techniques (Connor and Menzies, 1995, 1996; Morris et al., 1994a). Moreover, with disease, there is upregulation of TfRs on the oligodendrocyte, e.g., the periplaque region in multiple sclerosis plaques contains TfR-positive oligodendrocytes (Hulet et al., 1999). TfRs are markedly upregulated on macrophages and microglia following ischemia and hypoxia (Kaur and Ling, 1999) (Fig. 3).
DCT-1 (DMT-1, NRAMP-2)
The DCT-1 protein (called natural-resistance-associated macrophage protein [NRAMP]-2 in humans) (Conrad et al., 1999) was cloned using a functional expression system (Gunshin et al., 1997). Mutations in this gene appear to explain the microcytic anemia in mk mice and in the Belgrade b rat (Aisen et al., 1999). DCT-1 is a 561 amino acid protein with 12 membrane-spanning domains that couples iron transport with proton import. DCT-1 also transports other divalent cations, including Zn2+, Mn2+, Co2+, Cd2+, Cu2+, Ni2+, and Pb2+ (Gunshin et al., 1997). DCT-1 is ubiquitously expressed, including in the duodenum and the brain.
DCT-1 is upregulated by dietary iron deficiency and could be an important mediator of intestinal iron absorption. DCT-1 is likely regulated by way of an IRP-related mechanism (Aisen et al., 1999), which is consistent with the presence of one IRE in the 3′ UTR region of the DCT-1 mRNA (Gunshin et al., 1997). DCT-1 is a member of the “natural-resistance-associated macrophage protein” (NRAMP) family, and thus its properties may provide insight into how these proteins confer resistance to pathogens (Gunshin et al., 1997).
DCT-1 plays a role in iron transport by way of transferrin and TfR because DCT-1 localizes to endosomes and likely transports ferrous iron out of endosomes. DCT-1 is also expressed on the plasma membrane of many cells and could account for some proportion of TfR-independent transport of iron into the brain and among cells within the brain (Fillebeen et al., 1999) (Figs. 2 to 4). Two NRAMP proteins have been identified, NRAMP-1 and NRAMP-2, though only NRAMP-2 appears to transport iron. NRAMP-1 is found only on macrophages, and therefore might be localized to microglia as well.
The Belgrade, which has a mutation of DCT-1, has low iron levels in virtually all tissues in the body (Wood and Han, 2000). Iron is abnormal in neurons and oligodendrocytes but not astrocytes in the brain of this animal (Moos et al., 1999). This suggests that iron is transported into neurons and oligos by way of DCT-1, but that the DCT-1 is not found on astrocytes or is not prominent (Moos et al., 1999). Iron (and manganese) has been shown to enter brain astrocytes by way of a non–transferrin-dependent mechanism (Takeda et al., 1998). Therefore, iron transport into astrocytes could occur by way of SFT or the non–transferrin-related mechanisms described below. It is likely that the DCT-1 is also present on brain capillary endothelial cells because DCT-1 is a component of the Tf-TfR endosome transport pathway in capillary endothelial cells (Fig. 4).
SFT, like DMT-1, is localized to endosomes, although it may only transport FeII in endosomes. SFT expression stimulates iron assimilation from transferrin (Aisen et al., 1999). It appears that SFT can transport iron across endosomal membranes as well as across the external cell membrane (Aisen et al., 1999). Therefore, at least two cation transporters exist that can transport iron across cell and endosomal membranes. Because SFT is associated with TfR iron transport, it is probably localized to endothelial cells and neurons, although this has not been confirmed.
Brain ferritin: Possible extracellular transport function
The fact that TfRs are located mainly in gray matter, whereas white matter has the highest concentrations of iron, had remained an enigma until recent studies from the Connor group (Hulet et al., 1999). They discovered that there are ferritin binding sites in the human brain, that these are found primarily in white matter tracts, and therefore could provide a mechanism for delivery of iron to oligodendrocytes by way of extracellular iron bound to ferritin (Hulet et al., 1999). Ferritin binds to human brain tissue in a competitive and saturable manner with a dissociation constant of 0.35 nM and a binding site density of 116.7 fmol/mg protein (Hulet et al., 1999). Binding is much higher in white matter compared with gray matter in both human and mouse brain (Hulet et al., 1999). A cellular ferritin receptor could be very important because ferritin is capable of delivering 2000 times more iron than transferrin (Hulet et al., 1999).
There is transferrin bound iron, ferritin bound iron, and other small fractions of iron found in CSF (Moos and Morgan, 1998). This supports an extracellular role for ferritin as well as an intracellular role (Moos and Morgan, 1998). Moreover, it appears that transferrin bound iron in extracellular spaces would primarily enter neurons, whereas ferritin bound iron would primarily enter oligodendrocytes (Hulet et al., 1999).
Brain melanotransferrin and lactoferrin
MTf, discovered in melanoma cells, has one intact transferrin-like (FeIII) binding site but, unlike transferrin, is linked to cell surfaces with a glycosylphosphatidylinositol anchor (Wessling-Resnick, 1999). Over expression of MTf in Chinese hamster ovary (CHO) cells shows that it can be internalized in cells and transport iron intracellularly (Wessling-Resnick, 1999). Whether this protein is an important iron transporter in any normal tissue remains to be determined.
MTf (also called p97) is found on normal capillary endothelial cells (Qian and Shen, 2001). This protein appears to facilitate iron entry into endothelial cells by way of a pathway independent of the TfR. MTf can be membrane bound, i.e., on brain capillaries, but can also be found in a soluble form. MTf can transport iron through brain capillaries in a pathway parallel to the transferrin/TfR pathway. In the diseased Alzheimer brain, microglia also express MTf (Qian and Shen, 2001), perhaps in response to an overload condition.
Lactoferrin is another transferrin homologue found in milk, neutrophils, the intestine, and the brain (Wessling-Resnick, 1999). Lactoferrin, like transferrin, appears to bind ferric iron, binds to a lactoferrin receptor, and stimulates endocytosis and delivery of iron intracellularly (Wessling-Resnick, 1999). Lactoferrin receptor is a 105 kD glycoprotein similar to TfR (Qian and Shen, 2001).
Lactoferrin is found in brain capillary endothelial cells and transports iron into endothelial cells (Fillebeen et al., 1999). Cultured brain endothelial cells have lactoferrin receptors (Fillebeen et al., 1999). A specific unidirectional transport of iron occurs by way of a receptor-mediated process with no apparent intraendothelial degradation. Lactoferrin release of iron occurs in bovine brain endothelial cells (Fillebeen et al., 1999). The Lf transport of iron can be stimulated with TNF-α (Fillebeen et al., 1999). Lactotransferrin receptor has also been localized to capillary endothelial cells and to some neurons in the brain of humans, and lactoferrin receptor increases in the midbrain of Parkinson's patients.
AGE (protein modified by advanced glycosylated end-products) receptors are found on monocytes, macrophages, endothelial cells, pericytes, podocytes, astrocytes, and microglia. Advanced glycosylated endproduct-modified proteins also bind to lysozyme and lactoferrin, lactoferrin found in vessels, and in macrophages. Whether lactoferrin receptors are upregulated in brain microglia or macrophages remains to be determined, although there is one report of lactoferrin receptors on some glial cells (Thornalley, 1998).
Iron transport into mitochondria
Two proteins, called Mmt1p and Mmt2p, are proposed to be iron transport proteins in the yeast inner mitochondrial membrane (Goldman and Kranz, 2001). There must be similar transporters that shuttle iron from ferritin and possibly endosomes into the inner mitochondrial membrane of mammalian cells (Wessling-Resnick, 1999). The human ABC7 protein, a homologue of the yeast Atm1p mitochondrial iron transporter, may serve to transport iron into mitochondria in mammals (Goldman and Kranz, 2001).
IRON STORAGE: FERRITIN
Biology
Ferritin is the main iron storage molecule found in cells (Aisen et al., 1999). Mammalian ferritin is a heteropolymer of 24 subunits made up from H (for heart or heavy) and L (liver or light), being designated either in the organ where they predominate or for their molecular weights of 21,000 or 19,000. The ratio of H and L subunits varies among organs and in the brain varies between cell types. The H-chain subunit is involved in oxidizing ferrous ions to ferric ion, whereas L-chain subunits are more involved with pore formation and storage. Ferritins can contain as many as 4000 iron atoms as ferrihydrite (FeOOH), although they typically have around 2000 iron atoms (Aisen et al., 1999).
The H subunit has a ferrioxidase center that binds to ferrous dimers and catalyzes their oxidation by way of peroxidiferric intermediate. Fe(III) is then transported to the protein cavity, where core formation is initiated at carboxyl groups on glutamates of L chains. This completes the ferrous binding, oxidation, and core formation process. A growing core serves to promote iron oxidation. CP is a ferroxidase and may serve to oxidize the Fe(II) in ferritin in some cells (Aisen et al., 1999).
How ferritin relates to transport of iron into cells by way of Tf, TfR, DCT-1, SFT, and from the metabolism of heme is unknown. Possibly, upon its entry into the cytoplasm, iron is immediately bound by ferritins. Alternatively, transferrin or some other iron-binding protein could transfer iron from the transporters to ferritin.
Brain
Ferritin is the main storage protein found in the brain (Roskams and Connor, 1994). The highest levels of iron and ferritin are found in the globus pallidus, substantia nigra, red nucleus, and dentate nuclei (Koeppen, 1995). Of note, these are regions that often have calcium deposition seen on computed tomography and magnetic resonance imagery scans (Koeppen, 1995). Most of the soluble iron is associated with ferritin in the brain (Koeppen, 1995).
There are light and heavy chain ferritins found in the brain (Connor et al., 2001). Several genes for heavychain ferritin have been identified. In the normal adult human hippocampus, ferritin H-chain RNA with the novel 279 nt sequence localizes strongly to small nonneuronal cells, probably glia, to capillary endothelial cells, and to selected populations of neurons (granule cells of the dentate gyrus).
H- and L-chain ferritins are found in endothelial cells, microglia, and oligodendrocytes. Astrocytes must contain ferritin, but this appears to be at very low levels (Connor et al., 2001). Neurons contain mostly H-chain ferritin. H- and L-chains are found in endothelial cells, although the L-chain predominates (Figs. 2 and 4). The L-chain predominates in microglia and can be used as a marker for microglia in the human and rodent brain (Fig. 3). The H-chain predominates in oligodendroglia in the piglet brain and in other species as well (Connor et al., 2001). In rats, during development, there is a shift in ferritin-containing cell types from predominantly microglia at postnatal day (PND) 5 to predominantly oligodendrocytes by PND 30. At PND 5, microglia are found throughout gray and white matter areas of the brain, but only amoeboid microglia in discrete foci in the subcortical white matter are ferritin positive. At PND 15, some oligodendrocytes in the subcortical white matter express ferritin, but the majority of ferritin-containing cells within white matter are still microglia. By PND 30, the predominant ferritin-containing cell type within white matter are oligodendrocytes (Connor et al., 2001).
A connection between heme metabolism and iron metabolism is emphasized by the finding that administration of HO inhibitors (Sn protoporphyrin) to patients increases the ferritin levels in plasma (Berglund et al., 1999). Administration of protoporphyrin IX (heme without the iron) elevates ferritin H-chain and TfRs. Incubation of lung endothelium with heme or hemoglobin induces HO-1 and ferritin in these cells. The ferritin induction appears to confer resistance of the endothelial against heme mediated and oxidative stress mediated injury (Balla et al., 1992).
Ferritin concentrations increase with age in the brain, apparently increasing to bind the iron that increases with age as well (Connor et al., 2001). Ferritin is markedly upregulated in microglia following ischemia (Kondo et al., 1995). Ferritin is upregulated in microglia in Alzheimer's plaques, and the number of ferritin positive microglia increase in Parkinson's disease (Connor et al., 2001).
TRANSPORT OF IRON OUT OF CELLS
Very few mechanisms have been described for the egress of iron from cells. In fact, the major mechanism for limiting iron levels in many cells appears to be by limiting transport into cells (Wessling-Resnick, 1999). This may be particularly true in the brain where a wide variety of diseases, and just aging itself, are associated with iron overload. However, several transporters and processes have been recently identified that are responsible for export of iron out of intestinal cells and into the blood stream and out of blood endothelial cells and into the brain parenchyma.
Hephaestin
Once iron is transported into brain endothelial cells by way of Tf/TfR, DCT-1, SFT, or another mechanism, it must then be transported out of the endothelial cell into the brain. How this occurs is unknown. Sla mice appear to transport iron into intestinal epithelial cells but cannot transport iron out of these cells into the blood (Rolfs and Hediger, 1999; Wessling-Resnick, 1999). The sla mice lack the protein hephaestin, which is a membrane-bound homologue of CP that likely functions as a multicopper ferrioxidase. The protein contains only one transmembrane domain, suggesting that hephaestin is not the basolateral iron transporter. It is likely that this protein oxidizes Fe++ to Fe+++ and facilitates transfer to the transporter that moves iron from the intestinal epithelium to the blood (Rolfs and Hediger, 1999; Wessling-Resnick, 1999).
Ferroportin-1/Ireg-1/MTP-1
A transporter postulated to export iron across the basolateral surface out of the duodenal cells and into the circulation has recently been cloned and characterized by several groups in different species. The gene is referred to as Ireg-1 (Donovan et al., 2000) or MTP-1 35 in mice and ferroportin-1 in zebrafish. Zebrafish ferroportin-1 is required for the transport of iron from maternally derived yolk stores to the circulation. The protein product is a putative multiple membrane-spanning transporter that functions as an iron exporter when expressed in xenopus oocytes. These results are consistent with the ability of hypotransferrinemic mice to facilitate mucosal iron transfer, despite the lack of transferrin expression.
The localization of ferroportin-1/Ireg-1/MTP-1 in cells and tissues is consistent with its proposed function of exporting iron from cells. In the duodenum, ferroportin-1/Ireg-1/MTP-1 localizes to mature enterocytes and is absent from the crypts. The protein is also found in the liver, predominantly in Kupffer cells where iron is scavenged from red blood cells. It has an IRE in its 5′ UTR mRNA and is upregulated by iron deficiency. Studies of ferroportin-1 also indicated that CP was necessary for iron efflux, whereas apotransferrin was not. A similar ferroxidase and transporter must exist for the brain endothelial cell, where a ferrioxidase (perhaps hephaestin or a homologue) and a transporter (IREG-like protein or another family member) must collaborate to transport iron out of the endothelial cell where it can be bound by transferrin or ferritin in the extracellular space. It is likely that ferroportin-1/Ireg-1/MTP-1 function may be perturbed in mammalian disorders of iron deficiency or overload.
The structure of the ferroportin-1 messenger RNA might indicate that its translation into protein can be regulated by the interaction of an iron-regulatory element with iron-regulatory binding proteins. This issue, as well as the relative roles of transcriptional and translational regulatory mechanisms, needs to be clarified.
In a pedigree with atypical hemochromatosis inherited as an autosomal dominant trait, a surprising finding that the mutated gene, SLC11A3, codes for ferroportin-1 in these patients was observed (Ponka, 2002). The identified mutation (A77D) probably results in loss of ferroportin-1 function, suggesting that the affected individuals are haploinsufficient for this gene product. In a nearly similar pedigree with an autosomal dominant hemochromatosis, the phenotype of the iron overload is caused by an activating mutation, i.e., a gain rather than a loss of function. However, Fleming and Sly (2001) suggest that these mutations in ferroportin-1 result in its decreased function in reticuloendothelial cells, analogous to patients with defects in the CP gene resulting in impaired iron release from reticuloendothelial cells.
HO-1 and HO-2
HO-1 and HO-2 appear to play a role in transport of iron out of cells following metabolism of heme. Knockout of HO-1 results in the deposition of iron in many organs including the brain (Poss and Tonegawa, 1997a). Animals with HO-1 knockouts do not process intracellular iron normally, and cells from these animals are more sensitive to oxidative stress (Poss and Tonegawa, 1997b). A patient with a mutation in the HO-1 gene also had abnormal deposition of iron in many organs of the body including the brain (Yachie et al., 1999). These results suggest that not only does HO-1 metabolize heme, but that it serves a chaperone function of handing the iron off to other proteins, perhaps ferritin, that must transport the iron out of cells. An iron efflux pathway remains to be described.
Of interest is the recent finding that HO plays a role in the entry of non–transferrin-derived iron into mitochondria through the permeability transition pore of dopamine challenged astrocytes (Schipper, 2000). The iron derived from heme would appear to be cycled back into heme synthesis.
HO-2 also appears to play a role in chaperoning iron. Mice with HO-2 knockouts have accumulation of iron in lung cells (Dennery et al., 1998). This implies that hemederived iron must be recycled through HO-2, perhaps back to heme proteins in the mitochondria during cytochrome synthesis or perhaps being bound by ferritin.
HO-1 appears to be important in iron transport from cells (Ferris et al., 1999). Expression of HO-1 is linked to cellular iron efflux, thereby demonstrating a role for HO-1 in cellular iron mobilization. Iron accumulation in cells of mice with targeted deletions of the HO-1 gene induces cell death because it can be reversed with iron chelators. Thus, HO-1 provides cytoprotection by augmenting iron efflux.
ATP-requiring iron transporter
Recently, an ATP-requiring iron transporter localized together with HO-1 in the microsomal membrane fraction that mediates iron efflux was described (Baranano et al., 2000). The transporter is specific for ferrous iron, is temperature- and time-dependent, and detected only with hydrolyzable nucleotides. It differs from all known ATPases and appears to be a P-type ATPase and is greatly enriched in the spleen. Iron treatment markedly induces ATP-dependent iron transport in macrophages. Mice with HO-1 deletion have selective tissue iron accumulation and display augmented ATP-dependent iron transport in those tissues that accumulate iron.
Frataxin and mitochondrial iron transporters
Friedrich's ataxia is an autosomal recessive disease that has been mapped to chromosome 9. Frataxin protein is localized to mitochondria and has been shown to be an iron-transport mechanism for mitochondria (Rolfs and Hediger, 1999). Mutations in the frataxin gene resulting in an expanded GAA repeat produces an abnormal protein that results in iron accumulation in mitochondria, death of sensory neurons in the dorsal root ganglia, and death of neurons in Clark's column as well as other neurons, resulting in the clinical syndrome of Friedrich's ataxia. In addition, the human ABC7 protein, a homologue of the yeast Atm1p mitochondrial iron transporter, may serve to transport iron into mitochondria (Goldman and Kranz, 2001).
Ceruloplasmin
CP is a copper containing oxidase that transports copper and is also involved in tissue iron metabolism and likely in facilitating iron exit from a variety of tissues including some cells in the brain (Rolfs and Hediger, 1999). CP, a 132kD α2-glycoprotein, with six plastocyanin-type domains and six copper atoms, is found in high concentrations in serum. The CP protein transports copper in serum and in cells but also functions as a ferroxidase, amino oxidase, and antioxidant (Rolfs and Hediger, 1999).
The ferroxidase function of CP has now been shown to be crucial for egress of iron from many tissues including the brain (Qian and Shen, 2001; Rolfs and Hediger, 1999). Mutations in the CP gene results in progressive degeneration of the basal ganglia and retina in association with iron deposition in these regions (Qian and Shen, 2001; Rolfs and Hediger, 1999). Because CP cannot pass the blood-brain barrier, cells within the brain must synthesize this protein. It has recently been shown that brain astrocytes express a glycosylphosphatidylinositol-linked membrane-bound form of CP (Qian and Shen, 2001; Rolfs and Hediger, 1999). It is likely that this membrane-bound CP would serve as a ferroxidase to convert ferrous ion to ferric so that it can be transported out of astrocytes by way of an unknown transporter (Donovan et al., 2000) either to the extracellular space to be bound by transferrin or ferritin or to an iron importer on vascular endothelial cells like DCT-1 or SFT. A ceruloplasminemia should not be confused with Wilson's and Menkes diseases, which have mutations of a copper ATPase and a P-type ATPase copper transporter, respectively (Rolfs and Hediger, 1999).
DISORDERS OF IRON METABOLISM
Hereditary hemochromatosis
Hemochromatosis is an iron-overloading disorder that results from excess dietary iron uptake and deposition in multiple organs. The mutated gene in hereditary hemochromatosis is HFE (Feder, 1999). The product of this gene, HFE protein, is homologous to major histocompatibility complex class I proteins, but HFE protein does not present peptides to T cells. On the basis of recent structural, biochemical, and cell biological studies, it has been shown that the TfR is a ligand for HFE and lowers its affinity for iron-bound transferrin (Feder, 1999; Trent et al., 2001). In hemochromatosis, the Hfe mutation inactivates the protein and thereby increases the affinity between Tf and TfR. Although this process is not completely clear, this leads to an increase in iron deposition in cells because iron absorption is not down-regulated by iron stores (Wessling-Resnick, 1999). Thus, HFE protein is directly linked to TfR-mediated regulation of iron homeostasis. Although the binding of HFE to TfR is critical for the effects of HFE, at present, the details of HFE function remain to be determined. For the brain, however, this is a minor issue because the brain is not primarily affected in this disease.
Hallervorden-Spatz disease
In this autosomal, recessively inherited disease, adolescents and young adults develop a progressive, disabling movement disorder characterized by spasmodic and uncontrollable movements of the trunk and limbs and by distorted body positions. In autopsied brains, accumulations of iron in the globus pallidus and pars reticulata of the substantia nigra are profound. In patients with Hallervorden-Spatz disease, an underlying mutation in a gene that encodes pantothenate kinase-2 that is expressed solely in the brain has been detected (Ponka, 2002). This enzyme is essential in coenzyme A biosynthesis, and catalyzes the phosphorylation of pantothenate (vitamin B5) and related substrates. The product of this reaction, 4′-phosphopantothenate, is then converted to 4′-phospho-pantetheine in a reaction that consumes cysteine. The authors hypothesize that neurodegeneration may be related to cysteine accumulation that has previously been observed in the degenerating brain areas of patients with Hallervorden-Spatz disease. Cysteine is an iron chelator, and accumulations of cysteine-bound iron may promote iron-dependent oxidative damage.
Neuroferritinopathy
A previously unrecognized adult-onset neurodegenerative disease that is associated with iron accumulation and abundant spherical inclusions that contain ferritin in the basal ganglia has been described (Ponka, 2002). The affected individuals all have the same mutation insertion of an adenine within the portion of the gene that encodes the carboxy terminus of the L chain of ferritin, an ironstorage protein. The mutation leads to synthesis of a unique 22 amino acid carboxy terminus. Throughout the white matter of the brain, axonal swellings are immunoreactive for neurofilaments, ubiquitin, and tau, a characteristic of neurodegenerative diseases. Interestingly, it appears that only neurons develop significant pathology. Thus, the authors propose that the disorder should be referred to as “neuroferritinopathy.”
HEME AND IRON METABOLISM FOLLOWING INTRACEREBRAL HEMORRHAGE
Following ICH and SAH the brain tissue parenchyma and CSF compartment, respectively, are exposed to large numbers of red blood cells and their heme iron-containing protein, hemoglobin. An understanding of how hemoglobin, heme, and iron are removed following intracranial bleeds is important because of the well-known potential for these molecules to induce oxidative stress and tissue injury. As described above, it is likely that the same transport proteins for heme and iron metabolism that are present in other organs also are present in the brain. All of the prerequisite systems to convert hemoglobin iron to ferritin and the insoluble ferritin byproduct, hemosiderin, are present in the CNS. Indeed, the incorporation of iron into holoferritin likely provides neuroprotection by preventing iron-catalyzed lipid peroxidation following ICH. In addition, for the brain, there are both blood-brain and blood-CSF barriers that control iron uptake and export. After an ICH, these barriers, if intact, likely affect the course of heme and iron removal. On the other hand, opening of the blood-brain barrier following ICH also likely affects iron uptake by neurons and glial. We will review what is presently known about the brain's removal of hemoglobin/heme and its associated iron following ICH with the caveat that the specific details are incompletely defined.
Any description of the pathways and mechanisms of blood removal from the brain parenchyma also needs to consider the fact that, following ICH in addition to red cells, the blood's plasma protein component also enters the extracellular space. This is significant for several reasons. Albumin, the protein with the highest concentration in plasma, also possesses medium affinity for binding heme (Grinberg et al., 1999). Thus, albumin, which remains in the tissue for several days and can be taken up by neurons and glia (Del Bigio et al., 2000), may serve to trap heme released from hemoglobin after ICH. In our porcine lobar ICH model, we have observed an expanding outward movement of Evans blue-tagged albumin during the early hours in perihematomal white matter (Wagner and Broderick, 2001; Wagner et al., 1996). In addition to albumin, the full complement of plasma proteins enters the interstitial space during an ICH, including transferrin, haptoglobin, and hemopexin that could affect the metabolism of hemoglobin, heme, and iron. In animals studies with hematomas induced by whole blood versus packed red blood cells, plasma proteins activate resident microglia and promote macrophage entry into the brain earlier and to a greater extent than the hematoma's mass effect or the presence of hemoglobin (Koeppen et al., 1995).
Red blood cells and hemoglobin
In the subacute phase following ICH, red blood cells and their contents are removed by a carefully controlled process without rapid hemolysis. Morphologically, red cells maintain their biconcave shape for several days following ICH (2–3 in rats, 4–8 in dogs and 5–10 in humans) (Darrow et al., 1988; Huang et al., 2002; Wagner and Broderick, 2001; Xi et al., 1998b). Even in vitro, red cell lysis occurs slowly with the release of oxyhemoglobin starting after 2 days (Macdonald and Weir, 1991). In dogs, hemoglobin reaches its peak concentration by the second day following blood injections into the CSF (Marlet and Barreto Fonseca, 1982). A similar time course of hemolysis occurs in the subarachnoid space following SAH in humans (Macdonald and Weir, 1991). In our porcine ICH model (Andaluz et al., 2002; Wagner et al., 1996a), hematoma removal is delayed, with volumes remaining essentially unchanged for the first 7 days. Thereafter, the majority of the hematoma is removed by day 14 (Wagner and Broderick, 2001).
In the early hours following ICH, oxyhemoglobin is converted to deoxyhemoglobin as the red cells lose their oxygen. Deoxyhemoglobin, which is present for several days following its release from red cells, is spontaneously and nonenzymatically oxidized to methemoglobin as ferrous iron is converted to its ferric form (Bradley, 1993). As long as the iron is in its ferrous state, the heme and globin moieties of hemoglobin do not readily separate from each other.
A quantitative biochemical analysis of iron removal from the clot in a rat ICH model using 59Fe hemoglobin demonstrated that at 24 hours essentially all of the 59Fe remained in hemoglobin (Atlas and Thulborn, 1998). In addition, no labeled iron was found in ferritin nor was hemosiderin present. Furthermore, the Perl's Prussian blue staining reaction for Fe3+ in the hematoma was negative during the first day. In a rabbit ICH model, perihematomal hemoglobin and heme were demonstrated histochemically along the walls of capillaries at 24 hours (Koeppen et al., 1995). A few microglia showed iron staining at 24 hours after ICH; however, this number was considerably greater at 48 hours. Also, the number of iron-reactive microglia were greater, and their cell bodies and the proximal processes were larger in whole blood versus red cell infused brains. By 5 days, large collections of macrophages were present in lesions of both infusion types.
Packed red blood cells do not cause acute edema development after infusion into the basal ganglia or frontal white matter. However, they do cause delayed edema that appears to be related to release of hemoglobin (Xi et al., 1998a). This coincides with the peak in perihematomal edema development that occurs several days after spontaneous ICH in humans or whole-blood infusions in animal models. This edema can be substantial and deleterious, as suggested by a recent study showing that delayed post-ICH brain edema is associated with significant midline shift (Zazulia et al., 1999).
Interestingly, in contrast with intact red blood cells, infusion of lysed erythrocytes into the basal ganglia of rats (Xi et al., 1998a) or into the white mater of pigs results in marked brain edema formation by 24 hours (Wagner et al., 2000b). Hemoglobin injections into the basal ganglia induce marked edema at 24 hours (Xi et al., 1998a). Methemoglobin infusions at concentrations found in erythrocytes also markedly increase brain-water content (Xi et al., 1998a). Lysed red cell infusions into porcine white matter caused brain edema-induced deaths (Wagner and Broderick, 2001; Wagner et al., 2000b), possibly by vasospasm induced by oxyhemoglobin, a suspected spasmogen (Macdonald and Weir, 1991). This result, which is generally not seen in the porcine lobar ICH model produced by either whole blood or packed red cells, supports the concept that rapid controlled red cell removal without marked hemoglobin, heme, or iron release is present in the acute phase after an ICH (Wagner and Broderick, 2001).
This careful removal of hemoglobin following ICH is important because several studies have demonstrated its potentially deleterious effects. Intracortical hemoglobin administration produces chronic focal spike activity, cavity lesions, and gliosis at injection sites (Rosen and Frumin, 1979). Although brief exposures (1–2 hours) of hemoglobin to neuronal cell cultures are nontoxic, exposures for 1 day cause neuronal death (Regan and Panter, 1996). Specific mechanisms that may be involved in neuronal cell injury based on in vitro findings include: hemoglobin activation of lipid peroxidation, exacerbation of excitotoxicity, inhibition of Na+/K+ ATPase activity, and induction of neuronal depolarization (Sadrzadeh et al., 1987).
Hemosiderin and hematoidin
The major hemoglobin-derived pigments in brain tissue following ICH are hemosiderin and hematoidin. Hemosiderin is invariably present in the brains of ICH patients who survive. Temporally, approximately 6 days were required in humans and 5 days in a rabbit thalamic ICH model for the first appearance of hemosiderin granules near the lesion (Koeppen et al., 1995). Because hemosiderin is an inert molecule and is not removed, it is a classical neuropathological marker in autopsied brains of a previous hemorrhage.
Invading macrophages or activated microglia participate in blood removal from the brain following ICH and convert the heme iron released from the lysed red cells to hemosiderin. This process first occurs with iron storage as iron-ferritin and later as the coarse, orange pigment, hemosiderin, a process that requires several days. Hemosiderin is a water insoluble degradation product of ferritin and consists of more than 25% Fe3+ (Garcia et al., 1994). Iron in brain hemosiderin appears stable and cannot participate in metabolic processes. The persistence of hemosiderin-laden macrophages long after an “old hemorrhage” in which the hematoma is replaced by a fibroglial scar or a cavity is indicative of the brain's difficulty in removing this blood degradation product. Another hemoglobin breakdown product, hematoidin, consists of bilirubin. This yellow nonferrous pigment is usually extracellular and becomes visible 10 days after ICH in humans and in a mouse ICH model and is also poorly removed. Hematoidin does not contain iron and therefore does not react with Prussian blue.
Heme
The process through which heme is released from hemoglobin into the extracellular space following ICH and into the CSF following SAH is not known. In the gut, heme release from hemoglobin occurs by enzymatic digestion of the globin protein (Conrad and Umbreit, 2000). As described above, once released, hemopexin could bind heme released from hemoglobin. In intestinal cells, uptake of radiolabeled heme results in labeling of three proteins: a membrane component and two soluble proteins. The membrane component is believed to contain the heme receptor; the smaller water soluble component is mobilferrin, whereas the larger cytoplasmic component is the 520 kDa paraferritin protein complex (Uzel and Conrad, 1998). At present, whether similar proteins are present in the brain is unknown as is their role in heme removal after ICH.
HO-1 induction: hemorrhage and heme
Following hemorrhage, either hemoglobin itself or the released heme must be transported into the cells for metabolism. As described above, heme transport into vascular cells, microglia and macrophages, and possibly neurons occurs by way of the hemopexin/hemopexin receptor/endosome pathway. Following intracellular transport of heme, heme would induce HO-1 in cells that do not express HO-2, and heme would be metabolized by HO-2 in cells like neurons that express HO-2.
Following injections of whole or lysed blood in the SAH space of adult rats, there is diffuse induction of HO-1 in microglial cells throughout the brain (Sharp et al., 1999; Turner et al., 1998, 1999). This diffuse induction was hypothesized to be caused by uptake into microglia and heme-dependent induction of HO-1. To test this, animals had pure hemoglobin injected into the SAH space (Turner et al., 1998). SAH injections of pure heme also induced HO-1 in microglia throughout the brain, suggesting that free hemoglobin in the extracellular spaces was selectively taken up into microglia and that the heme directly induced HO-1 in the microglia (Turner et al., 1998). Therefore, the microglia serve as a hemeprotein scavenger. There were also focal regions of HO-1 induction following experimental SAH hemorrhage that appear to represent regions of focal ischemia where heat shock proteins are induced (Turner et al., 1999). These focal regions of heat shock gene expression, including HSP-70, HO-1, and HSP-47, are blocked by antioxidants (Turner et al., 1999). This suggests these are focal areas of ischemia caused by vasospasm with consequent heat shock protein expression in ischemic cells. Antioxidants did not block the generalized induction of HO-1 in microglia that is hypothesized to be caused by heme uptake (Turner et al., 1999).
Although the above studies helped delineate the metabolism of extracellular heme in brain, they did not address any role of heme and HO-1 in vessels as related to SAH hemorrhage-induced vasospasm. This has been done in an elegant study from Suzuki and colleagues (Suzuki et al., 1999). They showed that inhibiting HO-1 with antisense oligonucleotides worsened and prolonged the delayed vasospasm that occurred following experimental SAH hemorrhage in rats. This exacerbation of vasospasm was caused by HO-1 gene expression in vessels and associated meningeal tissues and not to brain HO-1 gene expression (Suzuki et al., 1999). Therefore, it would be of interest to examine the induction of HO by heme and the inhibition of HO by compounds such as chromium mesoporphyrin in SAH hemorrhage. The induction of HO-1 and ferritin protein in arteries following experimental SAH has been confirmed in monkeys (Abboud and Haile, 2000). An HO-1 polymorphism, identified for the HO-1 gene that is associated with susceptibility to emphysema (Yamada et al., 2000), could also be important for outcomes from ischemic or hemorrhagic stroke (Yamada et al., 2000).
Paradoxically, all products of HO activity can also be toxic to cells. Cellular iron can contribute to free radical formation and damage to DNA, proteins, and lipids (McCord, 1998). As indicated throughout this review, prooxidant iron is well-known to induce CNS damage. CO may amplify free radical generation within mitochondria (Zhang and Piantadosi, 1992). Thus, metalloporphyrin inhibitors of HO may be protective. Tin-mesoporphyrin, a potent HO inhibitor, is protective against in vitro hypoxic injury and trauma in hippocampal slices, hydrogen peroxide exposure in renal epithelial, and in astroglial cells (Dwyer et al., 1998). In this regard, we recently demonstrated tin-mesoporphyrin's effectiveness in vivo to significantly reduce both edema and clot volumes at 24 hours after hematoma induction in our porcine ICH model (Wagner et al., 2000a).
Transferrin and iron removal following ICH
Under normal physiologic conditions, apotransferrin released from oligodendrocytes is believed to capture extracellular iron transported through the blood-brain barrier. Thus, following ICH, free iron in the extracellular space would likely be taken up by transferrin from oligodendrocytes. In addition, the fraction of iron that circulates as non–transferrin-bound iron is expected to increase significantly. The ependymal cells lining the ventricles that contain high levels of the DMT-1 iron transporter could also remove free iron present in the CSF (Burdo et al., 2001). Thus, several proteins may participate in iron removal from the interstitial space and ventricular system following ICH. Should transferrin become fully saturated, other compounds in the CSF that may participate in iron transport include citrate and ascorbate. At present, the level of iron in the extracellular space after ICH and the involvement of these proteins and molecules in iron removal is not known.
Astrocytes may also participate in iron removal following ICH. The existence of TfR-dependent and independent iron transport pathways in cultured astrocytes has been described (Qian and Shen, 2001). Cultured astrocytes, which display properties similar to those expressed by reactive astrocytes, also express DMT-1. Thus, the expression of DMT-1 by reactive astrocytes may facilitate transition metal uptake following injury (Williams et al., 2000). Astrocyte activation has been observed by us and others in perihematomal brain regions following ICH (Wagner and Broderick, 2001).
Ferritin and iron removal following ICH
Several potential pathways exist for removal of iron generated by HO activity following ICH. Ferritin, which has a high capacity for binding iron, is likely to be an important storage site for binding the released iron from metabolized heme. Iron generated by HO activity can also stimulate ferritin biosynthesis. Because the levels of CSF ferritin are roughly 10% of those in serum (Earley et al., 2000), CSF ferritin could also remove free iron in the CSF. Because the total CSF volume is replaced about every 8 hours (Earley et al., 2000), CSF ferritin could provide a mechanism for iron removal following ICH. CSF ferritin levels following ICH have not been examined, but they may be elevated. Indeed, increases in CSF ferritin are present in several CNS diseases including infectious, vascular, and dementing disorders. Local synthesis and release of ferritin by cells of the choroid plexus may also contribute to CSF ferritin (Earley et al., 2000).
As described above, ferritin synthesis normally occurs in microglia that is dependent on the local concentration of iron and the presence of iron regulatory proteins. Ferritin immunoreactivity was significantly greater than iron histochemical staining in microglia in perihematomal brain tissue at 24 and 48 hours following ICH (Koeppen et al., 1995). Furthermore, whole blood produced a greater ferritin response than red blood cells alone, suggesting a role for plasma proteins in stimulating ferritin expression. Activated microglia were also more hypertrophic and were present at considerable distances from the lesion following whole blood infusions as compared with infusions of packed red cells. Thus, on the basis of findings in our porcine lobar hematoma model, clotderived plasma proteins that diffuse through the interstitial space following ICH (Wagner et al., 1996a) may serve as a stimulator for distant microglial activation.
Heme, which acts as a physiologic inducer of ferritin biosynthesis, may be a more important derepressor of ferritin mRNA than iron after ICH. Because ferritin is a precursor of hemosiderin, induction of ferritin biosynthesis likely involves a shift to increased concentrations of L-ferritin (the storage subunit) and to lower concentrations of H-ferritin (the transport subunit). Furthermore, uncontrolled ferritin biosynthesis appears to occur in hemorrhagic lesions. Two mechanisms that may be responsible for this excessive ferritin accumulation: (1) loss of IRPs or (2) stimulation of ferritin biosynthesis by cytokines (Koeppen et al., 1995). Because IRPs in the brain are localized to astrocytes, iron has to be transferred to microglia to control ferritin biosynthesis. Thus, disruption of connections between astrocytes and microglia by the hematoma and perihematomal edema development may cause a loss of this control, and ferritin biosynthesis may proceed unchecked irrespective of iron concentrations. Ultimately, these events may contribute to hemosiderin accumulation.
After cerebral hypoxia/ischemia, there is a marked increase in histochemically detectable iron in the brain (Palmer et al., 1999). The hypoxic/ischemic event also is associated with an appearance of a large number of microglia (Ohno et al., 1995), many of which are iron-laden (Palmer et al., 1999). Indeed, iron-laden microglia are seen in many neurodegenerative diseases such as AD (Connor and Menzies, 1995). This iron must immediately be sequestered to prohibit additional oxidative stress, and ferritin is the most likely candidate for this function. Physiologically, seizures are frequently followed by a hypoxic/ischemic event. Recently, upregulation of H-ferritin in the brain was reported as part of a protective mechanism in seizure-prone areas (Lakaye et al., 2000). Thus, it is critical to demonstrate the cellular response of ferritin and its distribution after brain damage as reported in this study. The importance of the relationship between ferritin, microglia, and iron has increased recently with the observation of ferritin receptors on oligodendrocytes (Hulet et al., 1999).
OTHER MECHANISMS OF INJURY FOLLOWING INTRACEREBRAL HEMORRHAGE
Recent studies point to other factors, in addition to heme and iron, that contribute to injury around ICH hemorrhages (Fig. 5). The major tissue damage from a hemorrhage includes marked edema (Wagner and Broderick, 2001; Wagner et al., 1996a, 1998, 1999a, 2000a), isolated neuronal cell death around the hemorrhage (Felberg et al., 2002; Matsushita et al., 2000; Qureshi et al., 2001; Wagner et al., 1999b), and variable damage to adjacent white matter (Wagner and Broderick, 2001). Although space precludes an in-depth review of these other factors, it is important to mention the roles of thrombin, IL-1β/TNF-α and other cytokines, and glutamate in mediating edema and cell death. Thrombin produces edema when injected into the brain (Lee et al., 1996). More importantly, thrombin is rapidly released following ICH, and thrombin inhibitors prevent the early edema that occurs following ICH (Lee et al., 1996). In addition, recent studies show that thrombin induces the HIF (Jiang et al., 2002). The induction of HIF is important because HIF is a transcription factor that induces vascular endothelial growth factor that increases blood-brain barrier permeability (Zhang and Chopp, 2002) and could contribute to the delayed edema around hemorrhages. In addition, HIF induces glycolytic enzymes that could contribute to increased glycolysis and production of lactate that occurs around hemorrhages (Bergeron et al., 1997; Wenger, 2002). Lastly, thrombin exacerbates the injury produced by activation of N-methyl-D-aspartate receptors and could therefore contribute to neuronal injury by way of glutamate actions on neurons (Gingrich et al., 2002).
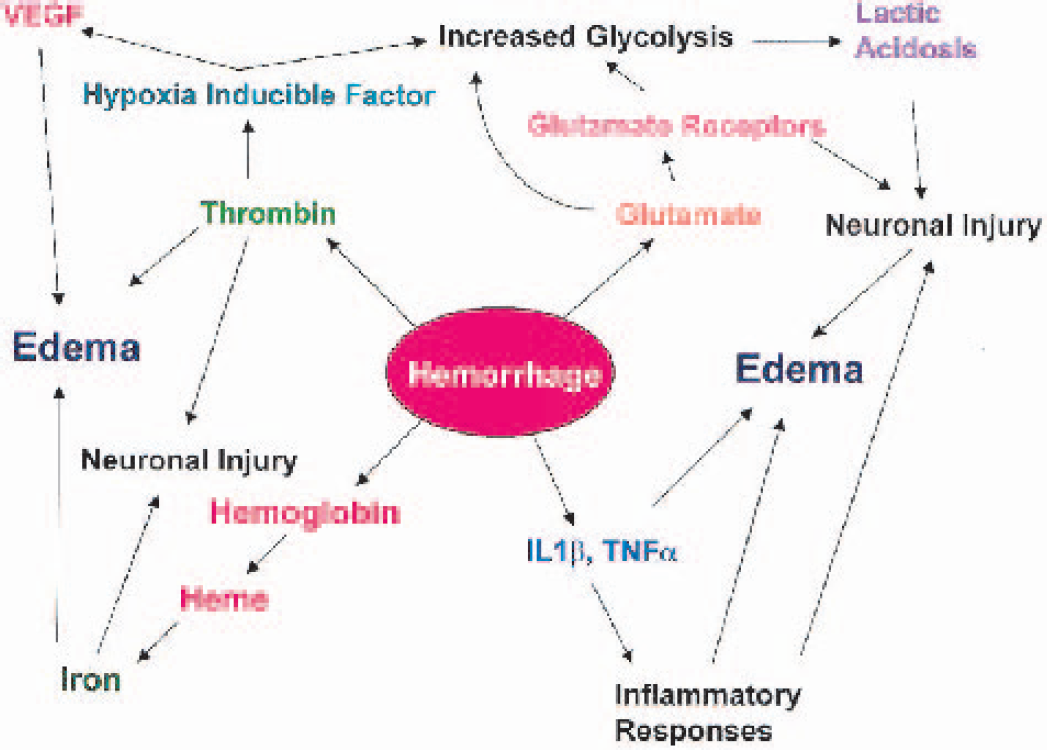
Recently published and ongoing studies support this putative scheme of events leading to brain injury following cerebral hemorrhage. Figure depicts the central roles of thrombin, glutamate, and oxidative stress-induced increases in pro-inflammatory cytokines on early edema formation and neuronal injury. Hemoglobin released from red blood cell breakdown contributes heme and heme-containing iron through heme oxygenase activity, which, although delayed, also participates in edema development and neuronal injury. VEGF, vascular endothelial growth factor; IL, interleukin; TNF, tumor necrosis factor.
Other studies have shown that pro-inflammatory molecules contribute to injury as well. Injections of IL-1β into the brain produce marked edema (Holmin and Mathiesen, 2000). IL-1β levels, as well as TNF-α levels, increase rapidly and dramatically following ICH (Wagner and Broderick, 2001; Wagner et al., 2001; Xi et al., 2001), and TNF antisense treatment improves ICH outcome (Mayne et al., 2001). Therefore, IL-1β and other pro-inflammatory cytokines likely contribute to the rapid and severe edema surrounding ICH hemorrhages (Wagner and Broderick, 2001). The transcription factor NF-κB that specifically regulates IL-1β and TNF-α is also rapidly activated following ICH (Hickenbottom et al., 1999; Wagner and Broderick, 2001; Wagner et al., 2001), possibly through early increases in oxidative stress (Wagner et al., 2002). Studies are underway to determine whether blocking NF-κB, IL-1β, and other cytokines decreases edema and cell death around hemorrhages. Our recent genomic studies demonstrate that a host of inflammatory and immune related molecules are induced in the regions around ICH and therefore likely play an important role in edema, cell death, and eventual removal of the hematoma from brain parenchyma. The inflammatory response to cerebral hemorrhage appears to be similar in some ways and different in others compared with ischemic stroke (Tang et al., 2002).
Lastly, glutamate may also contribute to injury around ICH. There are high concentrations of glutamate in the plasma that rapidly diffuses from the hemorrhage into the brain parenchyma and high concentrations of glutamate inside red blood cells that can potentially increase local glutamate levels. Our genomics study demonstrates that a large majority of the genes induced by the excitotoxin kainic acid are also induced around ICH, implicating excitoxicity in contributing to injury around the ICH (Tang et al., 2002). Glutamate could act on neuronal N-methyl-D-aspartate and alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA)/kainate receptors to produce neuronal injury, and the glutamate could act on AMPA/kainate receptors on oligodendrocytes to produce white-matter injury and edema (Matute et al., 2001). In addition, glutamate uptake into neurons and glia stimulates glycolysis and the production of lactate (Magistretti and Pellerin, 1999) that could help account for markedly elevated lactates around ICH hemorrhages (Mun-Bryce et al., 1993; Wagner et al., 1998). These elevated lactate levels could decrease tissue pH and contribute to neuronal injury as well. The actions of glutamate on the receptors in the brain could account for repeated reports of variably elevated blood flows around hemorrhages in both animal models and in patients with ICH (Miyazawa et al., 1998; Nath et al., 1987; Ropper and Zervas, 1982). The combination of mass effect, which would tend to decrease blood flow, and the actions of glutamate, which would tend to increase blood flow, likely explain the variably decreased and increased blood-flow findings reported around ICH in patients and likely explain the failure to see decreases of blood flow to levels that produce ischemia (Miyazawa et al., 1998; Nath et al., 1987; Ropper and Zervas, 1982). However, the combination of all of the above factors clearly could contribute to the edema, neuronal cell death, and white-matter injury caused by ICH.