
Abstract
Oxidative stress plays a significant role in secondary damage after severe traumatic brain injury (TBI); and melatonin exhibits both direct and indirect antioxidant effects. Melatonin deficiency is deleterious in TBI animal models, and its administration confers neuroprotection, reducing cerebral oedema, and improving neurobehavioural outcome. This study aimed to measure the endogenous cerebrospinal fluid (CSF) and serum melatonin levels post-TBI in humans and to identify relationships with markers of oxidative stress via 8-isoprostaglandin-F2α (isoprostane), brain metabolism and neurologic outcome. Cerebrospinal fluid and serum samples of 39 TBI patients were assessed for melatonin, isoprostane, and various metabolites. Cerebrospinal fluid but not serum melatonin levels were markedly elevated (7.28±0.92 versus 1.47±0.35 pg/mL, P<0.0005). Isoprostane levels also increased in both CSF (127.62±16.85 versus 18.28±4.88 pg/mL, P<0.0005) and serum (562.46±50.78 versus 126.15±40.08 pg/mL (P<0.0005). A strong correlation between CSF melatonin and CSF isoprostane on day 1 after injury (r=0.563, P=0.002) suggests that melatonin production increases in conjunction with lipid peroxidation in TBI. Relationships between CSF melatonin and pyruvate (r=0.369, P=0.049) and glutamate (r=0.373, P=0.046) indicate that melatonin production increases with metabolic disarray. In conclusion, endogenous CSF melatonin levels increase after TBI, whereas serum levels do not. This elevation is likely to represent a response to oxidative stress and metabolic disarray, although further studies are required to elucidate these relationships.
Keywords
Introduction
Traumatic brain injury (TBI) is the leading cause of death in individuals under 45 years. Patients surviving severe TBI remain with permanent neurologic and psychological disabilities, thus representing a significant social and economic burden.
Research has identified, among the sequelae of TBI, the increased production of free radicals via a number of pathologic cascades. After TBI, there is a net increase in excitotoxicity mediated by excitatory amino acids such as glutamate and N-methyl
Melatonin (N-acetyl-5-methoxy-tryptamine), a metabolite of the essential amino acid tryptophan, is secreted from the pineal gland in both daily and seasonal rhythms (Krauchi et al, 1997). Upon its release stimulated by nocturnal darkness, noradrenaline couples with β-adrenergic receptors, increasing intracellular cyclic adenosine monophosphate (cAMP) levels, eventually inducing the stimulation of arylalkylamine-N-acetyl-transferase (Reiter, 2003), and the production of melatonin. The pineal release of melatonin is not restricted solely to the systemic circulation (Tan et al, 2003). There is compelling evidence to suggest that melatonin is also released via the pineal recess directly into the third ventricle, resulting in cerebrospinal fluid (CSF) melatonin levels greater than those detected in the systemic circulation (Tricoire et al, 2002). A gradient of CSF concentrations between the third ventricle and downstream CSF has been proposed, supporting the hypothesis that melatonin is rapidly absorbed into the central nervous system (Tan et al, 2003).
The antioxidant activity of melatonin is a source of controversy. Early studies implied that melatonin is a highly-potent free radical scavenger (Tan et al, 1993), whereas others have restricted melatonin's role to a weak metal ion chelator, based on its direct antioxidant capability being mediated solely via its metabolites (Longoni et al, 1998). Despite these opposing theories, melatonin has been shown to exhibit indirect antioxidant properties through receptor-mediated actions. Melatonin upregulates mRNA levels of superoxide dismutase and stimulates the activities of the antioxidant enzymes glutathione peroxidase, glutathione reductase, and glucose-6-phosphate dehydrogenase (Rodriguez et al, 2004). It has been proposed that melatonin's antioxidant role is largely due to potentiation of other brain antioxidants, leading to further activation and attenuation of redox-sensitive transcription factors (Beni et al, 2004). Additionally, the beneficial effects of melatonin extend to improving mitochondrial respiration and ATP synthesis, as a result of stabilisation of the mitochondrial inner membranes against lipid peroxidation, and by significantly increasing the activities of complexes I and IV of the mitochondrial electron transport chain (León et al, 2005).
Melatonin, being highly lipophilic, readily traverses the blood—brain barrier and the phospholipid bilayer membrane, entering the cellular cytoplasmic and nuclear compartments (Menendez-Pelaez et al, 1993)—an essential prerequisite for any efficacious neuroprotectant. Furthermore, blood—brain barrier disruption is a common consequence of TBI, with blood cells and serum components leaking into the cerebral tissue, initiating a molecular cascade that activates the immune system and inflammatory response (Morganti-Kossmann et al, 2001), providing an additional source for the generation of free radicals. Preclinical data have shown melatonin to both decrease blood—brain barrier permeability and limit the extent of brain oedema (Gorgulu et al, 2001).
The deleterious effects of melatonin deficiency have been shown in preclinical models of excitotoxic seizures (Manev et al, 1996), ischaemia (Kilic et al, 1999), and TBI (Ates et al, 2006). Treatment with melatonin in animal models has been implicated in the attenuation of certain pathophysiologic states, including Parkinson's disease (Antolin et al, 2002), epilepsy (Lapin et al, 1998), and ischaemia (Kilic et al, 1999). Exogenous melatonin administration has also been shown to confer neuroprotective effects in preclinical models of TBI, reducing the degree of histologic damage and cerebral oedema, as well as improving neurobehavioral outcomes in mice and rats (Ates et al, 2006; Beni et al, 2004; Gorgulu et al, 2001; Sarrafzadeh et al, 2000).
Before any therapeutic application of exogenous melatonin for its neuroprotective potential in human TBI, the concentrations of endogenous CSF melatonin must be identified in the post-TBI setting, with respect to both absolute values and to the change in concentration over time. Therefore, this study aims to determine the changes of melatonin levels measured over 10 days in the CSF and serum of patients with severe TBI, and whether the concentrations correlate with markers of injury severity, oxidative stress, and neurologic damage, with a view to providing critical information required to further investigate the use of exogenous melatonin as a potentially therapeutic neuroprotective agent in this patient population.
Materials and methods
Patients and Sample Collection
This study was conducted in accordance with the National Health and Medical Research Council of Australia National Statement on Ethical Conduct in Research Involving Humans and had been approved by The Alfred Hospital Human Ethics Committee. Preadmission informed consent was waived and patients were enrolled in the study by the Intensive Care Unit (ICU) study coordinator, with delayed written consent obtained from next of kin while the patient was in intensive care.
Between October 2003 and September 2004, 39 patients (31 male and 8 female) with a severe TBI who were admitted to the ICU at The Alfred, Melbourne, were recruited into the study. The majority had TBI induced by vehicle accidents as either drivers or passengers (27/39), but some were pedestrians hit by vehicles (6/39). The other causes of TBI (6/39) were following: multistorey falls, a bicycle mishap, a physical assault, or a sudden collapse. Overall, 10/39 patients had essentially cranial injuries including brain haemorrhages, whereas the others also had multiple trunk and peripheral injuries. Details of the patients in terms of Glasgow Coma Scale (GCS), Injury Severity Score (ISS), Marshall CT Score at the time of admission (Marshall et al, 1991) and Extended Glasgow Outcome Score (GOSE) are given in Table 1. Additionally, 7/39 patients had hypotension (systolic blood pressure <90 mm Hg) and 9/39 had hypoxia (SaO2<90%). All the patients were under sedation, thus eliminating any light stimulus from the ICU environment and also decreasing brain electrical activity—no clinically apparent seizures were noted.
Traumatic brain injury patient demographics
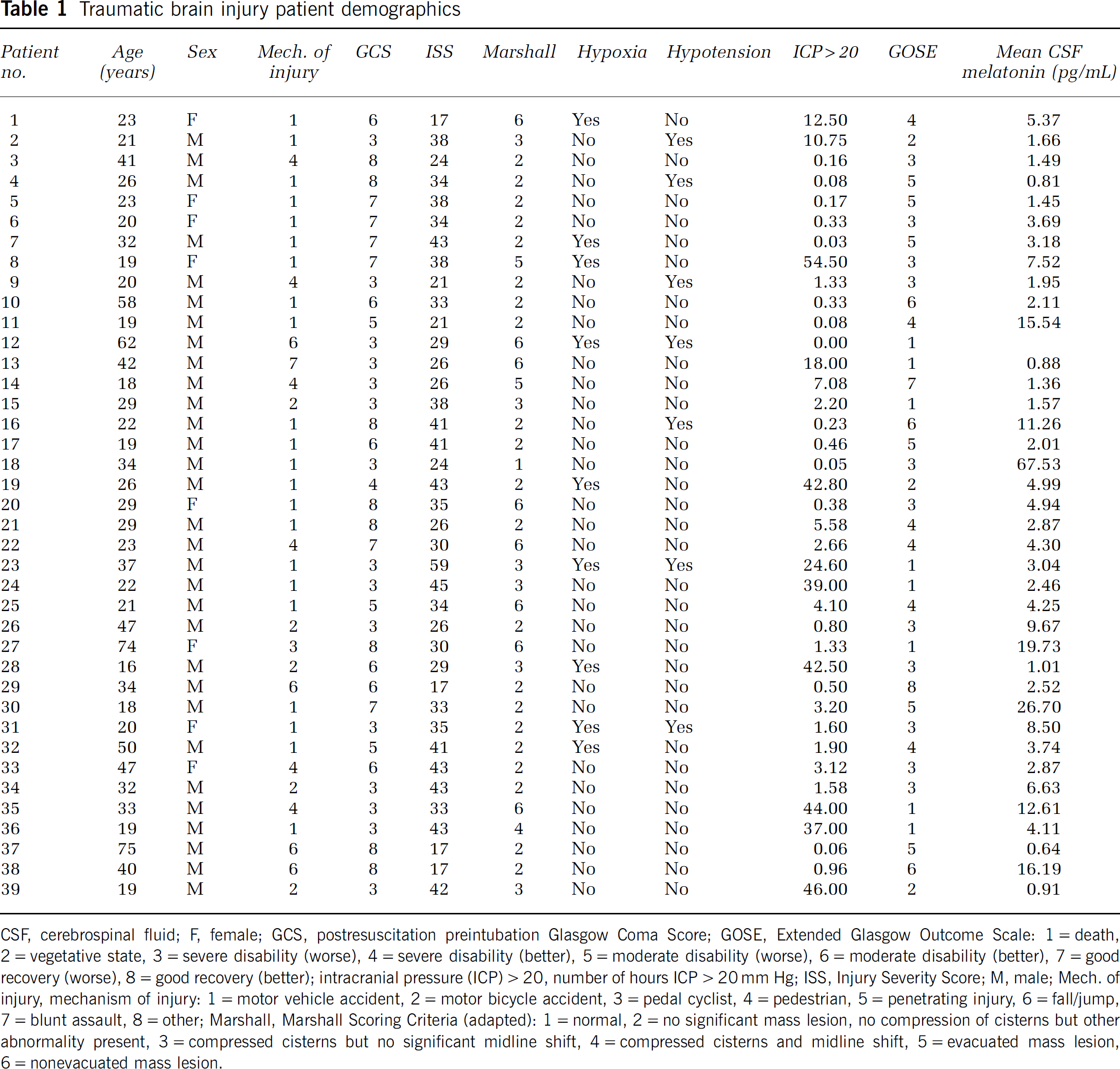
CSF, cerebrospinal fluid; F, female; GCS, postresuscitation preintubation Glasgow Coma Score; GOSE, Extended Glasgow Outcome Scale: 1=death, 2=vegetative state, 3=severe disability (worse), 4=severe disability (better), 5=moderate disability (worse), 6=moderate disability (better), 7=good recovery (worse), 8=good recovery (better); intracranial pressure (ICP)>20, number of hours ICP>20 mm Hg; ISS, Injury Severity Score; M, male; Mech. of injury, mechanism of injury: 1=motor vehicle accident, 2=motor bicycle accident, 3=pedal cyclist, 4=pedestrian, 5=penetrating injury, 6=fall/jump, 7=blunt assault, 8=other; Marshall, Marshall Scoring Criteria (adapted): 1=normal, 2=no significant mass lesion, no compression of cisterns but other abnormality present, 3=compressed cisterns but no significant midline shift, 4=compressed cisterns and midline shift, 5=evacuated mass lesion, 6=nonevacuated mass lesion.
Upon admission, all patients presented with a GCS of ≤8. After clinical and CT evaluation within 4 h, intraventricular catheters were placed in all patients to allow the continuous monitoring of the intracranial pressure (ICP) as well as drainage of CSF when the ICP exceeded 20 mm Hg. Increased ICP was defined as ICP>20 mm Hg and the duration of increased ICP was recorded. Any potential participants in this study who had slit ventricles or other reasons where a parenchymal catheter was used in preference to a ventricular catheter were not enrolled. Cerebrospinal fluid from the intraventricular catheter was collected via an extraventricular drainage system in which the collection bags were placed on ice. In most cases, CSF was collected continuously and the pooled sample was taken daily at 0900 hours along with a paired arterial blood sample. This continued on a daily basis until the CSF catheter was removed as part of the patient's routine management, in some cases up to 13 days after TBI. Cerebrospinal fluid and serum samples were centrifuged at 2,500 r.p.m. for 10 min at 4°C, aliquotted, and frozen at −70°C until analysed.
In assessing the patients, the Marshall Score of the admission CT scan was used to determine the radiologic type and severity of TBI (1=normal; 2=no significant mass lesion, no compression of cisterns but other abnormality present; 3=compressed cisterns but no significant midline shift; 4=compressed cisterns and midline shift; 5=evacuated mass lesion; and 6=nonevacuated mass lesion). Neurologic outcome was measured using the GOSE at 6 months after injury (Wilson et al, 1998). This assessment was based on a structured interview yielding information about consciousness, independence in activities of daily living, work, social, and leisure activities, family and friendships, and return to normal life. The same experienced interviewer performed the GOSE assessment for all subjects.
Control Cerebrospinal Fluid and Serum Samples
Serum samples were collected from 20 healthy volunteers (12 female and 8 male) with ages ranging between 21 and 55 years (Table 2). Cranial CSF samples were collected from the lateral ventricles, cisterna magna, or subarachnoid space in 10 non-TBI patients (five male and five female) with ages ranging from 30 to 74 years. These patients had a CSF sample collected as part of an elective surgical procedure unrelated to TBI, such as an insertion of a ventriculoperitoneal shunt to treat hydrocephalus.
Control patient demographics
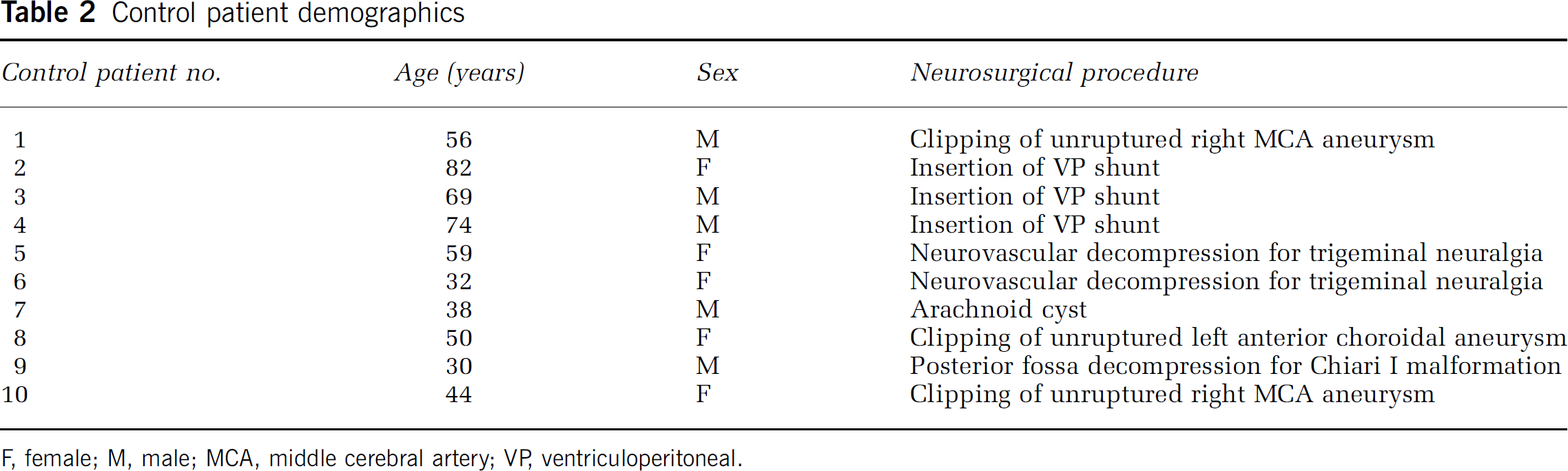
F, female; M, male; MCA, middle cerebral artery; VP, ventriculoperitoneal.
Assessment of Markers of Brain Metabolism in Cerebrospinal Fluid and Serum
Cerebrospinal fluid and serum samples were filtered using a 0.2 μmol/L syringe filter and measured for glucose, lactate, pyruvate, glycerol, and glutamate concentrations using an ISCUS clinical microdialysis analyzer (CMA Microdialysis AB, Sweden). Serum glutamate measurements were not performed. Serial dilutions were performed in parallel with the standard curve to verify accuracy. The minimum detectable concentrations for the ISCUS clinical microdialysis analyzer were as follows: glucose 0.1 mmol/L, lactate 0.1 mmol/L, pyruvate 10 μmol/L, glycerol 10 μmol/L, and glutamate 1 μmol/L. The intra- and intercoefficients of variance between the assays were calculated. The intracoefficient of variance was calculated between the TBI and control patient batch runs (n=2) and was found to be as follows: glucose 1.94%, lactate 4.99%, pyruvate 3.82%, glycerol 4.53%, and glutamate 10.07%. Intercoefficients of variance were calculated as glucose 5.21%, lactate 4.41%, pyruvate 5.30%, glycerol 3.78%, and glutamate 8.51%.
Quantification of Melatonin and 8-Isoprostaglandin F2α
Serum and CSF melatonin concentrations were measured using a commercially available melatonin competitive-binding ELISA (enzyme-linked immunosorbent assay) kit (IBL Immunobiological Laboratories, Hamburg, Germany). In eight experiments, the minimum detection limit for the assay was 0.91 pg/mL. The intra- and interassay coefficients of variance were 16.03% and 11.97%, respectively. The results of selected samples were verified independently using the Bühlmann Direct Melatonin radioimmunoassay (RIA) (Bühlmann Laboratories AG, Basel, Switzerland).
Serum and CSF isoprostane levels were measured using a commercially available 8-isoprostaglandin F2α ELISA kit (Cayman Chemical Company, Ann Arbor, MI, USA). In six assays, the minimum detectable limit was 1.78 pg/mL. The intra- and intercoefficients of variance were found to be 8.14% and 27.05%, respectively.
Statistical Analysis
Data are shown as mean±s.e.m. All data were analysed using SPSS (version 15.0, SPSS Inc., Chicago, IL, USA) or SigmaPlot 2000 (version 6.00, SPSS Inc., Chicago, IL, USA). Statistical analysis was performed with the data first grouped into TBI patient versus control patient groups and then ungrouped for analysis within the TBI patient population. Data were assessed for normality of distribution and, where possible, logarithmic transformations were used to normalise the data. Where appropriate, Pearson's or Spearman's correlations were performed to assess the degree of any relationship between variables. Correlations with P<0.05 were determined to be statistically significant.
Results
Patient Demographics
Thirty-nine patients sustaining TBI (Table 1) and 10 control patients undergoing elective neurosurgical procedures, with ‘normal’ cranial CSF (Table 2), were recruited in the study. The age of the TBI patients ranged between 16 and 75 years, with a mean age of 31.77±2.46 years, compared with 53.40±5.64 years for the control patients (P=0.001). Although males predominated in the TBI patient population (n=31, 79.5%) and genders were evenly divided among the control patient population, there was no significant gender difference across the two groups. Motor vehicle accidents resulted in the majority of cases (n=22, 56.4%), with 15.4% of TBI patients (n=6) involved in accidents as pedestrians and motor bicycle accidents accounting for 12.8% of cases (n=5). The median GOSE was 3, the mortality rate 20.59% (n=8), whereas 71.8% (n=28) of patients achieved unfavourable outcomes (GOSE≤4). Radiologic categorisation of injury revealed that 74.36% (n=29) of the patients suffered diffuse injury (Marshall Scoring Criteria categories I—IV) with the majority of patients (53.85%, n=21) sustaining diffuse injury category II, whereas 10 patients experienced focal TBI (Table 1).
Melatonin Profiles
Investigation into the post-injury temporal profile of endogenous melatonin levels aimed to provide insight into the impact of TBI on the production of melatonin. This characterisation will offer valuable information required as a foundation for future studies, both clinical and preclinical, investigating the administration of melatonin as a potential therapeutic neuroprotective agent. Cerebrospinal fluid samples obtained from TBI patients over a time course of up to 10 days were assayed for melatonin concentrations. Endogenous CSF melatonin concentrations gradually increased from the day of admission (day 0) to day 2, where the local maximum mean concentrations were detected in the TBI patient cohort. The mean concentrations decreased to a minimum mean concentration on day 5 and then increased to a maximum mean concentration on day 8, thus displaying a biphasic temporal profile (Figure 1A). The CSF melatonin concentrations were significantly elevated compared to controls overall (7.28±0.92 versus 1.47±0.35 pg/mL, P<0.0005) as well as on all days apart from days 0, 1, and 4 after injury. Similar to CSF melatonin, serum melatonin concentrations increased from day 0 to a maximum mean concentration on day 2, after which a gradual decrease to a minimum mean concentration on day 5 after injury was observed (Figure 1B). Serum melatonin levels of TBI patients (15.01±0.78 pg/mL), although higher than CSF concentrations, were not significantly elevated above controls (15.15±1.65 pg/mL).
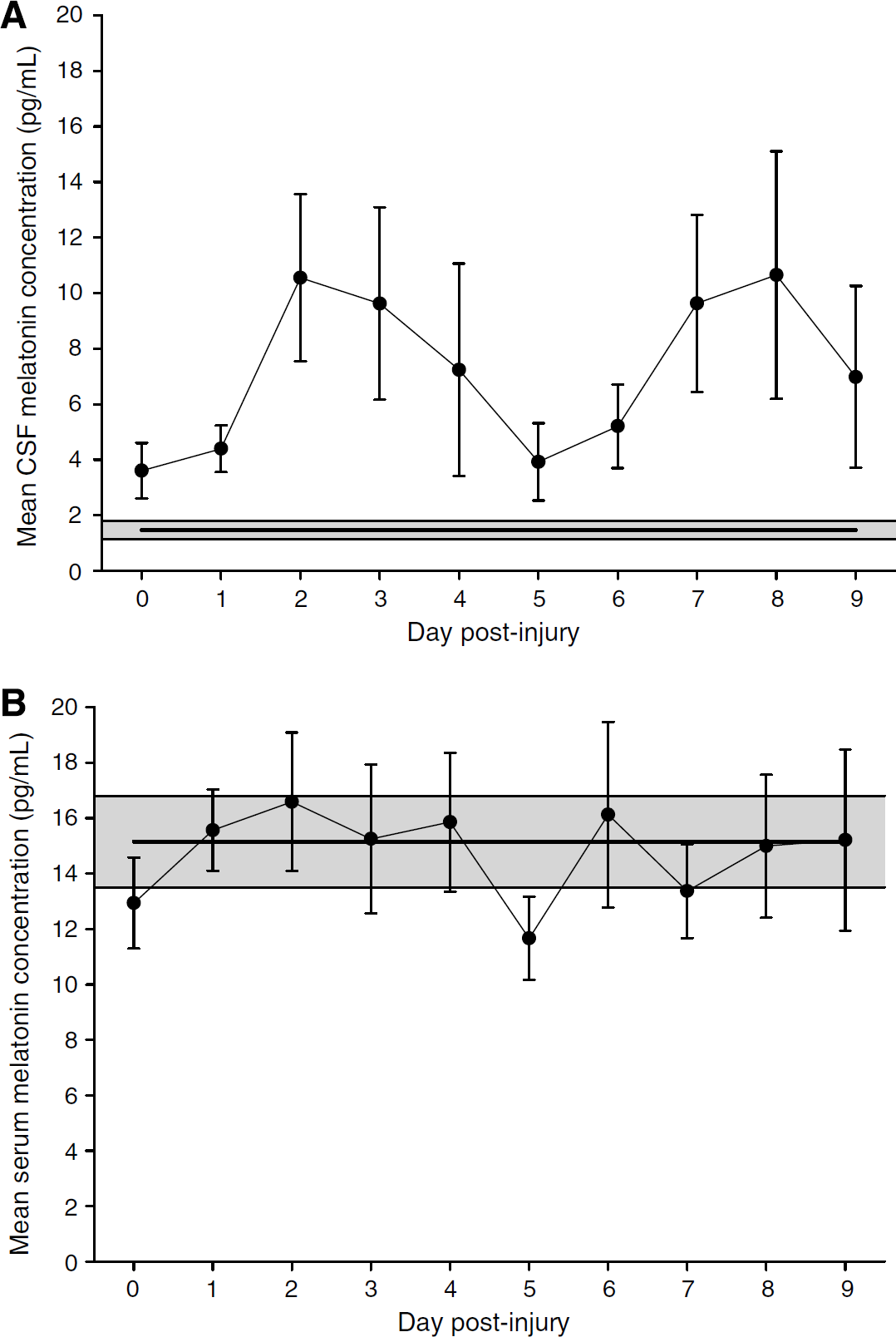
Temporal profiles of mean melatonin concentrations. (
Isoprostaglandin-F2α Profiles
Endogenous CSF isoprostane concentrations were found to increase to a maximum mean concentration on day 1 after injury (Figure 2A). Cerebrospinal fluid isoprostane was significantly elevated compared to controls (127.62±16.85 versus 18.28±4.88 pg/mL, P<0.0005). Serum isoprostane levels were higher than those measured in the CSF and decreased on day 1 after injury, after which a maximum mean concentration was observed on day 2 (Figure 2B). Endogenous serum isoprostane exhibited an overall mean of 562.46±50.78 compared with control levels of 126.15±40.08 pg/mL (P<0.0005).
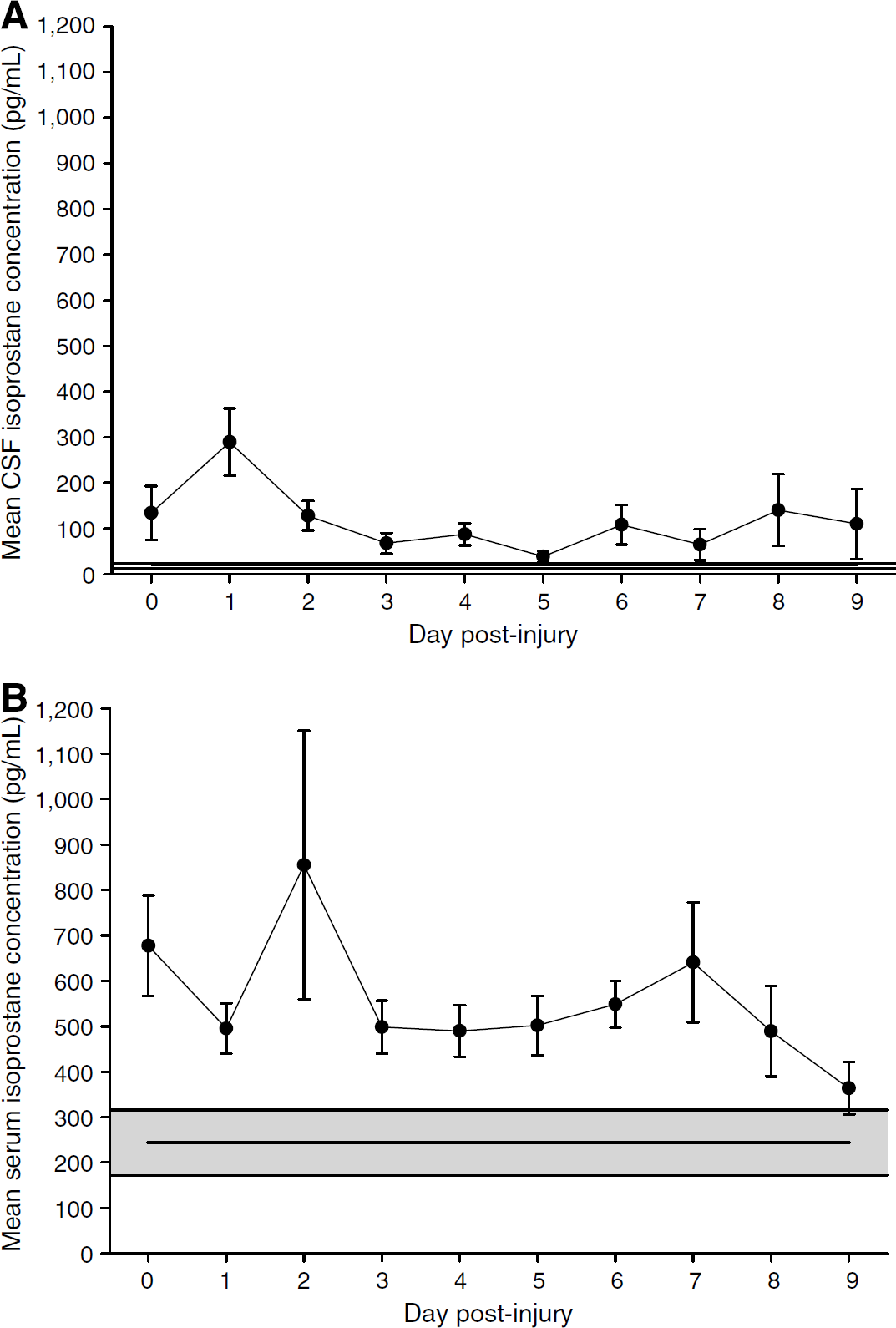
Temporal profiles of mean isoprostane concentrations. (
Profiles of Markers of Brain Metabolism
Characterisation of the profiles of selected markers of brain metabolism was conducted to establish the occurrence of a shift from aerobic to anaerobic metabolism, cell membrane damage, and excitotoxicity. Endogenous CSF glucose concentrations of severe TBI patients initially decreased on day 1, after which they gradually increased to peak on day 5 after injury (Figure 3A). Levels of CSF glucose in TBI patients displayed a significantly greater overall mean concentration as compared with control patients (4.29±0.10 versus 2.93±0.26 mmol/L, P<0.0005). Significant differences were detected between the TBI patient and control patient population means on all days apart from days 0 and 9 post-admission. Serum glucose measurements decreased to a minimum mean concentration on days 2 and 3 after injury, increasing to a peak on day 4 and gradually decreasing for the remainder of the measured time course (Figure 3B). Similar to CSF levels, serum glucose concentrations in TBI patients were significantly elevated when compared with those of control patients (6.44±0.15 versus 5.19±0.15 mmol/L, P<0.0005).
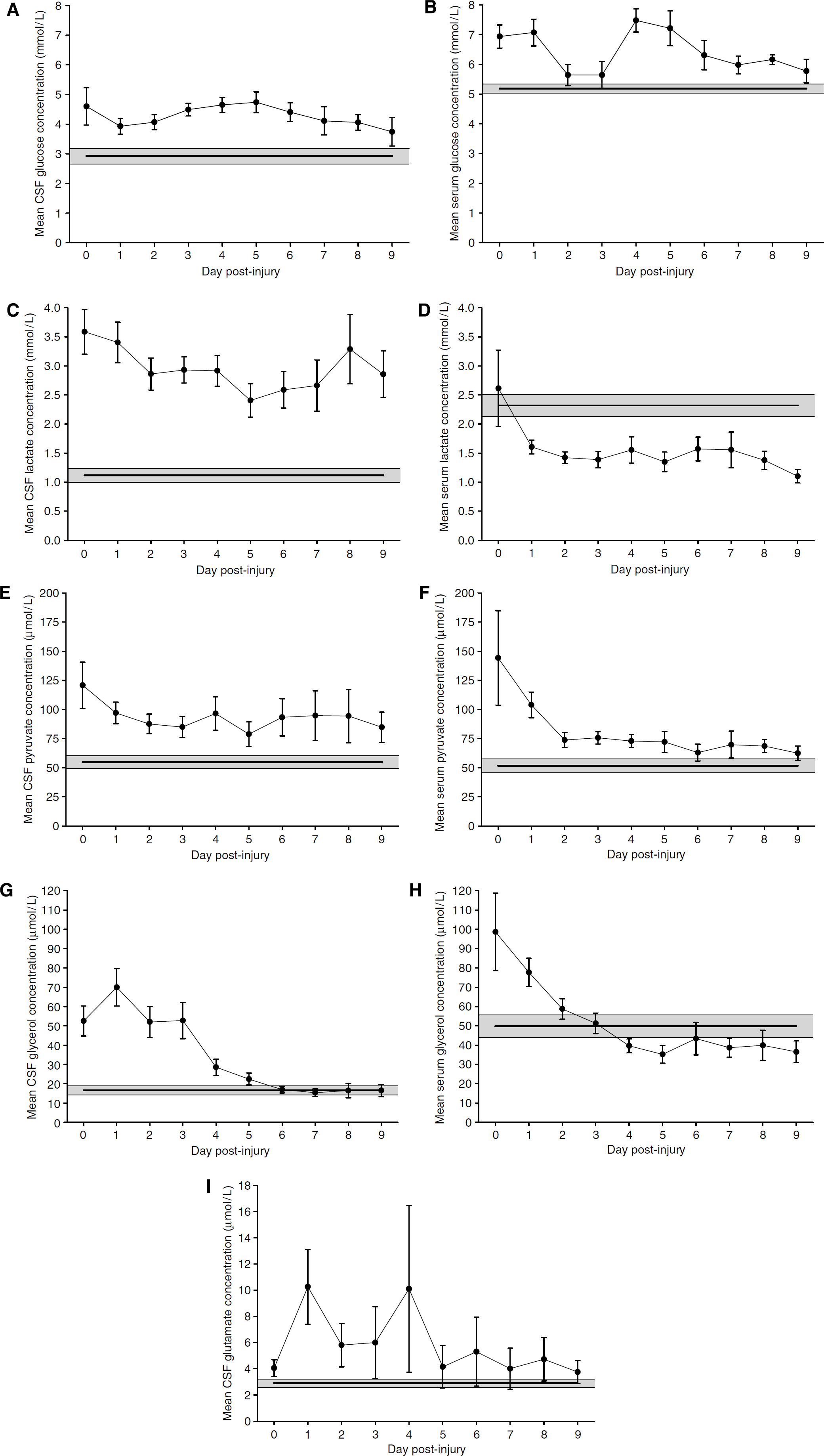
Temporal profiles of mean brain metabolism markers after injury. Cerebrospinal fluid and serum samples were filtered and then processed via the ISCUS clinical microdialysis analyzer for the measurement of glucose, lactate, pyruvate, glycerol, and glutamate. Control patient mean is shown by horizontal line, with shaded area representing s.e.m. (
Mean CSF lactate concentrations in TBI patients were maximal on the day of injury, after which they gradually decreased to a minimum mean concentration on day 5 after injury, rising to a secondary peak on day 8 (Figure 3C). Cerebrospinal fluid glucose concentrations were higher in TBI patients than the control patients (2.96±0.11 versus 1.12±0.12 mmol/L, P<0.0005). Significant differences were detected between the TBI patient and control patient population means on all days post-admission. Endogenous mean serum lactate levels exhibited their maximum concentrations on the day of injury, after which they tapered off gradually to a minimum concentration on day 9 after injury (Figure 3D). Regarding serum lactate concentrations, those in TBI patients were lower in comparison with controls (1.52±0.07 versus 2.32±0.19 mmol/L, P=0.001).
Similar to CSF lactate measurements, endogenous pyruvate levels in the CSF of severe TBI patients were initially maximal and gradually decreased to a minimum mean level on day 5 after injury (Figure 3E). Patients having sustained TBI displayed a significantly greater overall mean CSF pyruvate concentration than the control patients (92.50±4.11 versus 54.80±5.53 μmol/L, P<0.0005). Significant differences were detected between the TBI patient and control patient population means on days 0, 1, and 3 post-admission. Similar to serum lactate, endogenous serum pyruvate levels exhibited a maximum mean concentration on the day of injury, after which there was a gradual reduction in levels to a minimum on day 9 post-TBI (Figure 3F). Serum pyruvate levels were higher in TBI than in control patients (80.07±3.66 versus 51.75±6.03 μmol/L, P<0.0005).
As a marker of cell membrane damage, glycerol levels in the CSF of severe TBI patients increased to a maximum mean concentration on day 1 after injury, after which they descended to levels similar to those of control patients by day 6 post-injury, exhibiting a minimum mean level on day 7 (Figure 3G). Endogenous CSF glycerol was significantly higher in TBI than in control patients (40.75±2.98 versus 16.60±2.34 μmol/L, P=0.008). Significant differences were detected between the TBI patient and control patient population means for up to 4 days post-admission. Serum glycerol concentrations displayed a maximum mean level on the day of injury, following which they decreased to a steady concentration by day 4 after injury (Figure 3H). No statistically significant difference was noted between TBI and control patients' serum measurements (54.21±2.73 versus 49.80±5.93 μmol/L, P=0.505).
Glutamate levels in the CSF of severe TBI patients were found to peak on days 1 and 4 after injury, decreasing to a minimum mean concentration on day 9 (Figure 3I). Endogenous CSF glutamate exhibited a significantly elevated mean concentration (6.51±1.08 μmol/L) when compared with control patient values of 2.90±0.31 μmol/L (P=0.002).
To verify the accuracy of these measurements, serial dilutions were performed for each marker of brain metabolism in both CSF and serum samples. Regression values were calculated, and overall averages for CSF (r2=0.95) and serum (r2=0.94) measurements were obtained.
Correlation of Measurements between Cerebrospinal Fluid and Serum Samples
Cerebrospinal fluid melatonin displayed a positive relationship with serum melatonin levels (r=0.282, P<0.0005). In contrast, the levels of isoprostane measured in the CSF of TBI patients exhibited a weak negative correlation with serum isoprostane (r=−0.179, P=0.025).
Correlation of Melatonin with 8-Isoprostaglandin F2α and Markers of Brain Metabolism
To investigate potential relationships between melatonin and markers of both oxidative stress and brain metabolism, bivariate correlations were performed between these variables. Cerebrospinal fluid melatonin levels displayed a weak positive relationship with CSF isoprostane (r=0.208, P=0.007) and CSF glutamate concentrations (r=0.217, P=0.005); however, no significant correlations were found with overall endogenous CSF measurements of glycerol, lactate, or pyruvate. Melatonin values on each day were analysed against variables measured from the same sample. Further weak relationships were evident between CSF melatonin sampled on day 1 after injury and CSF isoprostane (r=0.563, P=0.002), CSF pyruvate (r=0.369, P=0.049), and CSF glutamate (r=0.373, P=0.046).
Serum melatonin levels were also found to exhibit a negative correlation with serum isoprostane (r=0.162, P=0.012), but displayed no relationships with markers of brain metabolism.
Correlation of Cerebrospinal Fluid and Serum Melatonin with Clinical Parameters and Outcome Measurements
No significant correlation was shown between CSF melatonin concentrations and GOSE. A weak positive correlation was shown between serum melatonin and GOSE (r=0.189, P=0.004), indicating that outcome measurements were higher in patients who exhibited higher levels of serum melatonin. When categorised into diffuse injury (Marshall Scoring Criteria categories I—IV), CSF melatonin exhibited a moderate negative relationship with radiologic evidence of diffuse injury (r=−0.433, P<0.0005). Cerebrospinal fluid but not serum melatonin displayed a weak negative relationship with duration of raised ICP (r=−0.171, P=0.028). No significant relationships were found between either CSF or serum melatonin and hypoxia or hypotension, respectively.
Discussion
Endogenous Cerebrospinal Fluid Melatonin Concentrations Increase after Severe Traumatic Brain Injury
Mean endogenous levels of CSF melatonin in patients having sustained severe TBI ranged from 3.60 to 10.65 pg/mL, with an overall mean of 7.28±0.92 pg/mL. Compared with the TBI patients, the CSF melatonin levels of control patients had a mean concentration of 1.47±0.35 pg/mL. The differences between daily melatonin levels and those of the control patients were shown to be significant on days 2 and 3 after injury, as well as from day 5 onwards in this data series. Thus, the data show that endogenous CSF melatonin concentrations increase post-TBI.
The control endogenous melatonin concentrations detected in this study are lower than those published previously, and reconciliation with those in the literature is required. The heterogeneous nature of the patient demographics, methods, and results of the current literature (Table 3) impede the definitive evaluation of physiologic endogenous concentrations of melatonin, and thus the reported levels are of limited value in determining accurate control measurements. Because of ethical considerations, despite the exclusion from the control patient population of individuals with deranged inflammatory markers or radiologic evidence of cancer, as well as those with a history of TBI or ischaemic events, these control patients still had pathologic examination requiring an invasive procedure and therefore cannot be considered entirely healthy ‘normal’ controls. Furthermore, the control patients in this study were significantly older than the TBI population (53.40±5.64 versus 31.77±2.46 years, P=0.001). However, to validate the value of the control patients included in this study, a number of factors in favour of their use must be brought to light. The control patients in this study were subjected to strict recruitment criteria, including screening for histories of previous TBI or ischaemic events. Moreover, samples were obtained from the cranial CSF, which is comparable to the ventricular CSF obtained from TBI patients. Other studies (Table 3) have not obtained cranial CSF for control patient use, with the exception of three studies which used the ventricular CSF obtained from post-mortem patients (Liu et al, 1999; Wu et al, 2003; Zhou et al, 2003), and one recent study using endoscopic sampling techniques (Longatti et al, 2007). In our study, the procedures for melatonin measurement were maintained consistently across the TBI and control patient populations, and hence the observed increase of endogenous CSF melatonin levels post-TBI is reliable. Accordingly, despite the inconsistencies shown between the control patients included in this study and those reported in the literature, the data suggest that endogenous CSF melatonin levels increase after TBI.
Physiologic concentrations of endogenous melatonin in cerebrospinal fluid
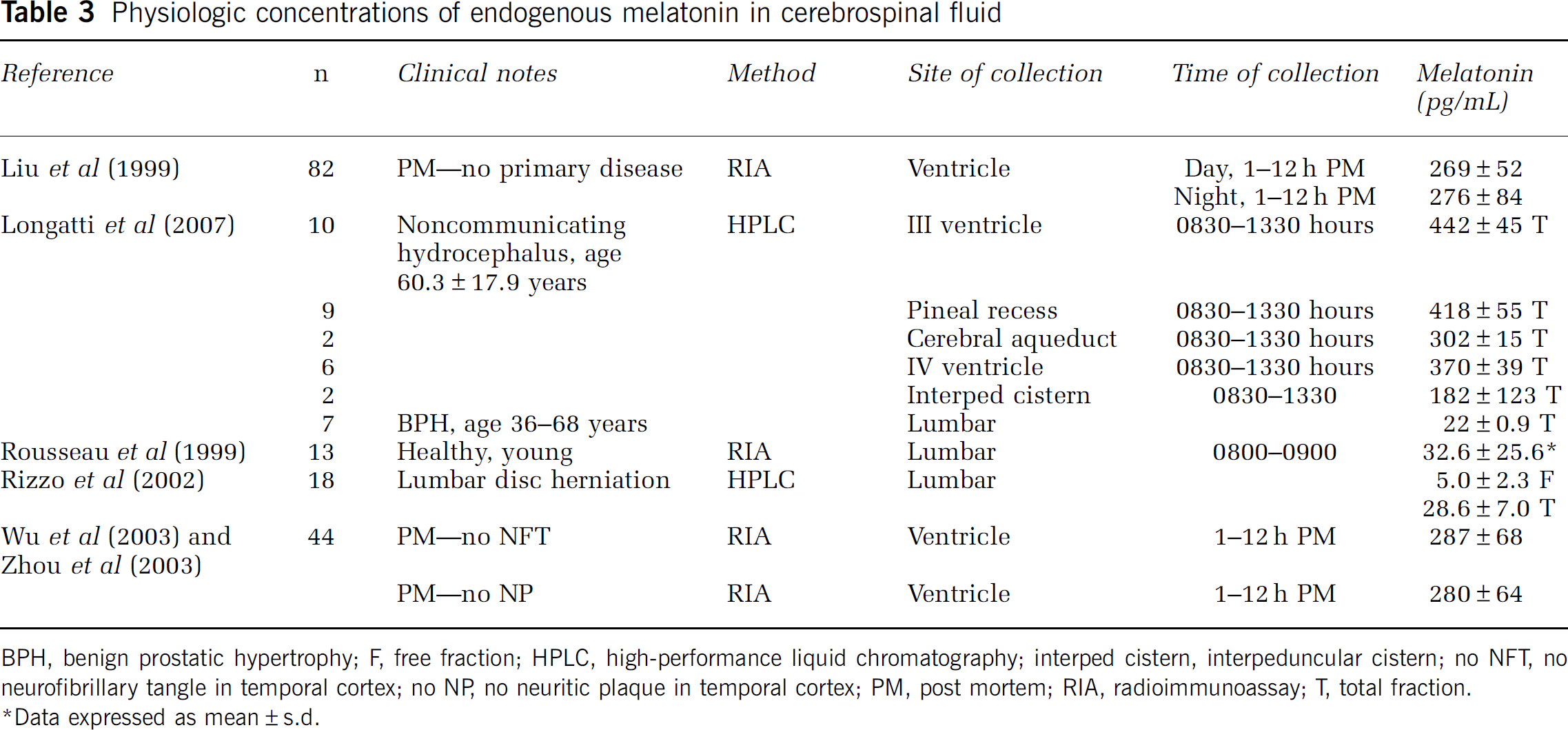
BPH, benign prostatic hypertrophy; F, free fraction; HPLC, high-performance liquid chromatography; interped cistern, interpeduncular cistern; no NFT, no neurofibrillary tangle in temporal cortex; no NP, no neuritic plaque in temporal cortex; PM, post mortem; RIA, radioimmunoassay; T, total fraction.
∗Data expressed as mean±s.d.
A further factor of consideration is the standard ICU management of patients suffering severe TBI. As part of this protocol, patients are administered noradrenaline as a peripheral vasoconstricting agent to maintain cerebral perfusion pressure (CPP)>60 mm Hg. As noradrenaline is a neurotransmitter involved in the release of melatonin from the pineal gland, any iatrogenic administration of noradrenaline might translate to an artificial increase in melatonin levels. Furthermore, sedation of patients in ICU would limit their exposure to light, thereby stimulating noradrenaline release and inducing a contrived rise in melatonin concentrations. A relationship between the amount of noradrenaline administered and melatonin concentrations would be suggestive of a confounding influence, yet this is not evident in our analysis (r=−0.152, P=0.115). Thus, we conclude that the increase in melatonin post-TBI is an endogenous response to the severe insult experienced by the patient, and not artificially derived.
Antioxidant Properties of Melatonin
The observed increase in melatonin concentrations may be rationalised in view of the antioxidant properties of melatonin. The hypothesis for this is that the deleterious oxidative stress resulting from TBI would induce a physiologic endogenous response, stimulating production of the antioxidant, melatonin. It is thought that these effects are mediated via the activator protein-1 (AP-1) pathway, and indeed this has been shown in preclinical animal models of TBI (Beni et al, 2004). This rationale is further supported by the weak positive correlation between CSF melatonin and CSF isoprostane (r=0.208, P=0.007), suggesting that melatonin production is increased in conjunction with lipid peroxidation and hence, oxidative stress.
The deleterious consequences of oxidative stress and specifically, lipid peroxidation, are significant in the injured brain. Indeed, the brain displays an increased susceptibility to damage via oxidative stress, exhibiting an elevated rate of metabolic activity with marked reactive oxygen metabolite generation (Reiter, 1998). Furthermore, its high levels of polyunsaturated fatty acids are a rich source for lipid peroxidation reactions, due to both the myelin content and the high membrane-to-cytoplasm ratio intrinsic to the brain (Varma et al, 2003). Besides from direct cellular damage, recent evidence suggests that free radicals also cause cellular damage and death indirectly via signalling pathways (Chan, 2001). Accordingly, oxidative stress has been associated with apoptosis, achieved via a number of pathways, including the activation of nuclear factor-κB (Martindale and Holbrook, 2002), as well as the mitochondrial release of cytochrome c and apoptosis-inducing factor (Chan, 2001). Apart from lipid peroxidation, oxidation of cellular proteins and nucleic acids also occurs (Bayir and Kagan, 2001; Chan, 2001). Previous research indicates that free radicals have been implicated directly in a number of pathologic events of the central nervous system, including increased blood—brain barrier permeability (Easton and Fraser, 1998), vasogenic oedema (Hall et al, 1992), post-traumatic microvascular damage (Kontos and Hess, 1983), and subarachnoid haemorrhage-induced vasospasm (Sano et al, 1980).
Melatonin and isoprostane, a specific product of lipid peroxidation known to increase both in animal models of brain injury (Roberts and Morrow, 2000) and in human TBI (Varma et al, 2003), exhibit weak positive relationships in both CSF and serum. As previously discussed, melatonin production may be stimulated in response to the oxidative stress of TBI. Indeed, upon viewing the temporal profiles of these two substances in concert (Figure 4), a potentially functional relationship between melatonin and isoprostane could be hypothesised. Accordingly, the shift in melatonin concentrations with a substantial increase on day 2 may be considered a response to the peak in endogenous CSF isoprostane observed on day 1 post-TBI. Although there is no direct evidence for a cause and effect relationship, these observations may be interpreted in favour of melatonin possessing antioxidant capabilities, and its production (and potentially, consumption) being altered as a consequence.
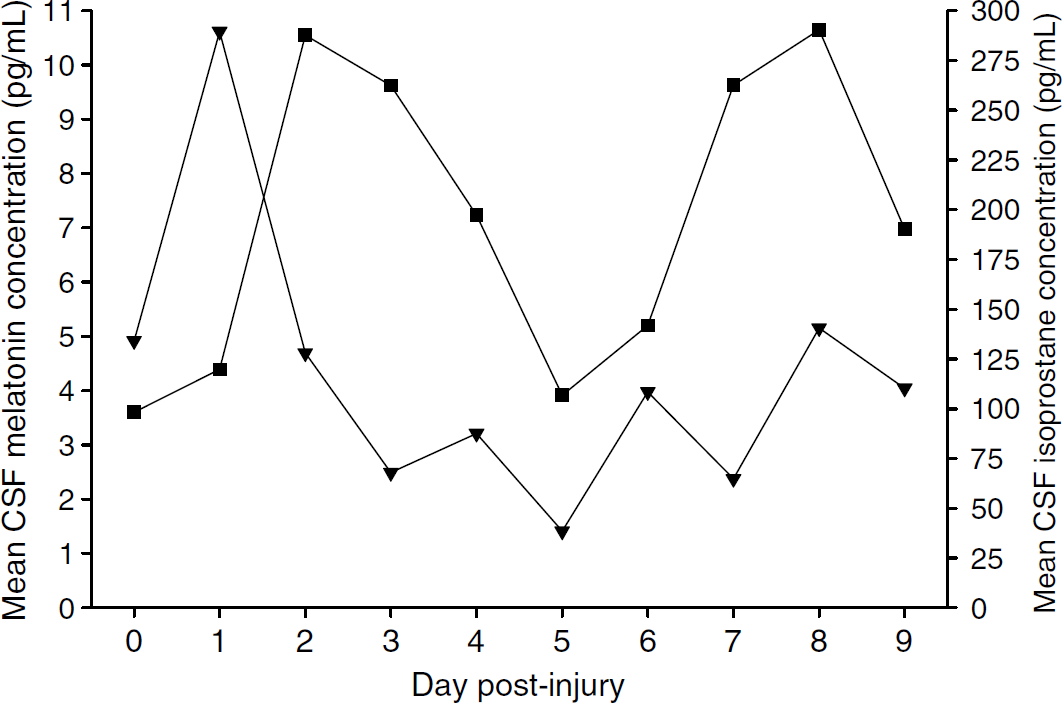
Mean CSF melatonin and CSF isoprostane distributions versus day after injury. Mean CSF melatonin levels in TBI patients indicated by (▪) and mean CSF isoprostane levels in TBI patients indicated by (▾). Melatonin and isoprostane are displayed concurrently to illustrate the potential phase shift, with a peak in melatonin concentrations on day 2 following a peak in isoprostane concentrations on day 1 after injury.
It is unclear, however, as to whether melatonin is released in sufficient amounts to significantly attenuate this process, as would be indicated by a strong negative relationship, or whether this correlation might be confounded due to the presence of other natural antioxidants in the brain, and this study design is of insufficient sensitivity to determine this with accuracy.
Anti-inflammatory Properties of Melatonin
Upon examination of the patients classified via the Marshall Scoring Criteria as having sustained diffuse injury (categories I—IV), a negative correlation was shown (r=−0.433, P<0.0005), implying that with increasing severity of injury, smaller concentrations of melatonin were detected in the CSF. Although there is no definitive evidence to support a cause and effect relationship, these data may indicate that the lower concentrations of melatonin in more severe cases of TBI might suggest greater consumption of melatonin to attenuate the degree of inflammation.
In this regard, melatonin has been proposed to possess anti-inflammatory capabilities by inhibiting the release of inflammatory cytokines (Lissoni et al, 1997). Currently, the literature attributes melatonin with both pro-inflammatory (Jiménez-Caliani et al, 2004) and anti-inflammatory effects (Mayo et al, 2005). Although melatonin is proposed to exert its anti-inflammatory effects both directly and via the actions of its metabolites, N1-acetyl-N2-formyl-5-methoxykynuramine and N1-acetyl-5-methoxykynuramine (Mayo et al, 2005), the precise mechanisms by which melatonin exerts a modulatory effect upon the inflammatory processes remain to be elucidated. Distinct similarities exist between melatonin and certain cytokines and, in particular, their temporal profiles after injury. When comparing the temporal profile of endogenous CSF melatonin to that of interleukin-6 (IL-6) (Kossmann et al, 1995), it is evident that melatonin peaks at approximately the same times as this cytokine after TBI. Indeed, preliminary data from the laboratory investigating IL-6 concentrations on days 1 to 5 following TBI reveal a trend towards a positive relationship between melatonin and IL-6 (r=0.223, P=0.052, n=77 samples) in CSF, but no significant correlation in serum measurements was found. When graphed together against the day post-trauma, a similar picture to that of melatonin and isoprostane is evident, with a shift of melatonin concentrations following those of IL-6. Although this does not provide conclusive evidence as to the anti-inflammatory actions of melatonin, it suggests that melatonin may exert these effects to a limited degree after TBI.
Serum Melatonin after Severe Traumatic Brain Injury
Analysis of the temporal profile of serum samples obtained from the same patient population shows that although endogenous serum melatonin levels did not increase significantly from those of controls, a similar temporal profile is observable. Endogenous serum isoprostane levels did, however, increase significantly, and displayed a sharp peak on day 2 after injury, with an otherwise comparable profile with that of CSF isoprostane. The weak negative relationship between serum melatonin and serum isoprostane might further lend credence to a functional relationship with melatonin being used in an antioxidant capacity. Accordingly, patients experiencing greater degrees of oxidative stress may have induced a larger extent of melatonin consumption and antioxidant activity. Similarly, there was a weak negative correlation between serum melatonin and diffuse brain injury, which might suggest anti-inflammatory properties of melatonin, properties reported widely in the literature. Finally, a weak positive relationship was observed between serum melatonin concentrations and GOSE scores, indicating that higher melatonin levels were correlated with better functional patient outcomes.
Limitations
Cerebrospinal fluid was collected on ice over a 24-h period. Although this allowed for a greater volume of fluid to be obtained for the study, a significant limitation of this procedure is that subtle molecular changes occurring over shorter periods cannot be reliably assessed. Therefore, any relationship shown represented the gross state over a 24-h period, and care must therefore be taken in extrapolating any findings using these samples. However, more frequent CSF sample collection would expose the patient to the external environment and increase the risk of infection. An additional factor involved in sample collection is the consistency in time of day at which the samples were collected. The diurnal pattern of melatonin, regulating the sleep—wake cycle, has been well described (Reiter, 2003). Melatonin levels have been shown to increase between 0200 and 0400 hours (Brzezinski, 1997), and to decrease towards their nadir during the day. Thus, the measurement of melatonin levels obtained in this study would fail to capture the diurnal variation of melatonin secretion, and levels obtained would merely represent a concentration averaged over the 24-h period between collections. Of note, after severe TBI, the natural diurnal pattern of secretion is suppressed due to the pharmacological induction of coma over the study period, wherein despite a diurnal variation in light intensity, the patient receives no light stimulus from the ICU environment, and thus our findings are valid and similar to those reported previously (Paparrigopoulos et al, 2006).
Conclusions
This study has shown that endogenous CSF melatonin concentrations increase after TBI, whereas serum levels fluctuate but remain within normal ranges. This elevation is likely to represent a response to oxidative stress and/or inflammation; however, owing to the design of this study, a causative effect cannot be shown. Therefore, further preclinical and clinical studies are needed to clarify the precise nature of any potential relationships between melatonin and oxidative stress and/or inflammation. Provided these beneficial roles of melatonin were further validated in animal models of TBI and in concert with existing data that show such an effect (Ates et al, 2006), a future direction in the study of melatonin post-TBI could include clinical administration of melatonin to these patients.
Footnotes
Acknowledgements
We thank Dr Nicole Bye of the National Trauma Research Institute for her role in CSF and serum sample collection, Dr Edwin Yan and Laveniya Satgunaseelan of the National Trauma Research Institute for permission to use the results of their cytokine assays in this study, and Associate Professor David Kennaway of The University of Adelaide, for the use of their direct melatonin RIA.
This work was partly supported by the Victorian Trauma Foundation.