
Abstract
Distinct neuronal populations show differential sensitivity to global ischemia, with hippocampal CA1 neurons showing greater vulnerability compared to cortical neurons. The mechanisms that underlie differential vulnerability are unclear, and we hypothesize that intrinsic differences in neuronal cell biology are involved. Dendritic spine morphology changes in response to ischemic insults
INTRODUCTION
Transient global ischemia occurs in patients after cardiac arrest or can be induced experimentally in animals by carotid artery occlusion. The resulting ischemia causes widespread depolarization of the neuronal plasma membrane, release of the excitatory neurotransmitter glutamate, overexcitation of ionotropic glutamate receptors, leading to sustained elevation of intracellular Ca2+, and consequently a delayed, selective cell death. 1 Pyramidal neurons in the CA1 hippocampal region are the most vulnerable, while their CA3 counterparts are resistant. Although cortical neurons are affected by ischemia, they are less vulnerable than those in hippocampal CA1 after a global insult.2,3 This suggests that different cell type-specific mechanisms are activated in response to insult. These mechanisms remain unclear, although recent work suggests that differential subunit-specific trafficking mechanisms that lead to the surface expression of Ca2+-permeable AMPA receptors (CP-AMPARs) may have a role. 4 However, the signalling pathways that are activated downstream of CP-AMPARs after oxygen/glucose deprivation (OGD) are unknown.
Dendritic spines are dynamic actin-rich structures that compartmentalize the postsynaptic machinery. The shape and size of dendritic spines influence synaptic function, and aberrant spine morphology has been linked to several pathologies that affect cognitive processes, including learning and memory.5,6 A number of studies from different groups have suggested that while cortical neurons exhibit OGD/ischemia-induced spine shrinkage and retraction, hippocampal neurons may exhibit spine growth after insult.7–9 The molecular mechanisms that underlie such changes in spine morphology are unknown. Importantly, a direct comparison of OGD-induced changes in spine size in cortical and hippocampal neurons is lacking. We hypothesize that the intrinsic molecular mechanisms that underlie regulation of spine size differs between neuronal types with differential vulnerability to OGD.
Regulation of the actin cytoskeleton has a crucial role in spine formation, elimination, and dynamic changes in spine morphology. 10 The Rho family of small GTPases is critically involved in the regulation of the spine actin cytoskeleton, for example, Rac1 and Cdc42 regulate dendritic spine maturation as well as structural plasticity of mature spines.11 The precise spatial and temporal regulation of small GTPases depends on their upstream regulators: the guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). 12 Disruption of the appropriate balance of GEF and GAP activity results in aberrant spine development and stabilization, and therefore abnormal neuronal function. 13
Rac1 has been implicated in OGD-induced pathways responsible for delayed neuronal death, neuronal degeneration, and cognitive dysfunction; however, a role in regulating dendritic spine morphology during OGD has not been explored.14,15 Furthermore, the GEFs or GAPs involved in regulating Rac1 during OGD are unknown. Tiam1 is a Rac-specific GEF, which is stimulated by NMDA receptor (NMDAR) activation in a Ca2+-dependent manner. 16 Tiam1 is required for spine and synapse development and it has been shown that a decrease in Tiam1 expression results in reduced spine density. 16
We recently showed that hippocampal and cortical neurons in culture exhibit a similar differential vulnerability to OGD as CA1 and cortical neurons
MATERIALS AND METHODS
Animal care and experimental procedures were conducted in accordance with British animal protection legislation and experimental protocols approved by the British National Committee for Ethics in Animal Research.
Cell Cultures and Transfection
Hippocampal and cortical neurons were prepared by dissection of E18 Wistar rat embryos of either sex. All procedures were approved by and performed in accordance with guidelines of Animals (Scientific Procedures) Act 1986 and the University of Bristol policy on working with animals. Hippocampal or cortical neurons were plated on poly-L-lysine-coated plastic dishes or glass coverslips (25mm diameter; VWR International, Radnor, PA, USA) depending on subsequent use. Primary cell cultures were used for experiments at 15 to 20 days
Oxygen/Glucose Deprivation
Neuronal cultures were washed three times with HEPES buffered saline (25 mmol/L HEPES pH 7.5, 137 mmol/L NaCl, 5 mmol/L KCl, 1.5 mmol/L CaCl2, 1.5 mmol/L MgCl2, 15 mmol/L sucrose for OGD condition or 15 mmol/L glucose for control condition) and incubated in a hypoxic chamber (MACS-VA500-microaerophilic workstation; Don Whitley Scientific, West Yorkshire, UK) for 20 minutes at 37°C, 95% N2 and 5% CO2. For OGD condition, buffer was previously bubbled with nitrogen. Control cultures were incubated under normoxic conditions at 37°C, 5% CO2 for the same time period as the OGD. For biochemical analyses, cells were lysed immediately after OGD in lysis buffer (25 mmol/L HEPES; 150 mmol/L NaCl; 0.5% Triton X-100; 1% protease inhibitors cocktail, Roche, Basel, Switzerland; pH 7.4). Also see ‘live imaging’ section.
Cell Death Assay
Hippocampal cultures were transfected with either the control vector pFIVEGFP or pFIV-shTiam1-EGFP. Five days later, cultures were subjected to 20 minutes of OGD and then returned to their original medium for 48 hours. Neurons were fixed and stained with 1 μg/mL Hoechst (Invitrogen) in phosphate-buffered saline (PBS) to identify fragmented nuclei as an indicator of apoptosis. Image analyses were performed with the experimenter being unaware of the experimental condition.
Active Rac1 and Active Tiam1 Glutathione-S-Transferase Pulldowns
Glutathione-S-transferase (GST)-fusion proteins were immobilized on glutathione agarose beads (Sigma Aldrich, St Louis, MO, USA) in HTG buffer (25 mmol/L HEPES, 150 mmol/L NaCl, 1% Triton X-100, 10% glycerol, pH 7.5) at 4°C for 1 hour. Beads were incubated with hippocampal or cortical lysate for 1 hour at 4°C in lysis buffer. After washing the beads with the same buffer, protein levels were detected by western blot using Rac1 antibody (BD Biosciences, East Rutherford, NJ, USA) or Tiam1 antibody (Bethyl, Montgomery, TX, USA). Glutathione-S-transferase bound to glutathione agarose beads was used as a negative control.
To analyse levels of active Tiam1 in synaptoneurosomes prepared from ischemic rat brains, Wistar rats of either sex at postnatal age 10 to 11 days were subjected to global ischemia. Animals were anesthetized with isoflurane (3% induction and 1% maintenance), 60% nitrous oxide, and 37% oxygen. After a midline incision, two 6.0 silk ligatures were placed around the left and right common carotid arteries and the arteries ligated. The arteries were permanently divided between the ligatures. Rats recovered from anesthesia for 10 to 20 minutes, before being euthanized. Sham-operated littermates were used as controls. Brains were removed immediately, and hippocampal and cortical regions were isolated. Crude synaptoneurosomal preparations were obtained using standard procedures; briefly, brain tissue was homogenized in 1 mmol/L HEPES pH 7.4, 0.32 mol/L sucrose, 1 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L NaHCO3 and protease inhibitors cocktail (Roche, Basel, Switzerland) in a glass-teflon homogenizer. Tissue was centrifuged at 1,000 g for 10 minutes at 4°C to remove the nuclear fraction. Resulting supernatant was centrifuged at 13,000 g for 15 minutes at 4°C to yield the crude synaptoneurosomal fraction, which was subsequently resuspended in lysis buffer. Glutathione- S-transferase pulldowns were then performed as described for cultured neurons.
Drug Treatments
1-Naphthylacetyl spermine (NASPM, 30 μmol/L; Tocris, Minneapolis, MN, USA) and D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5; 50 μmol/L, Tocris) were used to block CP-AMPARs or NMDARs, respectively. Oxygen/ glucose deprivation was performed in the presence of either drug (in the case of D-AP5, primary cultures were preincubated with 50 μmol/L of the drug for 5 minutes before OGD and for the duration of OGD). Rac1 or Tiam1 GST pulldowns were performed immediately after OGD. To block CaMKII activity, hippocampal cultures were preincubated with 10 μmol/L KN-62 (Sigma-Aldrich) 30 minutes before OGD and for the duration of the OGD. Tiam1 GST pulldowns were performed after OGD in the presence of the inhibitor.
DNA Constructs
Complementary oligos (Eurofins MWG Operon, Ebersberg, Germany) were targeted against bp 772 to 792 of the rat and mouse Tiam1. They were annealed and inserted in the BbsI/EcoRI sites of the pFIV vector: 5'-AAAGCGGCGGAATTTGGTGTCGGATATTCTCGAGAATATCCGACACCAAATT CCGTTTTTG-3' and 5'-AATTCAAAAACGGAATTTGGTGTCGGATATTCTCGAGA ATATCCGACACCAAATTCCGCCG-3'. EGFP was inserted in the sites
Immunocytochemistry
Neurons were plated on coverslips and analyzed at 15 to 18 days
Live Imaging
Live confocal images of dendrites were taken using a × 60 oil-immersion objective of a Nikon Eclipse Ti-E microscope (Tokyo, Japan). Neuronal cultures were placed on a heated stage set at 37°C and they were constantly perfused with HEPES buffered saline at 37°C at a flow rate of 3 mL/min. Z-stacks of 8 to 18 images were acquired every 10 minutes at 512 × 512 resolution with a z-stack step size of 0.4 μm. An initial z-stack was taken of the resting neuron in the presence of glucose and oxygen. The buffer was then switched to glucose-free HEPES buffered saline bubbled with nitrogen for OGD. Neurons were perfused with this buffer for 20 minutes and then the buffer was switched to glucose-containing HEPES buffered saline in normoxic conditions.
Maximum intensity projections were generated, and the cross-sectional area of dendritic spines was measured using the ImageJ software (NIH, CA, USA).
RESULTS
Oxygen/Glucose Deprivation Induces Differential Spine Shrinkage in Cortical and Hippocampal Neurons
Previous studies have shown that dendritic spine morphology is affected by OGD, but the details are unclear.7–9,17 We used live confocal imaging to directly compare OGD-induced changes in spine morphology on cultured hippocampal and cortical neurons. The spines of both cell populations rapidly shrank during OGD, with significant shrinkage observed after 10 minutes of OGD, and this effect was persistent after re-exposure to normal medium containing oxygen and glucose (Figures 1A to 1D). Interestingly, spine shrinkage on cortical neurons (∼54% shrinkage at 40 minutes time point compared with 0 minute) was significantly more pronounced than on hippocampal neurons (∼22% shrinkage at 40 minutes time point compared with 0 minute; Figures 1B to 1D). The initial spine shrinkage, observed at 10 minutes after the onset of OGD, was not significantly different between cell types. After this time, spines on hippocampal neurons were stable, whereas cortical neurons continued to shrink during OGD exposure, suggesting a divergence of mechanisms regulating spine size in the two neuronal populations. To investigate whether spine shrinkage during OGD resulted in a reduction in the total number of spines, we analyzed the linear spine density in both types of neuron. Spine density was not affected in hippocampal or cortical neurons during OGD (Figure 1E).
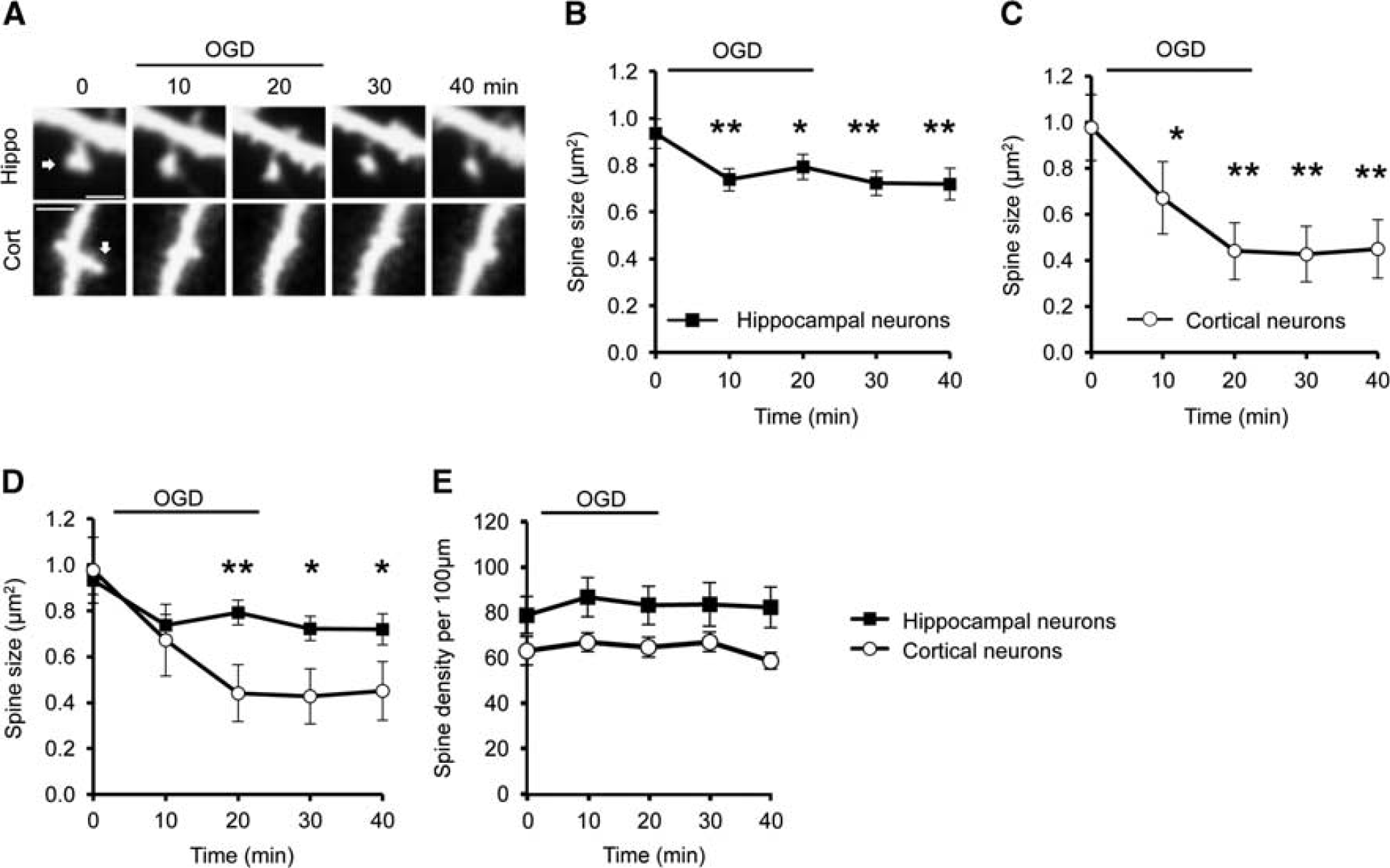
Oxygen/glucose deprivation (OGD) triggers differential spine shrinkage in cortical and hippocampal neurons. (
Rac1 and Tiam1 Are Differentially Regulated in Response to Oxygen/Glucose Deprivation in Cortical and Hippocampal Neurons
Since Rac1 and Cdc42 are involved in regulating dendritic spine morphology, 11 we hypothesized that differential GTPase activation might have a role in the observed spine remodelling in response to OGD. To analyze the endogenous levels of active GTP-bound GTPases in neurons in response to OGD, we used a GST fusion of the CRIB domain of p21-activated protein kinase 1 (PAK1), to specifically pull down the active state of Rac1 and Cdc42. Oxygen/ glucose deprivation caused a marked decrease in the proportion of activated Rac1 in cortical neurons, in contrast to hippocampal neurons, which showed a significant OGD-induced increase in activated Rac1 under the same conditions (Figure 2A). In contrast, OGD caused a decrease in the activated state of Cdc42 in both hippocampal and cortical neuronal cultures (Figure 2B). These results suggest a differential regulation of a Rac1 pathway in hippocampal and cortical neurons in response to OGD.
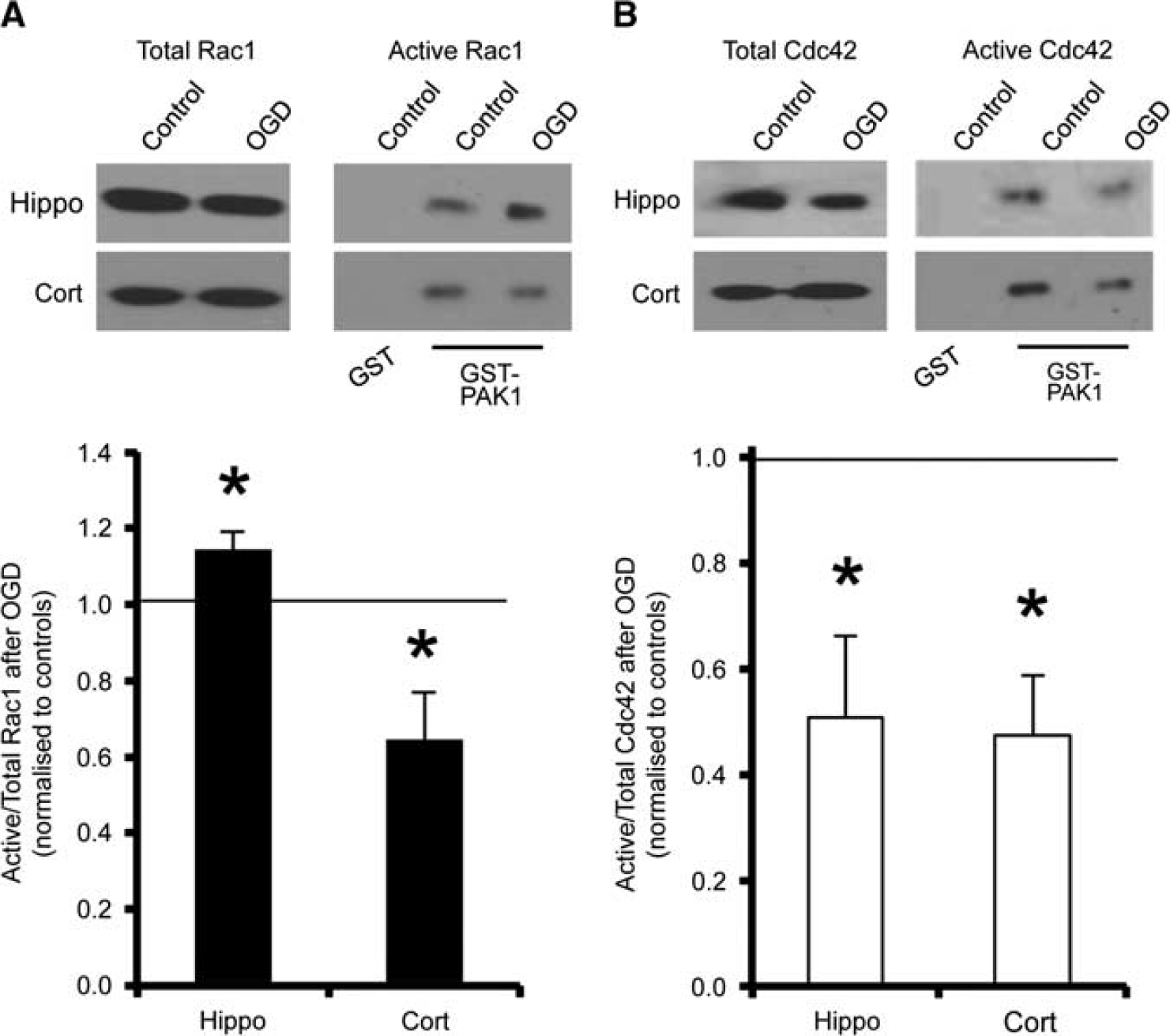
Oxygen/glucose deprivation (OGD) causes differential activation of Rac1, but not Cdc42, in cortical and hippocampal neurons. (
To investigate upstream regulators that control Rac1 activation during OGD, we focused on the Rac-specific GEF Tiam1, since a Tiam1/Rac1 pathway has previously been suggested to regulate spine morphology under basal conditions.
16
Initially, we analyzed endogenous expression of Tiam1 in hippocampal and cortical neurons by western blotting, which indicated that Tiam1 expression is significantly higher in hippocampal neurons compared with cortical neurons (Figure 3A). These results were in accordance with a previous study suggesting that Tiam1 is expressed at a higher level in the hippocampus compared with cortex in the mouse brain.
18
As well as total Tiam1 expression, we analyzed the proportion of Tiam1 activated in response to OGD. RacG15A is a nucleotide-free mutant of Rac1 that has a high affinity for activated Rac GEFs.
19
Glutathione-S-transferase pulldown assays using GST-RacG15A showed that Tiam1 was activated by OGD in total protein extracts from cultured hippocampal neurons but unaffected in cortical neurons (Figure 3B). To investigate whether Tiam1 activation is differentially activated in cortex and hippocampus after ischemia
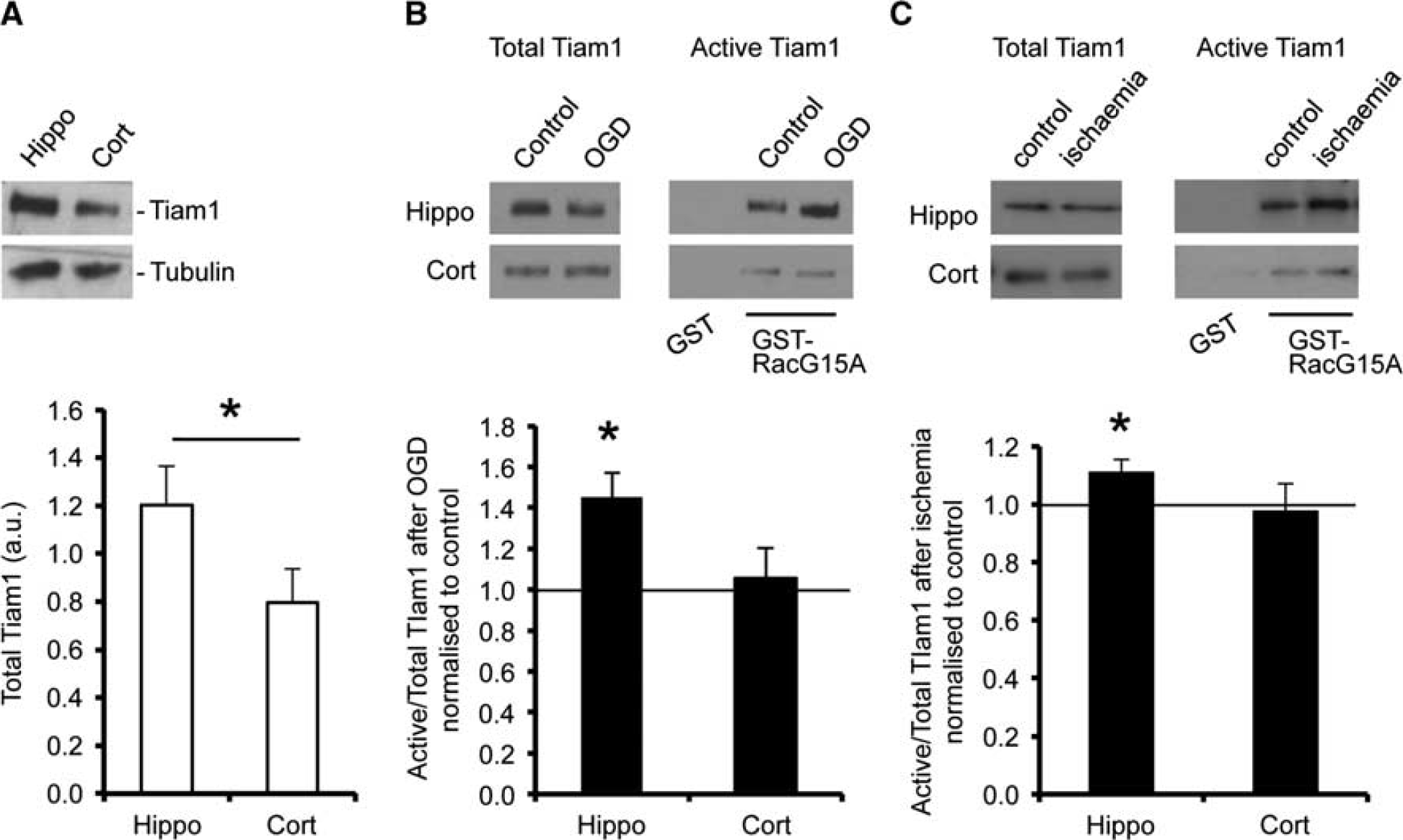
Tiam1 is activated in response to oxygen/glucose deprivation (OGD) in hippocampal, but not in neurons. (
Tiam1/Rac1 Are Activated by a Pathway Involving NMDA Receptors, Ca2+-Permeable AMPA Receptors, and CaMKII in Hippocampal Neurons During Oxygen/Glucose Deprivation
We previously showed that the rapid, NMDAR-dependent surface expression of CP-AMPARs during OGD differs between cortical and hippocampal neurons, and further that CP-AMPARs contribute to the excitotoxic effects of the insult in hippocampal neurons.4,20 We therefore hypothesized that the OGD-induced activation of Tiam1/Rac1 in hippocampal neurons may be downstream of CPAMPAR stimulation. To test this hypothesis, we investigated the effect of NMDAR or CP-AMPAR blockade on OGD-induced Tiam1 activation. When either NMDARs or CP-AMPARs were blocked with D-AP5 or NASPM respectively, the OGD-triggered activation of both Rac1 and Tiam1 in hippocampal neurons was abolished (Figures 4A and 4B).
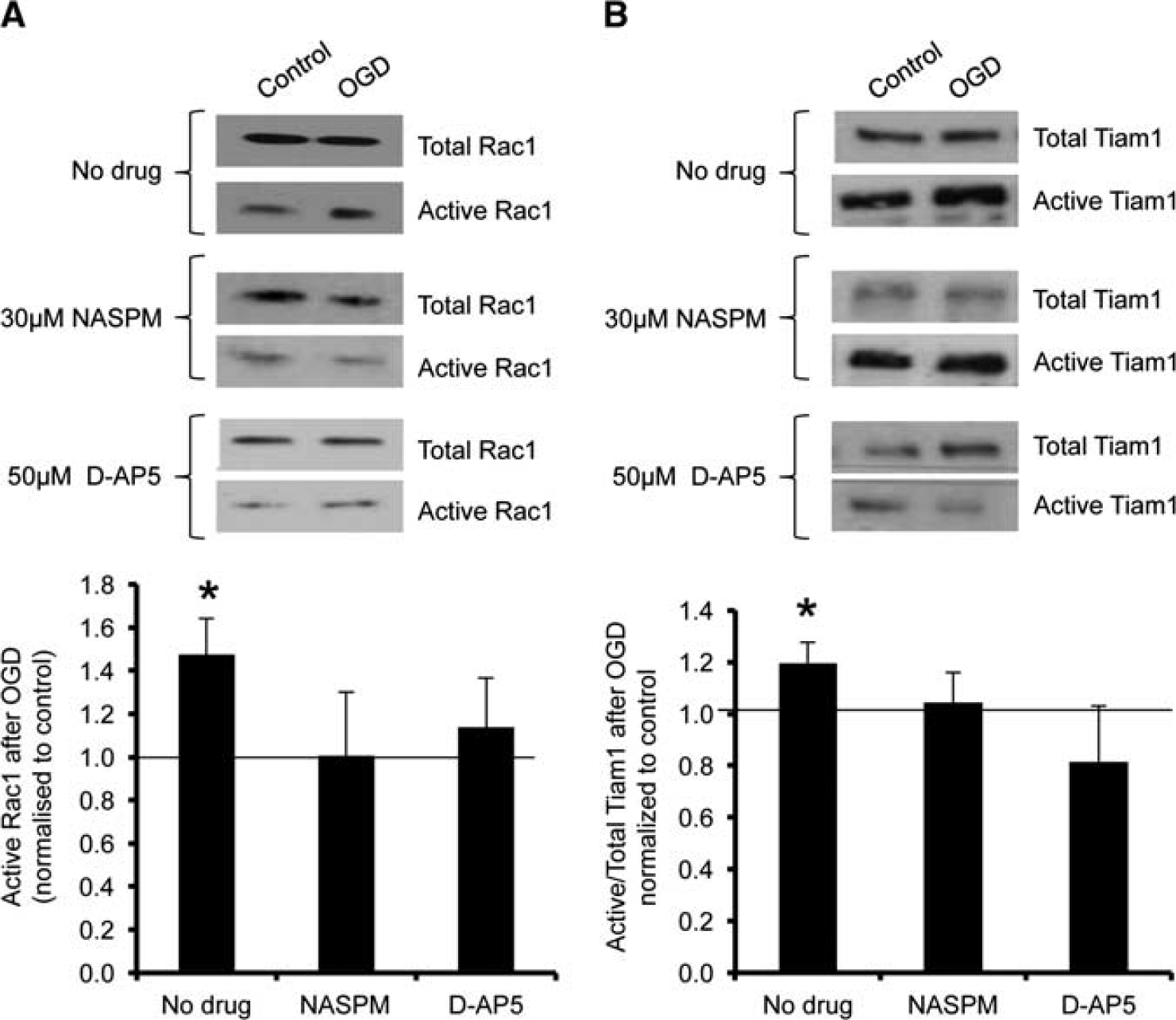
Oxygen/glucose deprivation (OGD)-induced activation of Tiam1 and Rac1 in hippocampal neurons is abolished after blockade of Ca2+- permeable AMPA receptors (CP-AMPARs) or NMDA receptors (NMDARs). (
Previous work in fibroblasts showed that CaMKII can phosphorylate and activate Tiam1, stimulating nucleotide exchange activity toward active Rac1. 21 Furthermore, CaMKII is activated by NMDAR mediated Ca2+ influx. 22 These observations suggested that CaMKII might be an activator of Tiam1 in hippocampal neurons in response to OGD, so we investigated the effect of pharmacological CaMKII inhibition on OGD-induced Tiam1 activation. Figure 5A shows that KN-62 completely blocks the increase in active Tiam1 after OGD. Moreover, endogenous expression of CaMKIIα was higher in hippocampal neurons compared with cortical neurons (Figure 5B), indicating that hippocampal neurons are better equipped to activate Tiam1 via this mechanism.
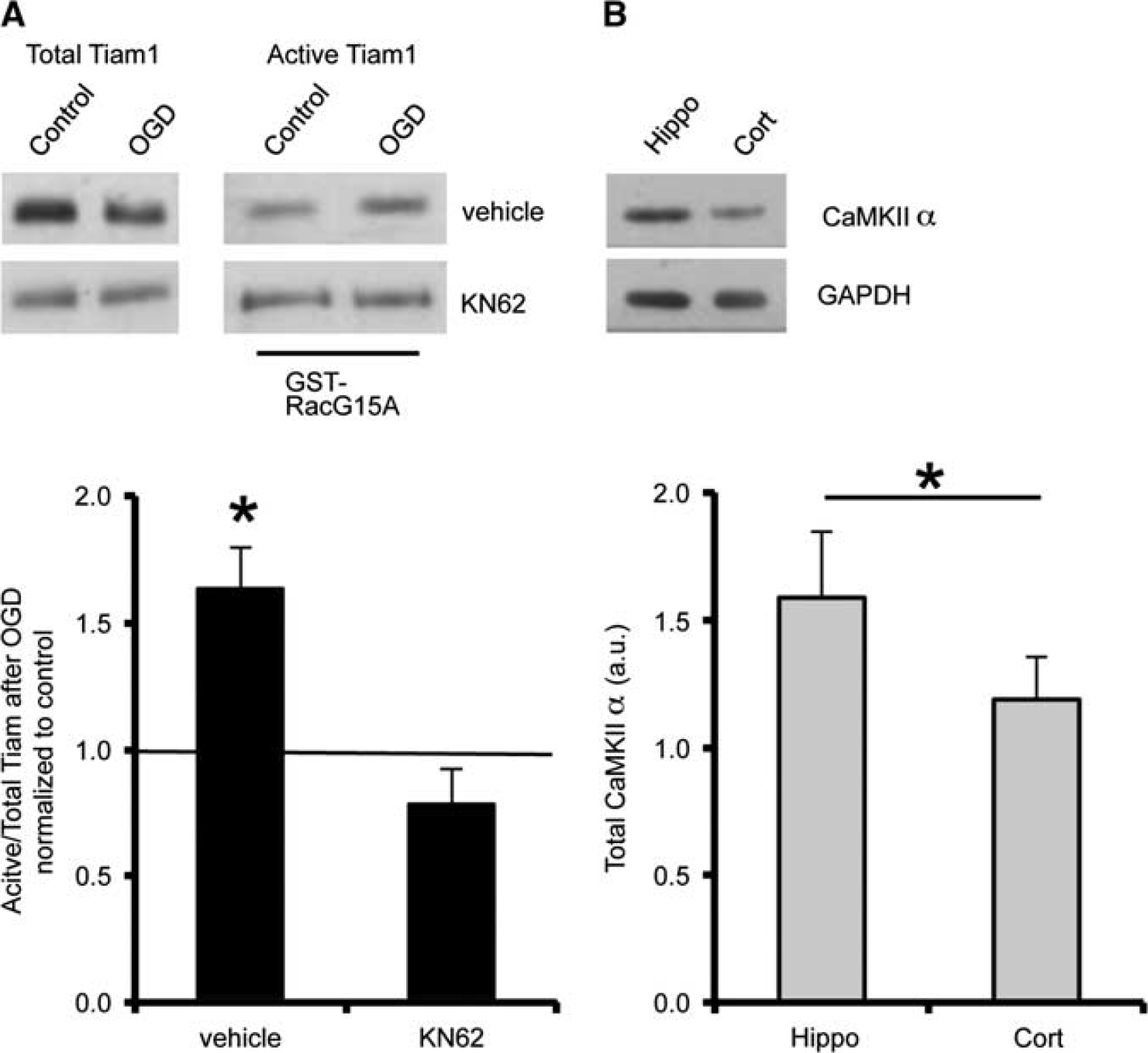
CaMKII activity is required for the oxygen/glucose deprivation (OGD)-induced activation of Tiam1 in hippocampal neurons. (
Tiam1 Knockdown Causes Further Spine Shrinkage in Response to Oxygen/Glucose Deprivation in Hippocampal Neurons
To test the hypothesis that high Tiam1 activity underlies the relatively small OGD-induced spine shrinkage in hippocampal neurons, we generated an shRNA construct to knock down Tiam1 expression. Figure 6 shows that Tiam1 shRNA caused an ∼ 66% knockdown of Tiam1 compared with a scrambled shRNA, 5 days after transfection in cultured hippocampal neurons and that Tiam1 expression is rescued by co-transfection with shRNAresistant Tiam1.
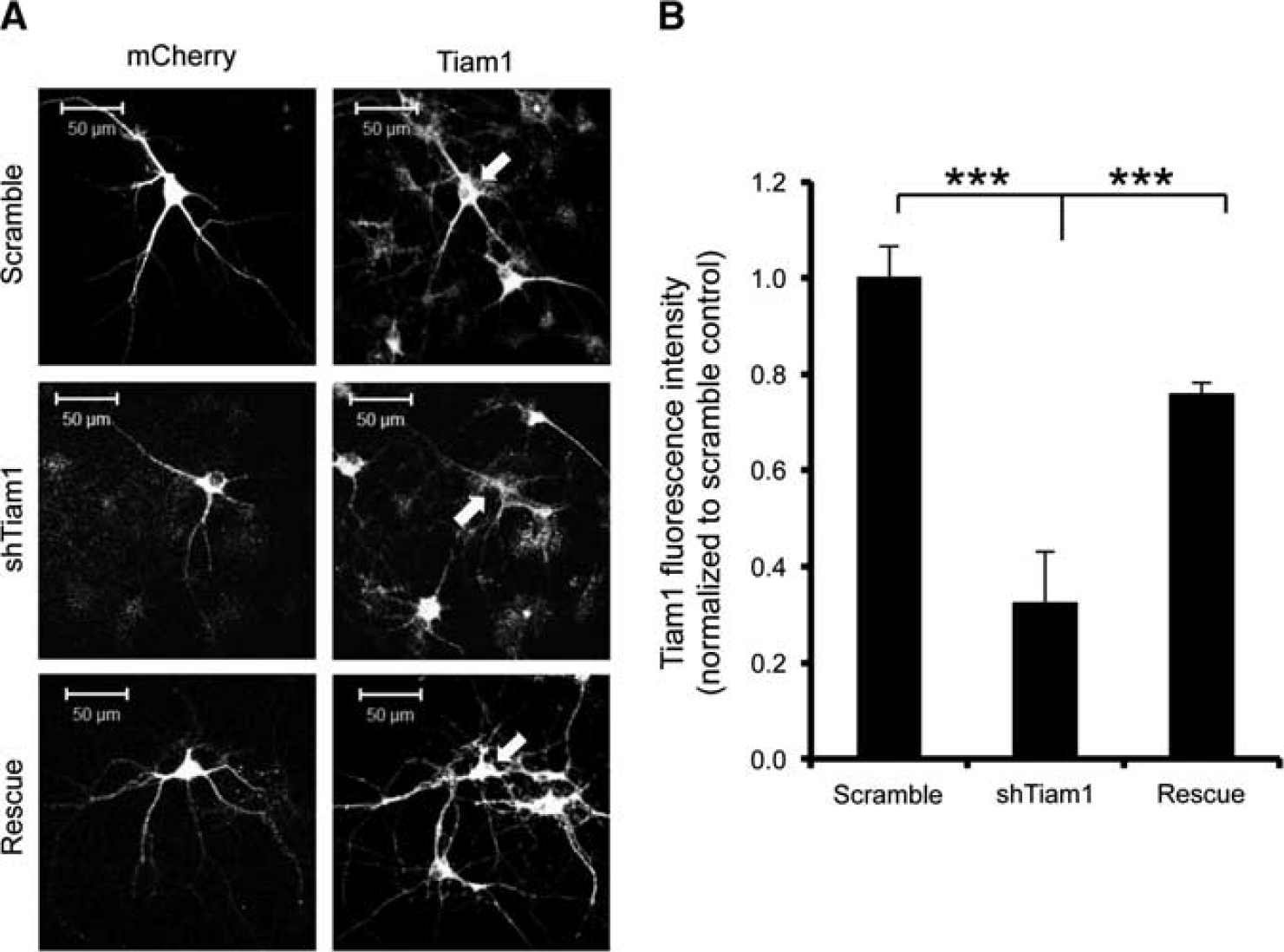
Validation of Tiam1 shRNA. Hippocampal neurons were cotransfected with pFIV-scrambled shRNA-mCherry plus GFP-IRES (control), pFIV-shTiam1-mCherry plus GFP-IRES (shTiam1), or pFIV-shTiam1-mCherry plus GFP-IRES-shRNA resistant Tiam1 (rescue) and Tiam1 expression was visualized by immunocytochemistry and confocal imaging. The mCherry signal was used as a mask and the average Tiam1 fluorescence intensity within that area was determined. Cells expressing Tiam1 shRNA showed a significant decrease in Tiam1 expression compared with controls. Arrows show transfected cells. (
We subsequently expressed Tiam1 shRNA in hippocampal neurons and analyzed OGD-induced spine shrinkage. Neurons expressing Tiam1 shRNA showed a significantly greater degree of OGD-induced spine shrinkage compared with control neurons expressing scrambled shRNA at the 40-minute time point, and the effect of Tiam1 knockdown was reversed by coexpression of the shRNA-resistant Tiam1 (Figures 7A and 7B). These results show that Tiam1 limits spine shrinkage in hippocampal neurons in response to OGD. To test the hypothesis that spine shrinkage reduces neuronal vulnerability to OGD, we compared neuronal death in hippocampal cultures expressing normal and reduced levels of Tiam1. The nuclei of apoptotic cells have a fragmented morphology, in contrast to healthy cells, which have intact, rounded nuclei with high contrast edges. 23 To visualize neuronal nuclei, we stained cultures with Hoechst reagent 48 hours after OGD. Control neurons expressing GFP show a fourfold increase in fragmented nuclei in response to OGD, indicating a high degree of vulnerability to insult. In contrast, the nuclei of neurons expressing Tiam1 shRNA are unaffected by OGD, showing that reducing Tiam1 expression is neuroprotective (Figures 7C and 7D).
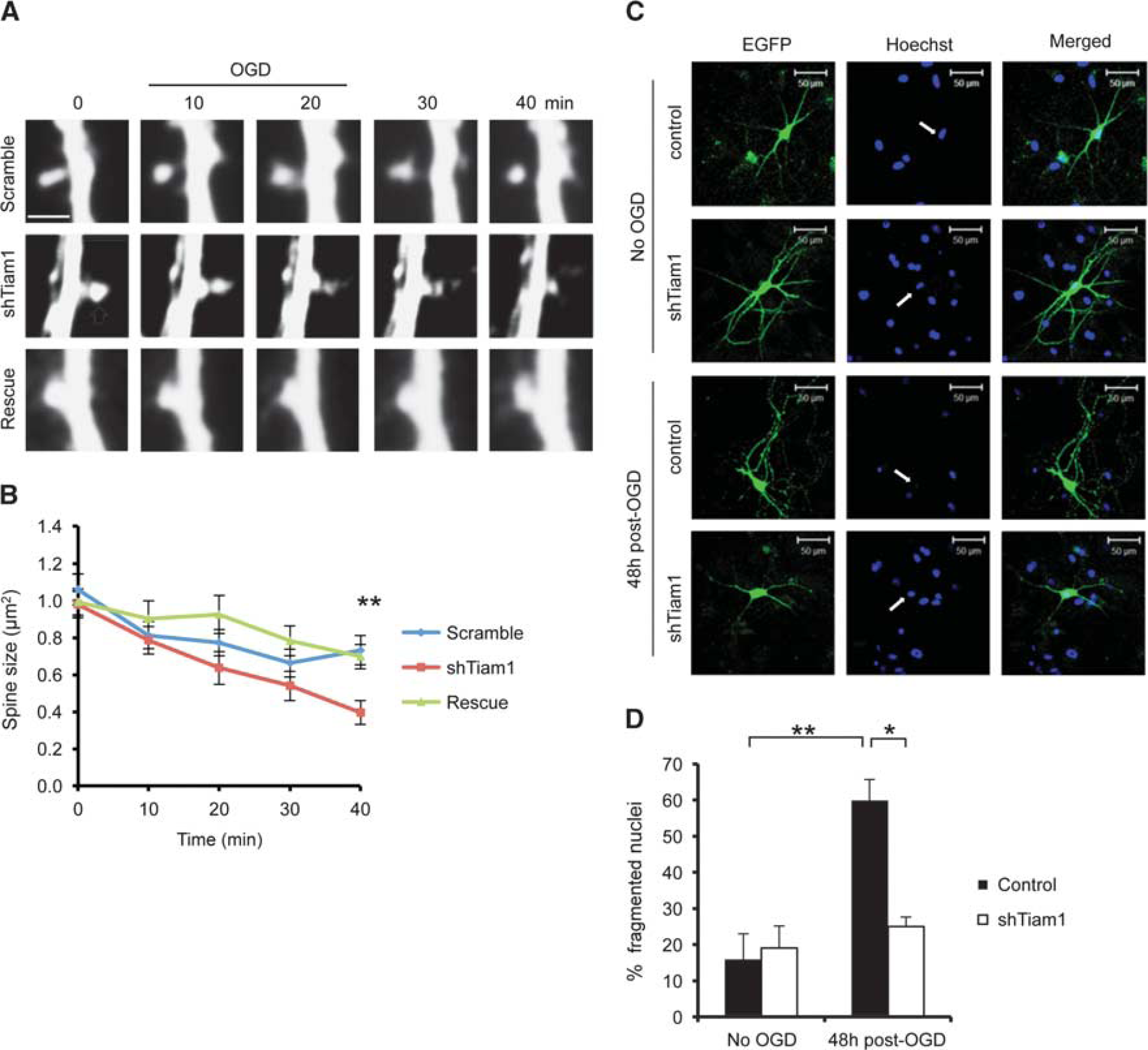
Tiam1 knockdown causes increased spine shrinkage and reduced cell death in hippocampal neurons in response to oxygen/glucose deprivation (OGD). (
DISCUSSION
Our results show that differential Rac1 activation via the Rac-specific GEF Tiam1 underlies distinct OGD-induced morphologic changes of spines in cortical and hippocampal neurons. Moreover, the pathways that regulate Rac1 activation appear to diverge in the two cell types. In hippocampal neurons, high CaMKII and Tiam1 activity lead to an increase in active Rac1 after CP-AMPAR stimulation during OGD. In contrast, Tiam1 activation is unaffected by OGD in cortical neurons, indicating that this pathway is not triggered during OGD in this cell type. Oxygen/glucose deprivation-induced spine shrinkage on hippocampal neurons is significantly less pronounced than on cortical neurons, and Tiam1 knockdown in hippocampal neurons causes an increase in spine shrinkage, showing a critical role for Tiam1 in determining the morphologic response to OGD. The effect of Tiam1 knockdown on OGD-induced spine shrinkage is seen at later time points (20 minutes after insult), but not during OGD exposure. In contrast, the difference between wild-type hippocampal and cortical neurons is evident during OGD. This indicates that the phenotype of hippocampal neurons with reduced Tiam1 is not identical to wild-type cortical neurons, suggesting the involvement of additional cell type-specific factors in OGD-stimulated spine shrinkage.
Activation of Rac1 has previously been shown to cause dendritic spine enlargement after synaptic stimulation or under basal conditions.16,24 In the light of these previous observations alone, our Rac1 activation results would predict an OGD-induced increase in spine size in hippocampal neurons, and a decrease in spine size in cortical neurons. However, our experiments show that hippocampal neurons exhibit a small decrease in spine size in response to insult. This suggests that an additional, Tiam1-independent pathway favoring spine shrinkage is recruited in both cell types in response to OGD. This unknown pathway might involve a Rac1 GAP as the critical regulator, which would account for the OGD-induced Rac1 deactivation observed in cortical neurons, or an alternative Rac1-independent pathway that drives spine shrinkage. The simultaneous increase in Tiam1 activation in hippocampal neurons offsets the effect of this additional pathway and consequently limits OGD-induced spine shrinkage. Further work will be necessary to define the pathway that drives spine shrinkage in response to OGD, and to elucidate the molecular machinery that leads to OGD-induced Rac1 deactivation in cortical neurons.
We show here that activation of the Tiam1/Rac1 pathway during OGD in hippocampal neurons requires NMDAR and CPAMPAR stimulation. Oxygen/glucose deprivation causes a rapid NMDAR-dependent trafficking of CP-AMPARs to synapses in hippocampal neurons that is required for delayed neuronal death, and we recently showed that the AMPAR subunit-specific trafficking mechanisms recruited in response to OGD are different in cultured hippocampal and cortical neurons.4,20 The results from the current study define the CaMKII/Tiam1/Rac1 pathway as a critical downstream effector of CP-AMPAR activation in hippocampal neurons after OGD.
Our results indicate that CaMKII is required for the OGD-induced activation of the Tiam1/Rac1 pathway in hippocampal neurons. It was previously reported that rat strains with a higher endogenous expression of CaMKII showed a higher vulnerability to excitotoxicity in the striatum. 25 A later study found no correlation between CaMKII protein expression levels and vulnerability to insult in different brain areas, but instead suggested that vulnerable striatal neurons exhibited greater CaMKII activity after ischemic insult compared with resistant cortical neurons. 26 These studies therefore support a role for CaMKII activation in ischemic cell death pathways and in the differential responses of distinct neuronal populations to ischemia. CaMKII is known to be activated by NMDAR stimulation. 27 Since we show that the OGD-induced Tiam1/Rac1 activation is CP-AMPAR dependent, our results suggest that under OGD conditions, CaMKII may also be activated by Ca2+ influx mediated by CP-AMPARs.
Changes in dendritic morphology have been previously reported at early stages of global brain ischemia.8,28,29 While a reliable cell type-specific pattern has not yet emerged from the published literature, it appears that cortical neurons generally respond to ischemia with spine retraction.28–30 In contrast, spine growth has been reported in response to ischemia in hippocampal neurons.8,17 Our results are broadly consistent with these observations, however we do not observe spine growth in hippocampal neurons, but instead a very modest shrinkage compared with the dramatic effect in cortical neurons. This discrepancy may reflect the different cellular environment of cultured neurons compared with neurons
Footnotes
The authors declare no conflict of interest.
Acknowledgments
The authors thank M Bass for GST-RacG15A and LZRS Tiam1 constructs, and K Wilkinson for assistance with shRNA design. This work was funded by SyMBaD—ITN Marie Curie Actions 238608.