
Abstract
Brain hypothermia is at present the most effective neuroprotective treatment against brain ischemia in man. Ischemia induces a redistribution of proteins involved in synaptic functions, which is markedly diminished by therapeutic hypothermia (33°C). Dendritic spines at excitatory synapses are motile and show both shape changes and rearrangement of synaptic proteins as a consequence of neuronal activity. We investigated the effect of reduced temperature (33°C and 27°C compared with 37°C), on spine motility, length and morphology by studying the distribution of GFP-actin before, during and after induction of in vitro ischemia. Because high-concentration actin filaments are located inside spines, dissociated hippocampal neurons (7-11DIV) from transgenic mice expressing GFP-actin were used in this study. The movement of the spines and the distribution of GFP-actin were recorded using time-lapse fluorescence microscopy. Under normal conditions rapid rearrangement of GFP-actin was seen in dendritic spines, indicating highly motile spines at 37°C. Decreasing the incubation temperature to 33°C or 27°C, dramatically reduces actin dynamics (spine motility) by approximately 50% and 70%, respectively. In addition, the length of the spine shaft was reduced by 20%. We propose that decreasing the temperature from 37°C to 33°C during ischemia decreases the neuronal actin polymerization rate, which reduces spine calcium kinetics, disrupts detrimental cell signaling and protects neurons against damage.
Introduction
Reduction of brain temperature by a few degrees, therapeutic hypothermia, dramatically influences outcome after experimental brain ischemia (Krieger and Yenari, 2004; Olsen et al, 2003), and after circulatory arrest in man (Bernard et al, 2002; HACASG, 2002). Therapeutic hypothermia is at present the most effective neuroprotective treatment against ischemic brain injury employed in clinical practice. The mechanisms by which hypothermia exerts its beneficial effect appear to involve multiple systems and have been extensively studied in vivo (Olsen et al, 2003; Yenari et al, 2003) and in vitro (Bossenmeyer-Pourie et al, 2000; Bruno et al, 1994; Shuaib et al, 1992, 1993). Some of the processes involved occur at synapses, including elevation of glutamate in the extracellular space (Busto et al, 1989a) and aberrant cell signaling (Kamme and Wieloch, 1996). Additionally, disruption of receptor—scaffold protein interactions (Aarts et al, 2002) or actin polymerization (Endres et al, 1999) attenuates ischemic brain injury. This strongly suggests that interactions among cytoskeleton elements in general and in the dendritic spine in particular play an essential role in the pathogenesis of ischemic brain injury, and are thus possible sites of action of the neuroprotective therapeutic (33°C) hypothermia.
Dendritic spines are small (<2 μm in length) morphological protrusions on neuronal dendrites, with a distinct head (0.5–1.5 μmol/L) and narrow neck (0.5 μm in width) (Harris and Kater, 1994). They are found in >90% of excitatory synapses (Gray, 1959), have high calcium inductances (Yuste et al, 2000) and a complex protein—protein signaling machinery, suggesting them playing an essential role in nerve cell communication and establishment of cellular memory (Hering and Sheng, 2001; Kennedy, 2000). Dendritic spines are highly motile, and may emerge and disappear within seconds or minutes (Dailey and Smith, 1996; Dunaevsky et al, 1999; Fischer et al, 1998). Dynamic changes in spine morphology are thought to be due to the unique cytoarchitecture of the spines, involving rapid turnover of actin polymers regulated by actin-binding and signaling proteins (Matus, 2005; Pollard and Borisy, 2003).
Approximately 80% of the actin is in a dynamic state (Star et al, 2002), and its turnover is highly regulated by proteins that either stabilize the actin monomer pool (thymosin or profilin), nucleate initiation sites for actin polymerization (arp2/3) or that stabilizes the actin polymer (contractin). Capping proteins (cofilin and gelsolin) prevent polymerization and severe polymers into smaller actin fragments and actin monomers or promote depolymerization (cofilin) (Pollard and Borisy, 2003). Under normal conditions, actin turnover in motile cells is ATP dependent (Atkinson et al, 2004). G-actin binds ATP, and the ATP—G-actin binds profilin in a complex that allows actin polymerization at barbed ends. Excess ATP—G-actin is sequestered into an inactive pool by binding to thymosin. Hence, when cellular ATP levels are high, most G-actin is either bound to thymosin or profilin, and actin self-polymerization will not occur randomly because critical levels of free G-actin (ATP and ADP associated) will not be reached (Pollard and Borisy, 2003).
Dendritic spines are sites for early damage after brain ischemia (Johansen et al, 1984) and focal swellings along dendrites are observed in the CA1 and CA3 regions during recovery after a transient ischemic episode (Hori and Carpenter, 1994; Hsu and Buzsaki, 1993). In cell cultures dendritic swellings rapidly appear after oxygen and glucose deprivation, with extensive spine loss, which is partly NMDA receptor mediated (Hasbani et al, 2001). Toxic NMDA receptor activation also decreases spine density (Hasbani et al, 2001) and AMPA receptor activation dramatically diminishes spine motility (Fischer et al, 2000). Also, deletion of gelsolin, an actin-capping and -severing protein, increases ischemic damage and activates mitochondria-induced cell death processes (Harms et al, 2004).
Spinogenesis is modulated by diverse extrinsic (trophic factors and synaptic activity) and intrinsic factors (calcium and ATP) (Bonhoeffer and Yuste, 2002; Fiala et al, 2002; Tashiro and Yuste, 2003), and is affected by temperature (Kirov et al, 2004; Roelandse and Matus, 2004). Given the dramatic effect of therapeutic hypothermia on neuronal survival after brain ischemia, and the importance of spine processes in ischemic cell death, we aimed to investigate the effect of in vitro ischemia (IVI) and mild hypothermia (33°C) on actin dynamics using time-lapse microscopy.
Materials and methods
Cell Culture Preparations
All animal experiments were approved by the Ethical Committee at Lund University. Transgenic mice expressing γ-cytoplasmic actin-GFP (Fischer et al, 1998, 2000) were used. Primary hippocampal mice cultures were prepared as described earlier (Brewer, 1997), with slight modifications from 2-day-old mouse pups. The animals were killed by decapitation, and the brain carefully transferred to ice-cold dissection medium containing Hibernate E medium (BrainBits Springfield, IL, USA), B27 supplement, 0.5 mmol/L
Microscopy and Image Acquisition
Light microscopy images were captured using a cooled digital camera (F-view) mounted on an Olympus IX-81 fluorescence microscope fitted with an additional ‘goggle lens’ for increased resolution. Images were analyzed with Soft Imaging System SIS (AnalySIS) software, which also controlled computer and microscope settings, optimized to prevent excessive photo bleaching. Chroma GFP filters and neutral density filters were used to further minimize bleaching and phototoxicity.
Cell Culture and In Vitro Ischemia
Before image acquisition, the cover glasses with the primary cultures were quickly mounted with silicon grease in a RC-21BR ‘closed bath imaging chamber’ (volume 260 μl), and placed on a PH2 ‘heated platform’. The temperature of the platform was regulated by a TC-344B ‘Dual Chamber system heater controller’. All media were preheated to 30°C on a hot plate to avoid bubbles in the perfusion tubes. The media entering the imaging chamber were then heated with a SH-27B ‘in-line solution heater’. The temperature was measured in the outflow media as well as in the mounting platform and calibrated to 37°C and 33°C±0.1°C, respectively, by using the TC-344B unit. All products mentioned above were from Warner Instrument Corp., Hamden, CT, USA. The incubation chamber was perfused with medium using a perfusion pump, MS-Reglo (Ismatec SA, Glattbrugg, Switzerland), set at a flow rate of 260 μL/min. The oxygen content of the perfusion medium was measured with a Clark electrode (Consort z921, Tumhout, Belgium).
During oxygen glucose deprivation (OGD), cells were incubated in culture medium similar to that of artificial cerebrospinal fluid, but with the pH and K+ and Ca2+ concentrations changed to levels comparable to those in the extracellular space of an ischemic brain (Rytter et al, 2003). Using this medium, ischemic CSF (iCSF), we exposed cultures to increasing durations of OGD better mimicking the conditions of ischemia in vitro, and for this reason referred to as IVI. In vitro ischemia was induced in cell cultures by switching to iCSF perfusion medium (in millimolar concentrations; 0.3 CaCl2, 70 NaCl, 5.25 NaHCO3, 70 KCL, 1.25 Na2PO4, 2 MgSO4, 40 sucrose and pH 6.8) bubbled with a 90% N2,5% CO2,5% H2 gas mixture. Cultures were exposed to 9, 12, 15, 20 or 25 mins of IVI, at 37°C and compared with 33°C or 27°C and 22°C, and maintained for a further 2, 24 or 48 h. The extent of neuronal death was assessed by staining with propidium iodide (PI) (10 μg/mL), added to the media before IVI (Rytter et al, 2003).
Calculation of Spine Shape and Motility
Changes in spine motility over time in stubby and mushroom-shaped spines were evaluated using computer-generated profile outlines (Figure 1; Fischer et al, 2000). Consecutive micrographs were captured every 20 secs over 10 mins (30 time points) at perfusate temperatures of either 33°C and 37°C or 27°C and 37°C. After the outlines of the spines were generated at different time points, the outlines were stacked to illustrate the movement of the spines over time (Figures 1B and Figure 1C). Multiple, clearly observable, single outlines in the stack signify a high motility of the spine, whereas overlapping outlines indicate less motility. To quantify the motility of the spine, a ratio between the distances of two lines, placed perpendicular to each other, was calculated for each captured picture frame (Figure 1D). One distance measured the width of the spine head at approximately its widest point, and the other measured the distance from the top of the spine to the spine base where the shaft joins the dendritic branch. The lines within the spine outline, formed a cross entirely enclosed within each outlined spine of the 30 analyzed picture frames (see Figure 1D). The change in the width/length (w/l) ratio was normalized and plotted over time. A change in ratio means that either the length or width or both change, which means that the plasma membrane moves, hence the spine is moving. The resulting graph shows a variability in the ratio over time indicating the degree of movement. To obtain a numerical value of the variability in spine (w/l) ratio, the difference between the highest and the lowest ratio over a 3-min period for each spine was calculated.
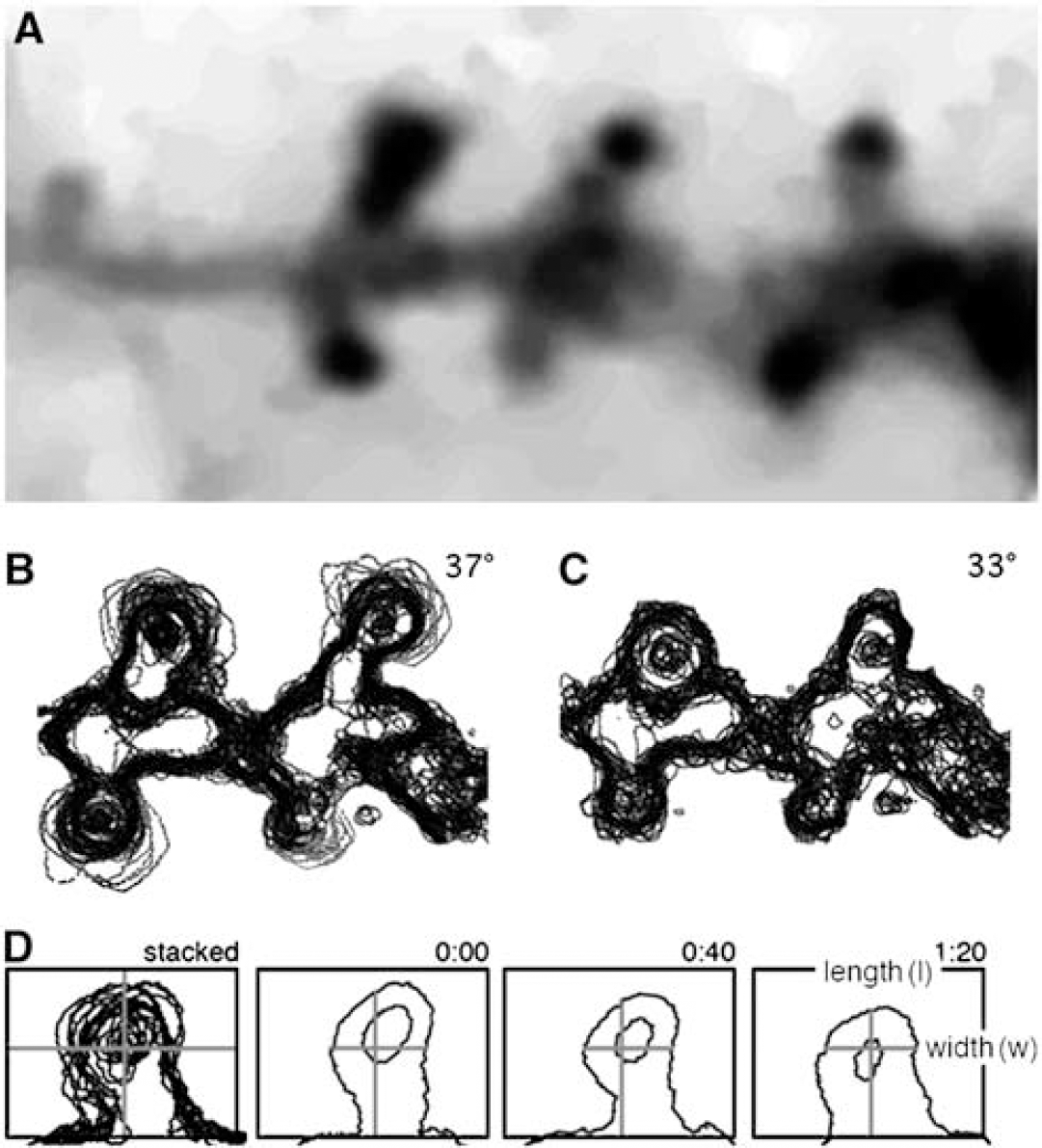
(
The temperature sensitivity of motility was assessed by its Q10 values.
Q10 = motility at temperature 37°/motility at temperature 27°.
Results
Temperature Effect on Cell Survival after In Vitro Ischemia
In cultures exposed to IVI for 9 mins, only low level of cell death was seen, whereas extensive cell death occurred in cultures exposed to more than 15 mins of IVI (Figure 2). Damage after 15 mins of IVI progressed slowly, requiring 48 h to develop fully. After 20 or 25 mins of IVI, cells degenerated faster so that all cells had died by 24 h. These data are in accord with our findings in organotypic hippocampal slices, where a 15 mins ischemic insult under IVI conditions caused intense damage to the CA1 region (Rytter et al, 2003). It is also similar to the temporal development of damage in vivo, where 10 to 15 mins of global cerebral ischemia induces complete degeneration of CA1 neurons (Olsson et al, 2003; Smith et al, 1984). However, the time to ischemic damage after IVI was substantial shorter than when OGD was induced in dissociated cultures in a glucose-free medium (Meli et al, 2002; Velly et al, 2003; Wie et al, 1999).
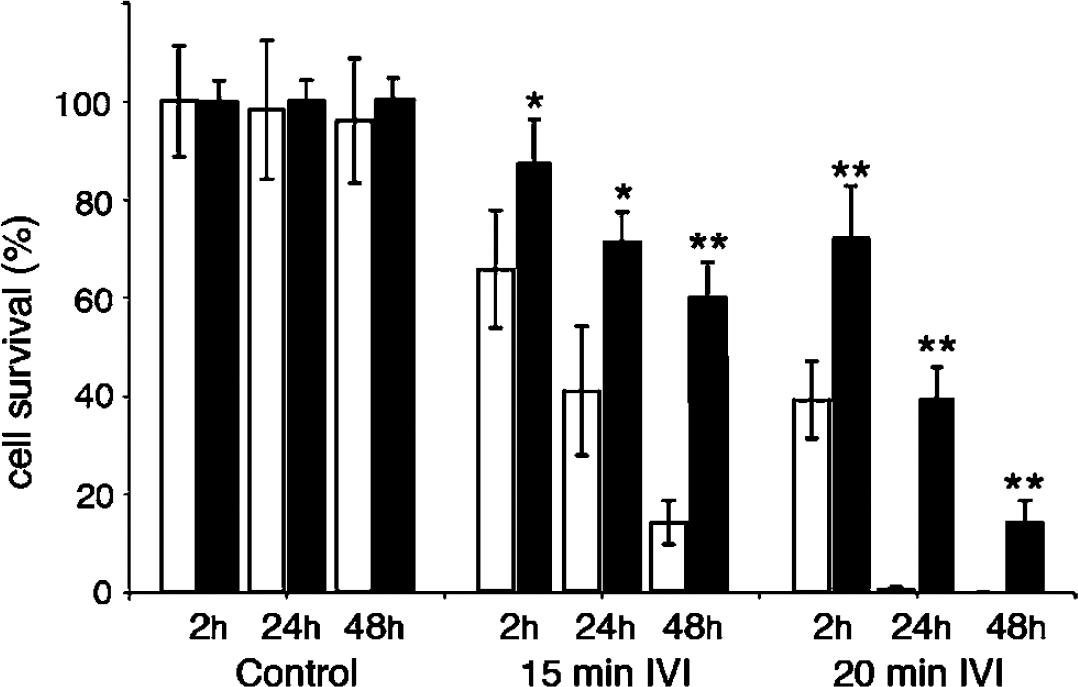
Cell survival after 15 and 20 mins of IVI at 2, 24, and 48 h of recovery. In vitro ischemia was performed at 37°C (white bars) and 33°C (black bars) in comparable cultures. The mean values at each time point were compared using Students t-test. *P<0.05, **P<0.01. Data are mean of±s.d. n = 3 to 7.
Decreasing the temperature of the medium to 33°C during IVI dramatically diminished cell death so that cell survival after 15 mins of IVI increased from 14%±4% to 60%±5% at 48 h of recovery (Figure 2 and Figure 3). There was also significant protection after 20 mins of IVI. After both 15 or 20 mins of IVI at 22°C and 27°C, no cell death could be discerned after 2 h of recovery, indicating substantial protection from acute damage. However, at later recovery times (24 and 48 h), the extent of cell death increased at these relatively low temperatures (data not shown).
Spine Motility and Temperature
GFP-actin-containing spines displayed the typical range of morphologies; thin, stubby, mushroom- and cup-shaped spines (Hering and Sheng, 2001). The majority of the spines were stubby and mushroom-shaped. Decreasing the temperature from 37°C to 33°C or 27°C markedly decreased the motility of the spines. Six spines (stubby or mushroom-like) were examined at 37°C and at 33°C in 30 consecutive time frames (Figure 4A and Figure 4B). An additional set of six spines was examined at 37°C and 27°C (Figures 4C and Figure 4D). The variations in the w/l spine ratio at 37°C were much larger than that from spines at 33°C or 27°C (Figures 4A and Figure 4D). When the spines perfused with medium at 33°C were returned to 37°C, spine motility recovered within minutes (data not shown). The mean values of variability in spine w/l ratio, between the highest and lowest ratio over a 3-mins period, are displayed in Table 1. Compared with 37°C, spine motility decreased by approximately 50% at 33°C and by 70% at 27°C. From the data displayed in Table 1, the Q10 value of the temperature dependence of the motility was calculated to approximately 3.9. The mean spine length calculated from 10 consecutive time-lapse images of 20 different spines from four different neurons is shown in Figure 5. Spine length decreased from 1.8±0.3 to 1.4±0.3 μm, when the temperature was decreased from 37°C to 33°C. It did not decrease further at 27°C (Figure 5) or 22°C (data not shown).

Phase-contrast micrographs of control hippocampal neuronal cultures (
Effect of temperature on spine motilitya
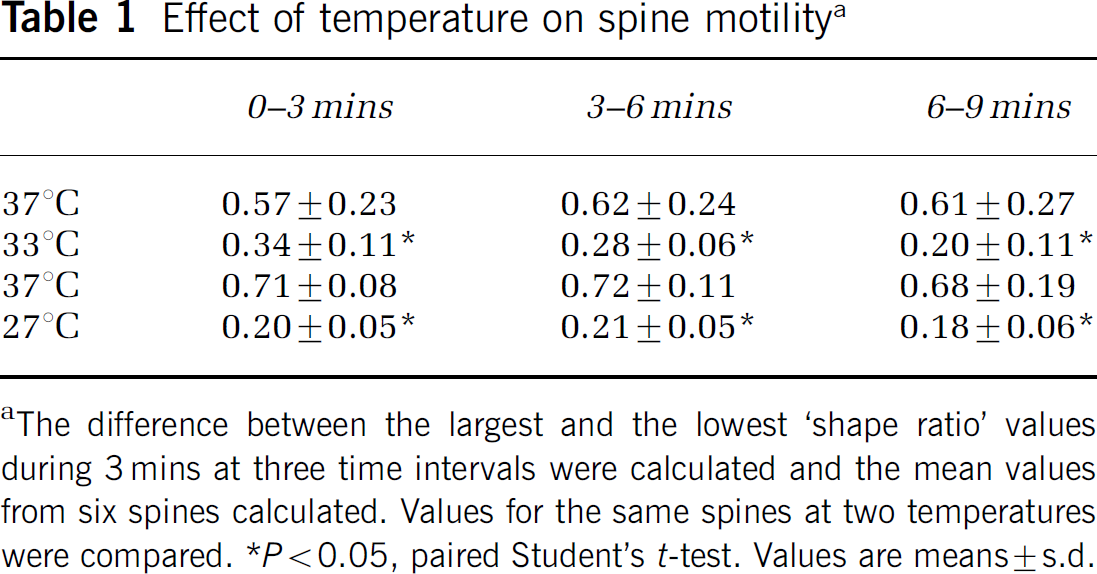
The difference between the largest and the lowest ‘shape ratio’ values during 3 mins at three time intervals were calculated and the mean values from six spines calculated. Values for the same spines at two temperatures were compared.
P<0.05, paired Student's t-test. Values are means±s.d.
In Vitro Ischemia-Induced Spine Loss
The spine number per 10 μm was counted over time in seven different segments during and after 15 mins of IVI insult at 37°C and 33°C, respectively (Figure 6 and Figure 7). Under control conditions (no IVI) spine density was the same (6 spines/10 μm) at 37°C as at 33°C. During IVI induced at 37°C, spine density decreased to 2 spines/10 μm after 9 mins and to 1 spine/10 μm after 12 and 15 mins of IVI. At 33°C, the decrease in spine density after IVI was significantly slower than at 37°C. There was no significant effect after 7 mins of IVI; moreover, spine loss never reached the levels seen at 37°C. During the recovery phase, spine density returned to normal levels 48 h after IVI at 33°C, wheras after IVI at 37°C, spines never recovered. We also found some dendritic protrusions that were stable, with little or no detectable motility, but with similar morphology and GFP distribution as the motile spines. Most of these stable spines did not retract in response to IVI.
In vitro ischemia also induced spine length reduction, although to a lesser extent at 33°C than 37°C (Figure 8). In a time-dependent manner, IVI also induced complete spine retraction. Before IVI, the mean spine length was 1.8±0.1 μm at 37°C and 1.2±0.05 μm at 33°C. During IVI the length of the spines, which had not fully retracted, decreased to 0.9±0.2 and 0.7±0.05 μm at 37°C and 33°C, respectively. At both temperatures, the number of spines on 50 μm long dendritic segments decreased from 25 to 3.5 after 15 mins of IVI. After IVI at 33°C, spine length and number recovered within 48 h whereas after IVI at 37°C spine length recovered, but only one-third of lost spines re-emerged.
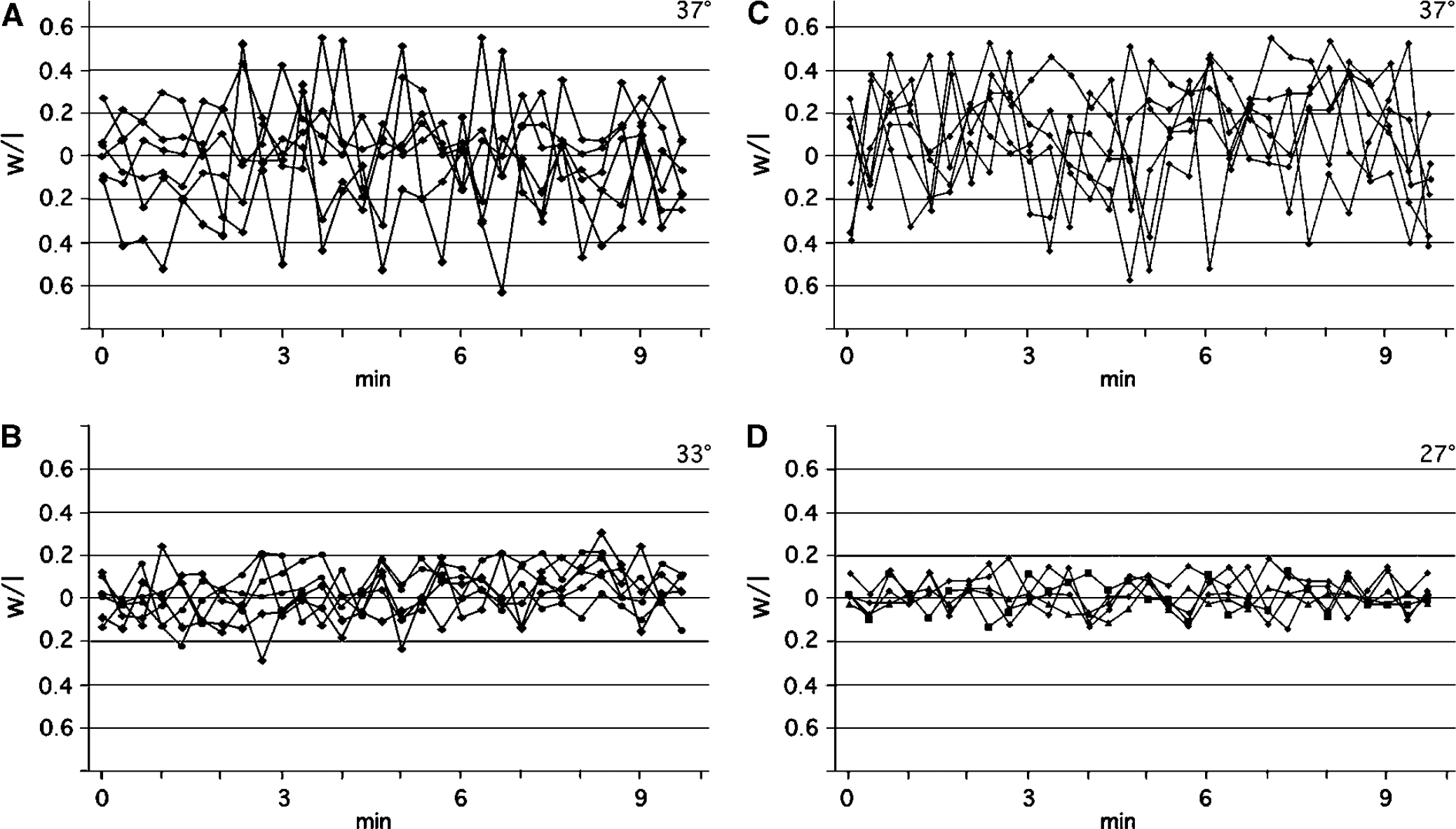
The effect of temperature on spine motility under normal oxygen and glucose conditions. Normalized ‘shape ratios’ plotted against time for six individual spines at two different temperatures are shown in (
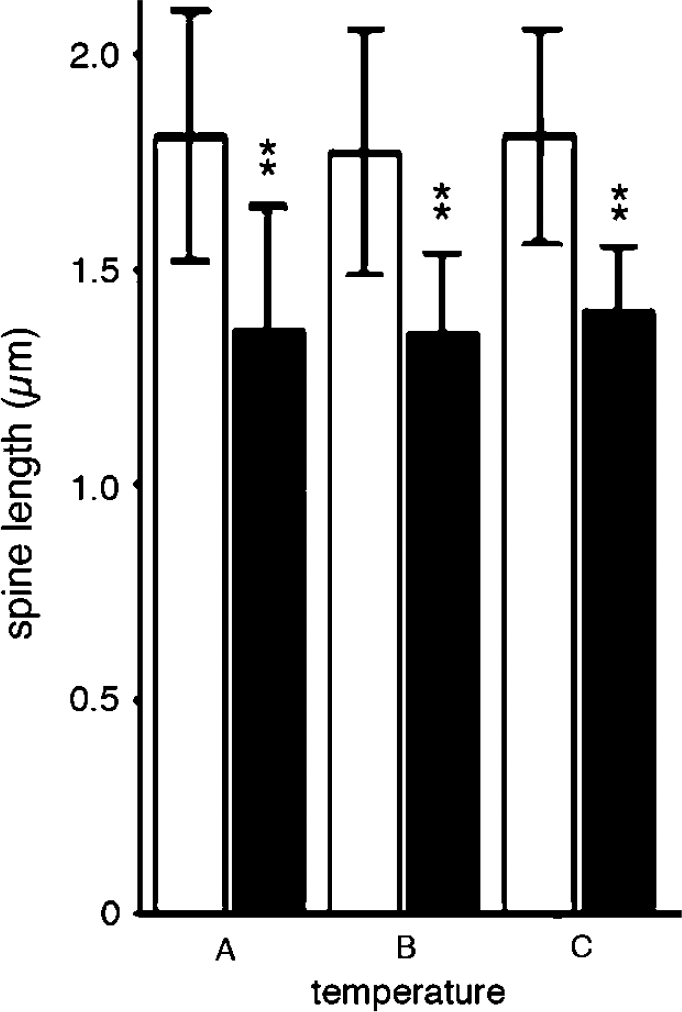
Spine length and temperature under normal oxygen and glucose conditions. The mean length of 20 spines from four different neurons measured at (
Effect of In Vitro Ischemia on Spine Motility and Morphology
In addition to the loss of spines during IVI, the neurons underwent dramatic actin-based morphological changes, including filipodia sprouting as well as the formation of bead-like swellings on the dendrites (Figure 7). Many of these bead-like GFP-actin-containing swellings appear where spines were lost during IVI. Filopodia were also frequently formed at these swellings. The dendritic segments between the swellings were often markedly thin and with no spines present. During IVI at 33°C, dendritic swellings were less pronounced or absent but, still, GFP-actin accumulated in the main dendritic trunk in proportion to the number of spines that had been retracted. During recovery, GFP-actin that had accumulated in the dendrite shaft dispersed and spines reappeared, albeit not always at the same position as before IVI. Spine motility was slightly reduced during IVI at 37°C (Table 2), whereas IVI at 33°C did not reduce motility further.
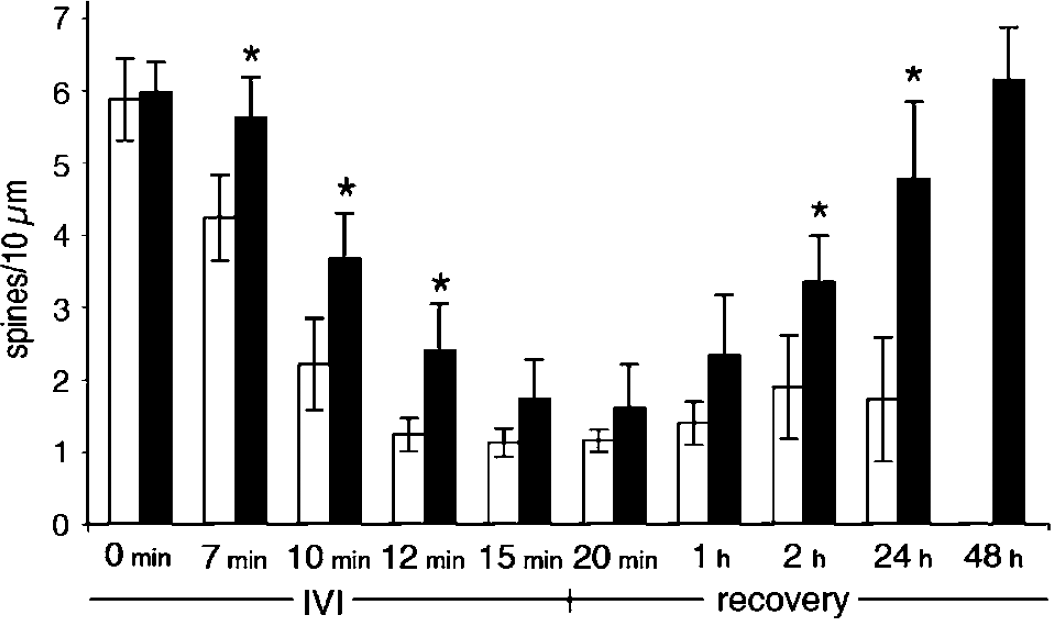
Spine loss after IVI. The number of spines/10 μm was calculated during IVI and during recovery after a 15-min insult at 37°C (white bars) and 33°C (black bars). Values are means calculated from seven different 100-μm-long dendritic segments at both temperatures. *P<0.05 using paired Student's t-test. Values are mean±s.d.
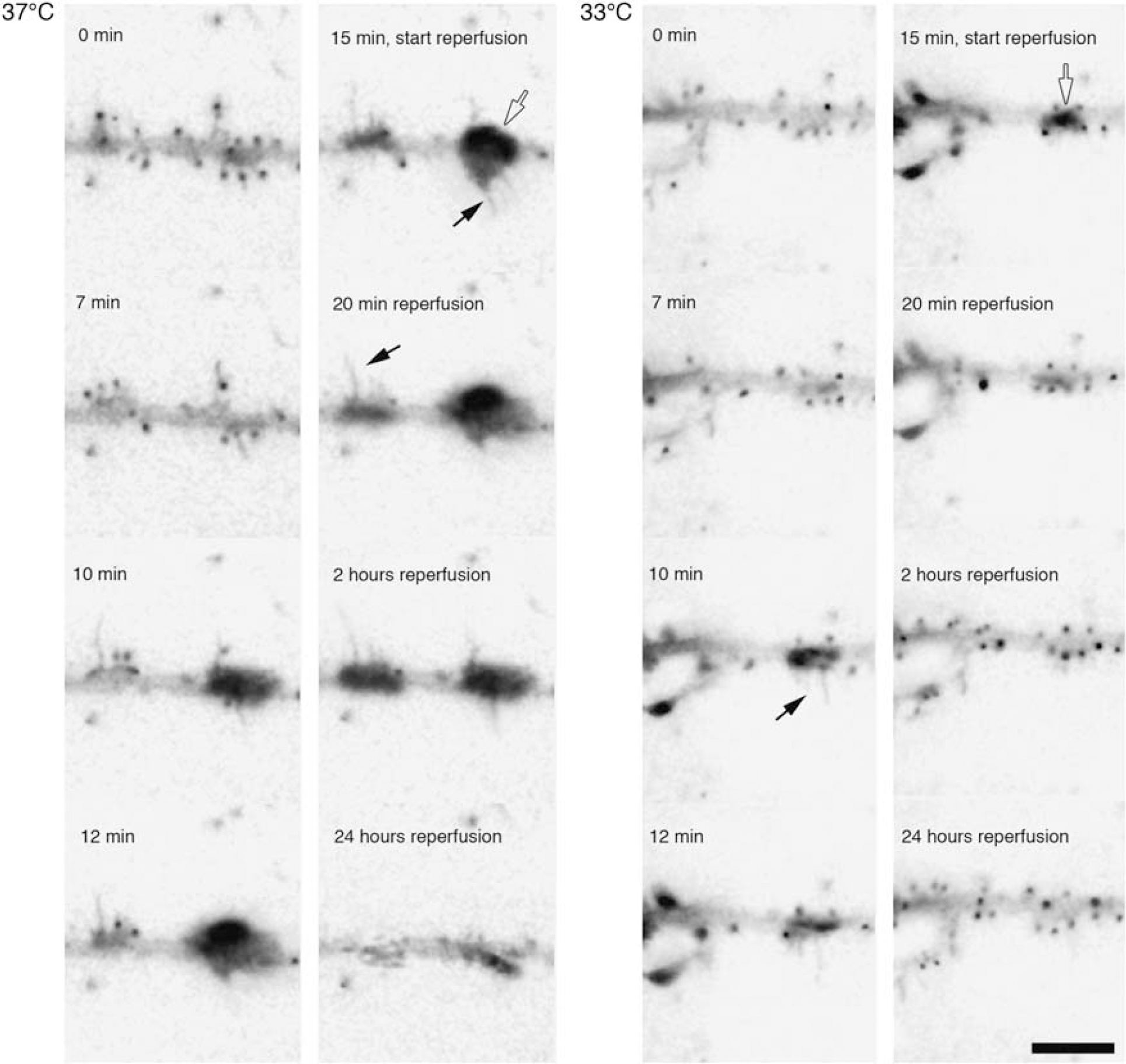
Morphological changes in dendrites induced by IVI. Two series of inverted micrographs showing changes induced during and after 15 mins of IVI at 37°C and 33°C. Polymerized actin in spines appear as black dots along the dendrites and in dendritic swellings (open arrows). Black arrows indicate IVI-induced filopodia. Scale bar, 10 μm.
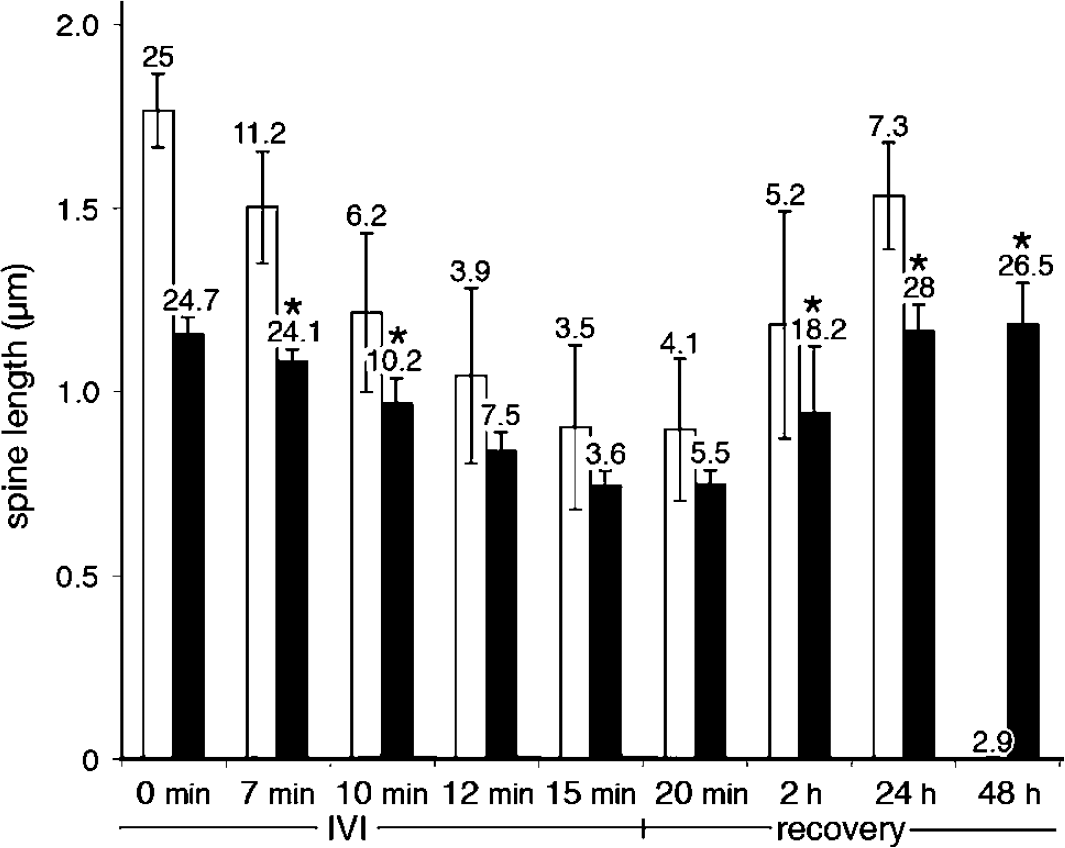
Spine length and density during and after 15 mins of IVI. Values are means of the lengths of visual spines from four 50-μm-long dendritic segments at either 37°C (white bars) or 33°C (black bars), during and after the IVI insult. In vitro ischemia induces spine loss and the numbers denoted above the bars indicate the number of measurable spines. The mean values for the number of spines at each time point were compared. *P<0.05, using Student's t-test. Values are mean±s.d.
Effect of in vitro ischemia on spine motility at 37°C and 33°Ca
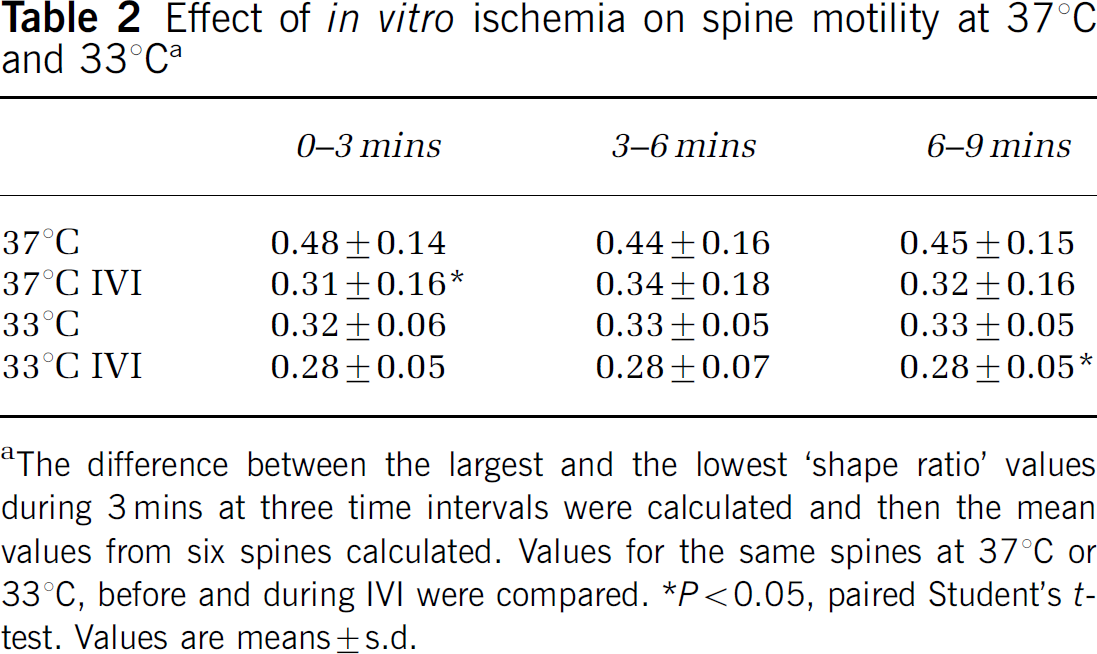
The difference between the largest and the lowest ‘shape ratio’ values during 3 mins at three time intervals were calculated and then the mean values from six spines calculated. Values for the same spines at 37°C or 33°C, before and during IVI were compared.
P<0.05, paired Student's t-test. Values are means±s.d.
Discussion
In this study, we show that small changes in temperature dramatically affect actin dynamics and dendritic spine remodeling. We found that a decrease in temperature from 37°C to 33°C, a magnitude that protects neurons against damage, diminishes spine motility and decreases spine length.
Spine Motility, Cell Death, and Temperature
Actin-based cell motility is highly temperature dependent. In keratocytes, motility is low at 30°C and increases sharply in the temperature range of 30°Cto38°C (Hartmann-Petersen et al, 2000). Also, lymphocyte movement decreases by 60% when temperature decreases from 36°C to 28°C (Miller et al, 2002). Evidently, temperature strongly influences mechanisms that regulate actin polymerization—depolymerization. From the motility data of keratocytes, a Q10 value of cell motility of approximately 4 can be calculated (Hartmann-Petersen et al, 2000), which is in the same magnitude as the calculated Q10 value (3.9) of spine motility. The effect of temperature on neuronal death is well established. Decreasing the temperature by 4°C to 33°C during ischemia provides full protection against damage (Boris-Moller et al, 1998), and hypothermia instituted at 6 h into the recovery phase is also markedly protective (Coimbra and Wieloch, 1994). Hence, the protective effect of hypothermia appears to be strongly correlated with the actin polymerization—depolymerization cycle.
Mechanisms Regulating Spine Motility and Cell Death
The decrease in spine motility and length at low temperature is not due to a decrease in glutamate release (Bruno et al, 1994, 1989b), because spine motility is inhibited by AMPA and NMDA receptor activation (Fischer et al, 2000). Rather, the temperature effect on spine motility and cell death could be mediated by the Rho and Rac system (Bonhoeffer and Yuste, 2002; Tashiro and Yuste, 2004; Threadgill et al, 1997). Transfection of constitutively active mutant forms of Rho reduces the number of spines on dendrites, while constitutively active forms of Rac enhance spine outgrowth (Luo et al, 1996; Nakayama and Luo, 2000; Tashiro et al, 2000). Interestingly, in cutaneous arteries, the Rho/Rho kinase system is activated by cooling (Bailey et al, 2004). The Rho inhibitor C3 as well as the Rho kinase inhibitor Fasudil decrease ischemic brain injury (Gertz et al, 2003; Laufs et al, 2000; Satoh et al, 2001). Changes in spine morphology are strongly linked to calcium responses, and calcium-binding proteins, such as gelsolin, are involved in actin dynamics (Star et al, 2002; Tashiro and Yuste, 2003). It has been suggested that small elevations in spine calcium decrease spine length, while moderate elevations causes spine elongation, and large influx of calcium cause spine retraction (Bonhoeffer and Yuste, 2002; Harris, 1999; Segal and Andersen, 2000). In spines with long necks, [Ca2+] rises faster and decays slower than in spines with short necks (Korkotian and Segal, 2000; Majewska et al, 2000; Volfovsky et al, 1999). Therefore, a shorter spine would provide less calcium load during an ischemic insult, and contribute to the protection mediated by hypothermia.
During IVI, GFP-actin was accumulated in the dendritic trunk at sites where spines have collapsed. This is in accord with other studies showing that actin depolymerization—repolymerization occurs during ATP depletion (Hinshaw et al, 1988, 1991; Jahraus et al, 2001). When ATP is depleted, ADP levels increase and ADP—G-actin is formed, releasing ATP from ATP—G-actin, thereby mobilizing ATP for cellular use. The affinity of ADP—G-actin for thymosin is 100 times lower than that of ATP—G-actin. Hence, under ATP-depleting conditions, the levels of free ADP—G-actin increase, and, once levels favorable for polymerization are reached, F-actin is formed (Atkinson et al, 2004). This explains the disappearance of GFP-actin in spine heads and the subsequent formation in the dendritic swellings during IVI at 37°C and 33°C. At 33°C, accumulation occurs to a lesser extent, indicating that ATP levels are also markedly diminished. This is in accord with the observation that mild hypothermia does not prevent the decrease in ATP levels during ischemia (Busto et al, 1989b), though the speed of ATP depletion is slower (Welsh et al, 1990).
The accumulations of F-actin formed during IVI at 33°C redispersed during recovery after IVI (Figure 7) and spines reappear at similar density, albeit in some cases at different locations. The inability of the accumulated dendritic GFP-actin to dissolve and repolymerize in spines during recovery after IVI at 37°C could be due to the loss of homeostasis regulating formation of actin cytoskeleton, such as mitochondrial ATP generation, calcium homeostasis and imbalance in the activation of regulatory proteins.
Consequences of Actin Depolymerization for Cell Survival
The re-organization of actin during mild hypothermia appears to disrupt detrimental signaling in a similar manner as actin depolymerization by cytochalasin D prevents apoptosis (Trapp et al, 2001) and ischemic damage (Harms et al, 2004), and gelsolin diminishes ischemic brain damage (Harms et al, 2004). Generally, hypothermia may affect protein—protein interactions in spines including receptor-scaffold protein binding. Disrupting NMDA receptor—PSD95 interactions diminishes cell death after OGD both in vitro and in vivo (Aarts et al, 2002). Thus, the decrease in spine motility and spine length during mild hypothermia, and the retraction of spines in response to IVI appear to be a protective response (Piccini and Malinow, 2001). In addition, in the reperfusion phase, postischemic hypothermia in vivo may diminish actin-based lymphocyte motility, thereby diminishing the inflammatory response. In conclusion, a decrease in brain temperature during and after ischemia decreases the actin polymerization rate, affecting spine and lymphocyte motility, thereby providing protection against ischemic damage.
Footnotes
Acknowledgements
This work was supported by the Swedish Research Council (grant no 8466), the Bergendahl Foundation, and King Gustav the V and Queen Victoria Foundation.