
Abstract
Inhibitors of HMG-CoA reductase (statins) are potent cholesterol-lowering drugs. Large clinical trials have shown that statins reduce the incidence of cerebrovascular events, which might be surprising because cholesterol is not an established risk factor for stroke. In addition to their cholesterol-lowering properties, statins exert a number of pleiotropic, vasculoprotective actions that include improvement of endothelial function, increased nitric oxide (NO) bioavailability, antioxidant properties, inhibition of inflammatory responses, immunomodulatory actions, regulation of progenitor cells, and stabilization of atherosclerotic plaques. In fact, statins augment cerebral blood flow and confer significant protection in animal models of stroke partly via mechanisms related to the upregulation of endothelial nitric oxide synthase. Retrospective clinical evidence suggests that long-term statin administration may not only reduce stroke risk but also improve outcome. Early secondary prevention trials are underway to test the hypothesis that statin treatment initiated immediately after an event improves short-term outcome. Lastly, recent evidence suggests that sudden discontinuation of statin treatment leads to a rebound effect with downregulation of NO production. Withdrawal of statin treatment may impair vascular function and increase morbidity and mortality in patients with vascular disease.
Keywords
HMG-CoA Reductase Inhibitors (Statins)
Pharmacology and Mechanism of Action
Hydroxymethylglutaryl coenzyme A (HMG-CoA; see Table 1 for a list of abbreviations) reductase, the rate-limiting enzyme of the mevalonate pathway for cholesterol biosynthesis, is present in the liver and nonhepatic tissues, catalyzing the early conversion of HMG-CoA to mevalonic acid (Reinoso et al, 2002) (Figure 1). Via their structural homology to HMG-CoA (as determined by X-ray crystallography; Istvan and Deisenhofer, 2001) statins competitively inhibit HMG-CoA reductase activity. Thus, statins reduce cholesterol content in the liver, which leads to negative-feedback low-density lipoprotein (LDL) receptor upregulation and subsequent lowering of total serum cholesterol levels. While all statins share HMG-CoA reductase inhibition as their common mechanism of action, they differ in absorption, binding, excretion, and solubility and exhibit variable dose-related efficacy in reducing LDL-cholesterol (for details, see Furberg, 1999; Vaughan and Gotto, 2004) (Table 2). Pravastatin and rosuvastastin are very hydrophilic and hardly penetrate cells; they are taken up by the liver via a specific receptor. Simvastatin or lovastatin, however, are relatively lipophilic and also penetrate the blood–brain barrier (BBB), which might be relevant for some of their pleiotropic effects (vide infra).
Abbreviations
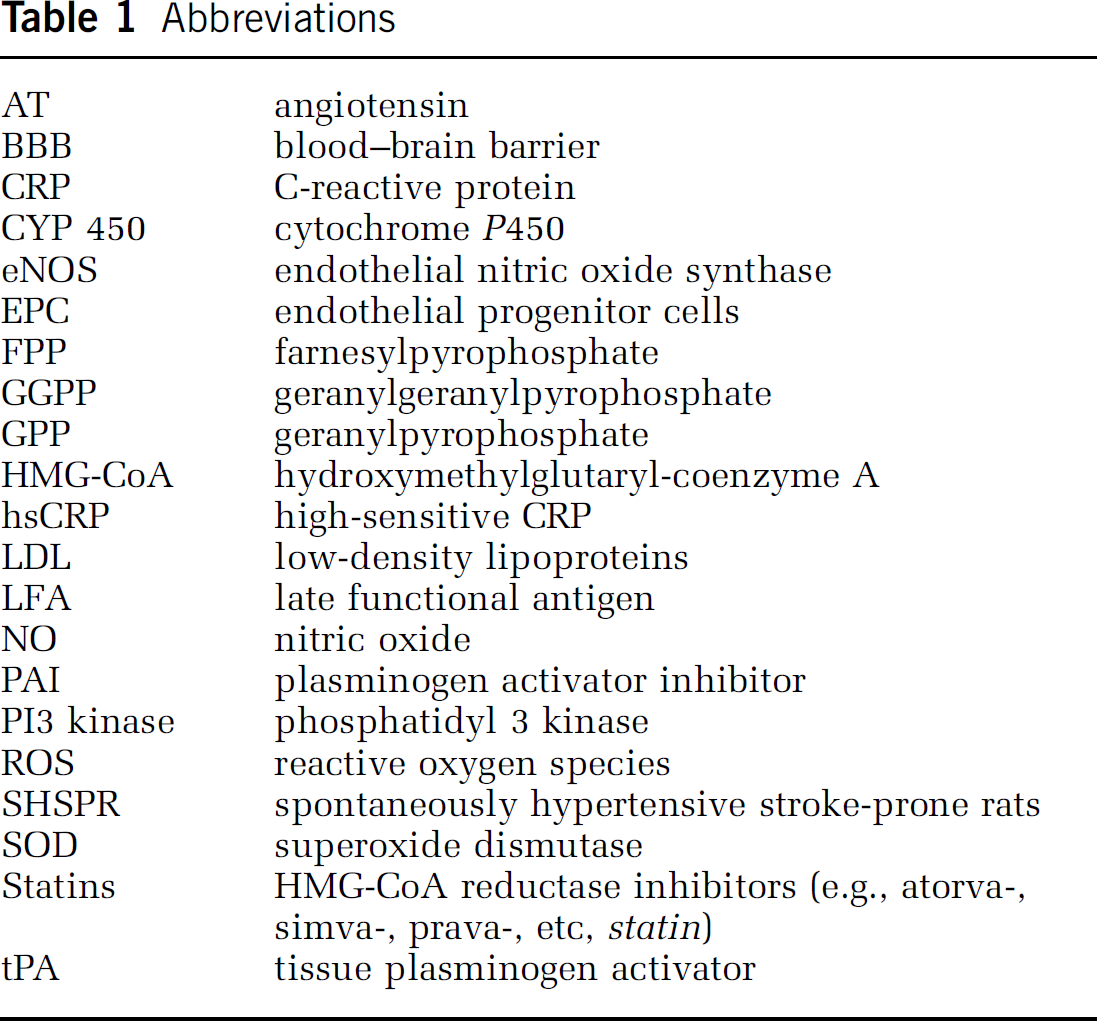
Comparative efficacy and pharmacology for various statins
Modified from Vaughan and Gotto (2004); Laufs (2003); McTaggart et al (2001); Buckett et al (2000) and Dobrucki et al (2001).
IC50 measured in rat hepatocytes.
NO release is the peak NO concentration after stimulation of bovine endothelial cells with 1 μmol/L statin compared as a percentage decrease from the peak concentration after stimulation with 1 mmol/L of the calcium ionophore A23187; CYP = cytochrome P450.
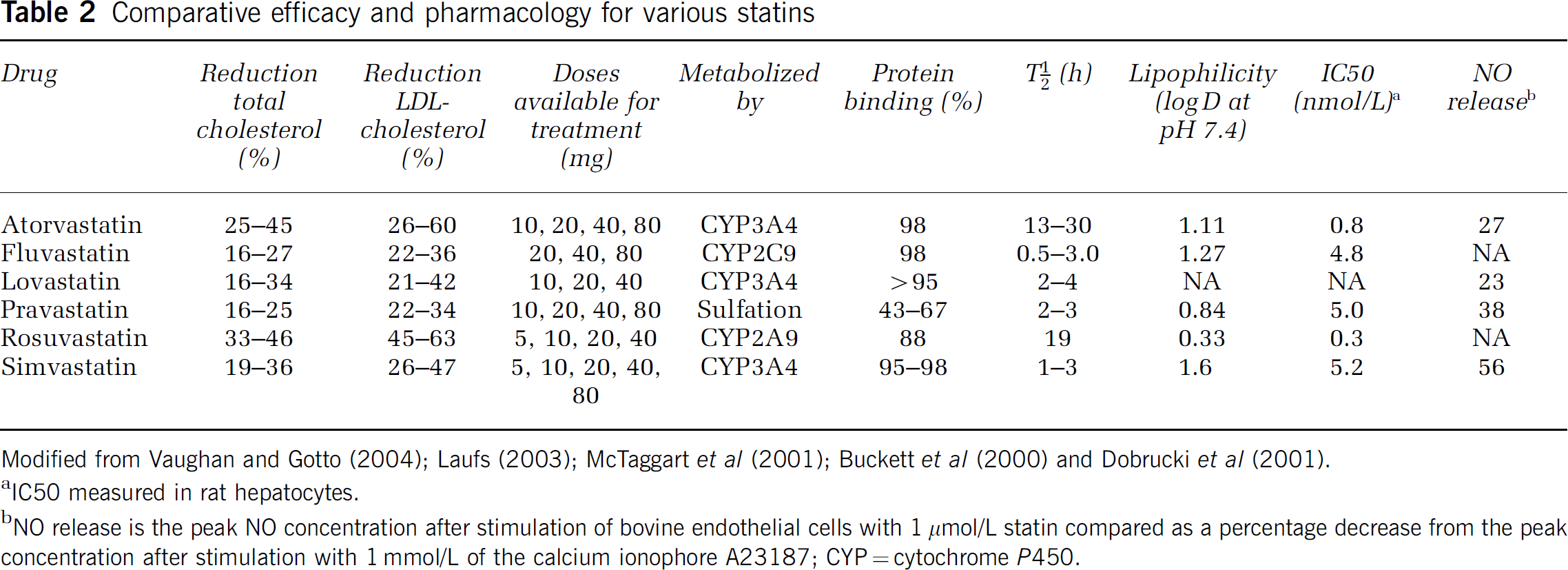
Mevalonate pathway for cholesterol synthesis. Hydroxymethylglutaryl-coenzyme A reductase inhibitors (statins) block the conversion from HMG-CoA to mevalonate. Thereby, not only the synthesis of cholesterol but also of isoprenoid intermediates is inhibited. Inhibition of geranylgenanylation inactivates small G-proteins Rho and Rac. Inhibiting Rho GTPase leads to upregulation of endothelial NO synthase (eNOS), inhibiting Rac GTPase inactivates NADP(H) oxidase (see text for details). CoA, Coenzyme A; PP, pyrophosphate (modified from Endres and Laufs, 2004).
Pleiotropic Effects
In addition to their cholesterol-lowering properties, statins exert a number of ‘pleiotropic’ effects. Pleiotropic effects of a drug are actions other than those for which the agent was specifically developed. Many pleiotropic effects of statins are beneficial, and most of them are because of HMG-CoA reductase inhibition. Inhibiting HMG-CoA reductase not only blocks the synthesis of cholesterol but also of a number of isoprenoid intermediates, which themselves have important biologic functions. Isoprenoids include geranylpyrophosphate (GPP), farnesylpyrophosphate (FPP), geranylgeranylpyrophosphate (GGPP), and squalene (Figure 1). Particularly, FPP and GGPP serve as important lipid attachments for the posttranslational modification of proteins including heterotrimeric G-proteins and small GTP-binding proteins. Isoprenylation converts small GTPase from a cytosolic (inactive) state to a membrane-bound (active) state. Hence, decreased levels of these isoprenoid intermediates have far-reaching biologic consequences. Pleiotropic actions of statins include improvement of endothelial function, increased nitric oxide (NO) bioavailability, antioxidant properties, inhibition of inflammatory responses, immunomodulatory actions, and stabilization of atherosclerotic plaques (for a recent review, see Davignon, 2004) (Figure 2). In addition, there are a few cholesterol-independent effects of statins that are not mediated by blocking the mevalonate pathway, such as binding to a novel regulatory integrin site that inhibits leukocyte function (Weitz-Schmidt et al, 2001).
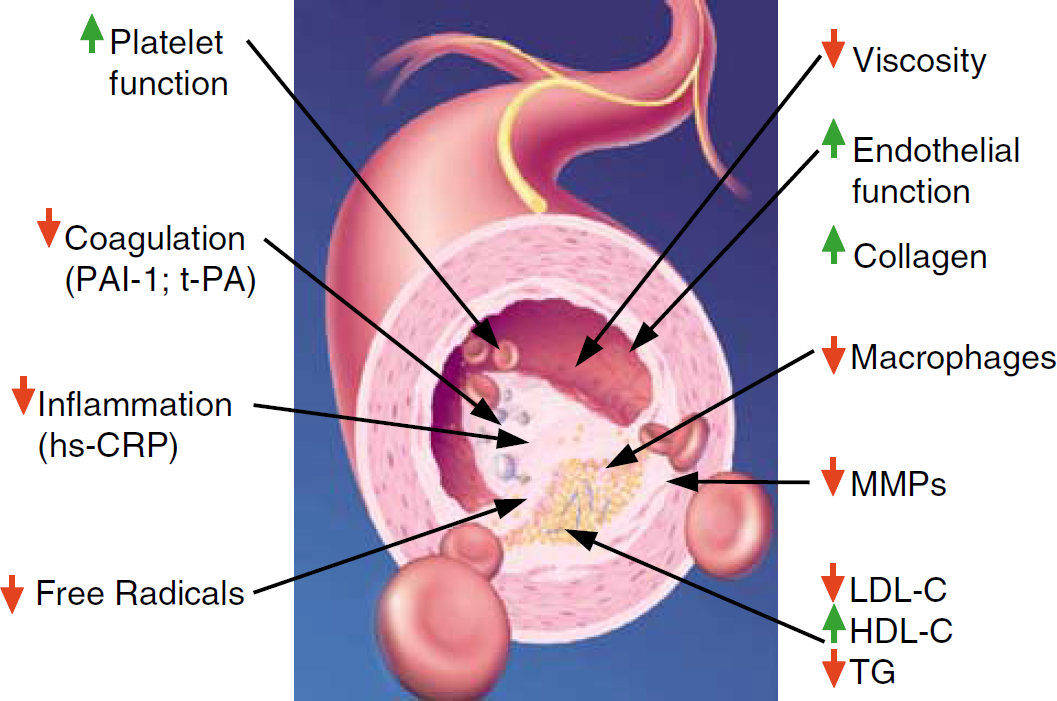
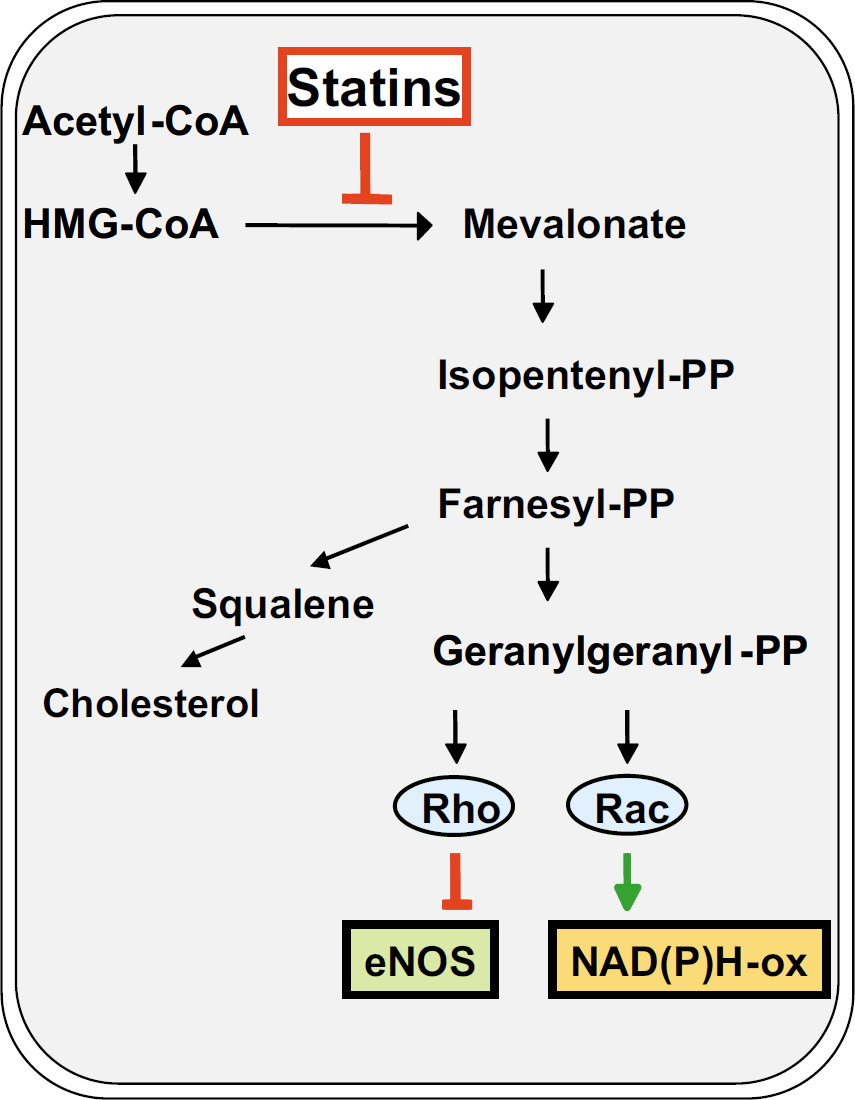
Pleiotropic effects of statins. Cholesterol-independent vasoprotective effects of statins. PAI-1, plasminogen activator inhibitor-1; t-PA, tissue-plasminogen activator; MMPs, matrix metalloproteinases; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; TG, triglycerides.
Safety and Drug Interactions
In general, the large number of clinical statin trials have largely dispelled doubt about the safety and tolerability of statins. Common adverse effects are relatively mild and often transient (gastrointestinal symptoms, headache, rash) but may also include CNS side effects, such as sleep disturbance (Barth et al, 1990). The most important adverse effects, however, are myopathy and increases in hepatic transaminases. In 2001, cerivastatin (Lipobay, Baycol, Bayer, Leverkusen, Germany) was withdrawn from the market because of several fatal cases of rhabdomyolysis (Staffa et al, 2002). Muscle toxicity of statins is dose-dependent and apparently related to HMG-CoA reductase inhibtion in the musculature. Inhibition of isoprenoid and cholesterol synthesis decreases coenzyme Q levels and cholesterol membrane content in the muscle cells. However, the exact mechanisms of muscle toxicity by statins are still not precisely known and seem to relate to a complex interaction between drug, disease, genetics, and concomitant therapy.
A number of important drug interactions with statins have been described that may increase the risk of muscle toxicity. Several statins are metabolized by cytochrome P450 (CYP450) isoenzymes and may therefore interact with CYP450 inhibitors (such as macrolide antibiotics, HIV protease inhibitors, cyclosporin, selective serotonin reuptake inhibitors, or grapefruit juice). In addition, fibrates (i.e., nonstatin lipid-lowering drugs), especially gemfibrozil may increase statin levels partly via inhibiting the glucuronidation and subsequent biliary excretion of statins. This may be mediated via interaction with UDP-glucuronosyltransferase (Prueksaritanont et al, 2002). Therefore, concomitant use of certain interacting drugs or nutrients can increase blood levels of statins and, consequently, the risk for myopathy (Gotto, 2003; Bellosta et al, 2004; Dreier and Endres, 2004).
Recent studies suggested some statins that are metabolized via CYP450 isoenzyme 3A4 (i.e., atorvastatin, simvastatin, lovastastin) may inhibit the antiplatelet activity of clopidogrel, which is an inactive prodrug that requires liver metabolism and activation by CYP450 (Lau et al, 2003, 2004). While some trials described an increase in cardiovascular morbidity and mortality in patients on clopidogrel prescribed atorvastatin versus other statin therapies (Wienbergen et al, 2003; Brophy et al, 2004) other clinical studies failed to show an effect on antiplatelet activity after concomitant use of statins with clopidogrel (Müller et al, 2003; Mitsios et al, 2004) and secondary analysis of randomized, placebo-controlled clopidogrel trials also did not show any adverse clinical interaction of clopidogrel with atorvastatin (Saw et al, 2003; Serebruany et al, 2004). Currently, there are no clear reasons to exclude the coadministration of these two classes of drugs in patients who are at high risk for coronary events (Bellosta et al, 2004).
Stroke and Cholesterol: The First Paradox
While cholesterol is a prominent risk factor for cardiovascular disease and myocardial infarction, the association between cholesterol and stroke has remained controversial (Taylor and Landau, 1990). Epidemiologic and observational studies have not shown a clear association between cholesterol levels and all causes of stroke. For example, neither the Framingham study nor the Honolulu Heart Study demonstrated any association of serum cholesterol with cerebrovascular disease (Dawber et al, 1965; Kagan et al, 1980). Similarly, a large meta-analysis of 45 prospective studies including 13,000 strokes in 450,000 individuals did not show any relationship of serum cholesterol levels with stroke incidence (Prospective Studies Collaboration, 1995) (Figure 3). It should be noted that in most of the trials included in the meta-analysis ischemic strokes were not adequately differentiated from hemorrhagic strokes. Moreover, there was no differentiation between different stroke subtypes such as atherothrombotic, lacunar, or cardio-embolic strokes (Prospective Studies Collaboration, 1995). In line with the notion that there may be no causal link between stroke and cholesterol, intervention trials with nonstatin lipid-lowering drugs (such as fibrates, cholestyramine, etc.) have collectively failed to show a reduction in stroke incidence (Atkins et al, 1993; Hebert et al, 1997). This is in contrast to the fact that (i) cholesterol levels were significantly lowered by these interventions and (ii) the incidence of myocardial infarctions was reduced.
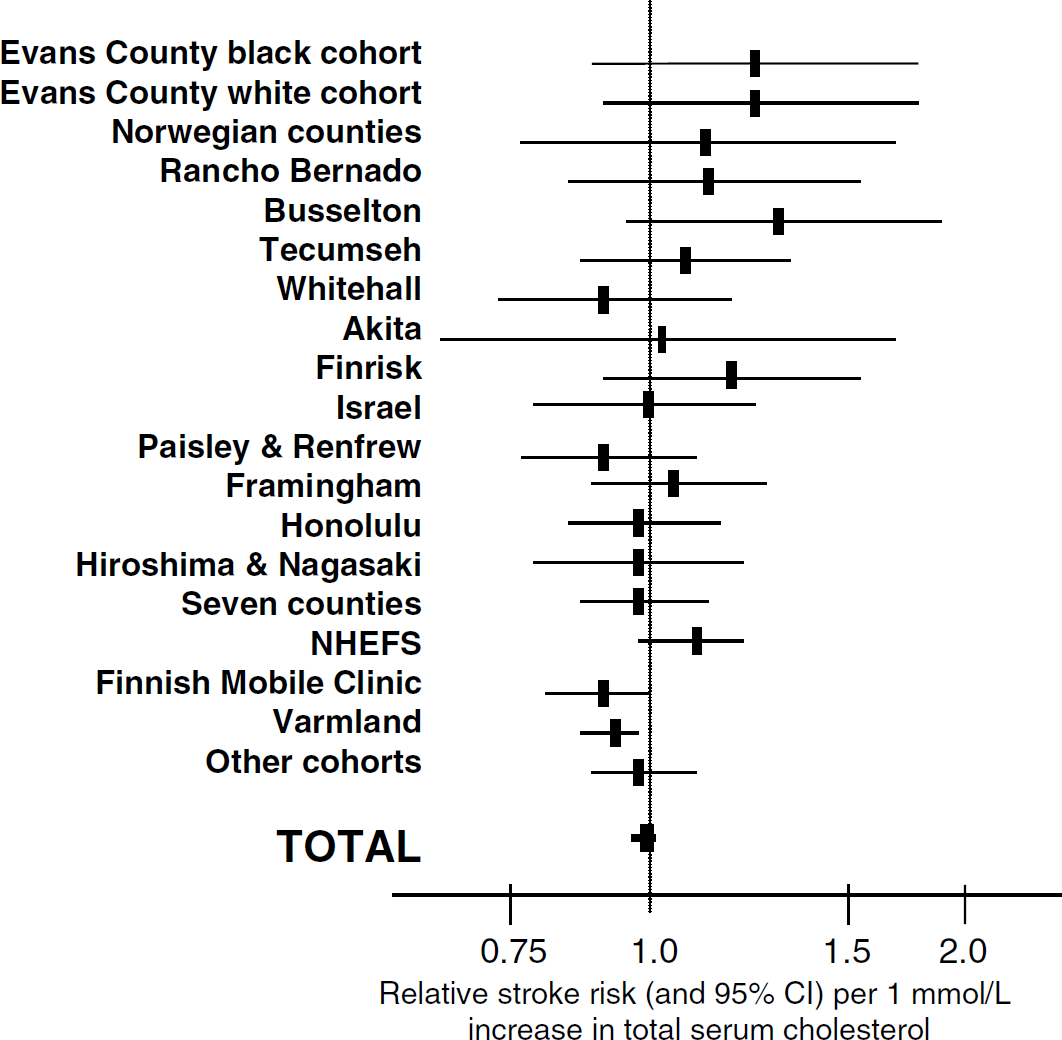
Cholesterol and stroke risk. Relative stroke risk and 95% confidence interval per 1 mmol/L increase in serum cholesterol levels. Data are derived from the Prospective Studies Collaborations (1995) which analyzed cholesterol, diastolic blood pressure and stroke (total of 13,000 strokes, 450,000 individuals, 45 prospective studies).
Conversely, some studies suggest that high cholesterol may be associated with ischemic strokes of atherothrombotic origin. For example, the Multiple Risk Factor Intervention Trial (MRFIT) with 351,000 middle-aged men that were followed for 6 years showed that the risk of death from ischemic strokes increased with very high cholesterol levels (Iso et al, 1989). Similarly, the Eastern Stroke and Coronary Heart Disesase Study with 70,000 participants showed a lower risk of nonhemorrhagic strokes with lower cholesterol levels (Eastern Stroke and Coronary Heart Disease Collaborative Research Group, 1998). Hachinski et al (1996) presented a case–control study with 180 patients in which stroke and transient ischemic attacks (TIA) of purely atherothrombotic origin indeed associated with higher levels of total and LDL cholesterol. Vice versa, low cholesterol levels might be associated with brain hemorrhages (hemorrhagic stroke), particularly in individuals with high blood pressure. For example, in the MRFIT the risk for hemorrhagic stroke was increased with low cholesterol levels (<160 mg/dL total cholesterol, the so-called ‘MRFIT cutoff') suggesting a J-shaped overall relationship between cholesterol levels and stroke (Iso et al, 1989). Similar results were obtained in the Eastern Stroke and Coronary Disease Collaborative Research Group (1998).
The relationship between stroke and cholesterol becomes even more complex with the notion that stroke outcome might be negatively associated with cholesterol levels. For example, the Lausanne Stroke Registry demonstrated that stroke patients with higher cholesterol levels had better outcomes after 1 month (Vauthey et al, 2000). In agreement with this, a retrospective study performed by Dyker et al (1997) found more patients dead or disabled after stroke with low cholesterol levels at stroke onset (Dyker et al, 1997). Taken together, there are many open questions regarding the relationship between cholesterol and stroke. The American Heart Association (AHA) does not list cholesterol as a risk factor for stroke.
Statins and Stroke: The Second Paradox
Clinical Evidence
Eight large, placebo-controlled and randomized studies show that statins reduce vascular events in primary and secondary prevention of coronary heart disease (4S Group, 1994; CARE Investigators, Sacks et al, 1996; LIPID Study Group, 1998; WOSCOPS Group, 1998; Byington et al, 2001; HPS Collaborative Group, 2004; Shepherd et al, 1995, 2002; Sever et al, 2003) (see Tables 3 and 4). Most of these trials included stroke and TIA as pre-defined primary or secondary events. Despite the fact that (i) cohort studies show no association between serum cholesterol concentrations and stroke, and that (ii) nonstatin lipid-lowering intervention trials did not show stroke protection (vide supra), these intervention trials showed that statin treatment reduces the incidence of stroke by 25% to 30% (relative risk reduction), which was termed the ‘statin stroke paradox’ (Law et al, 2003; Amarenco et al, 2004).
Trial acronyms
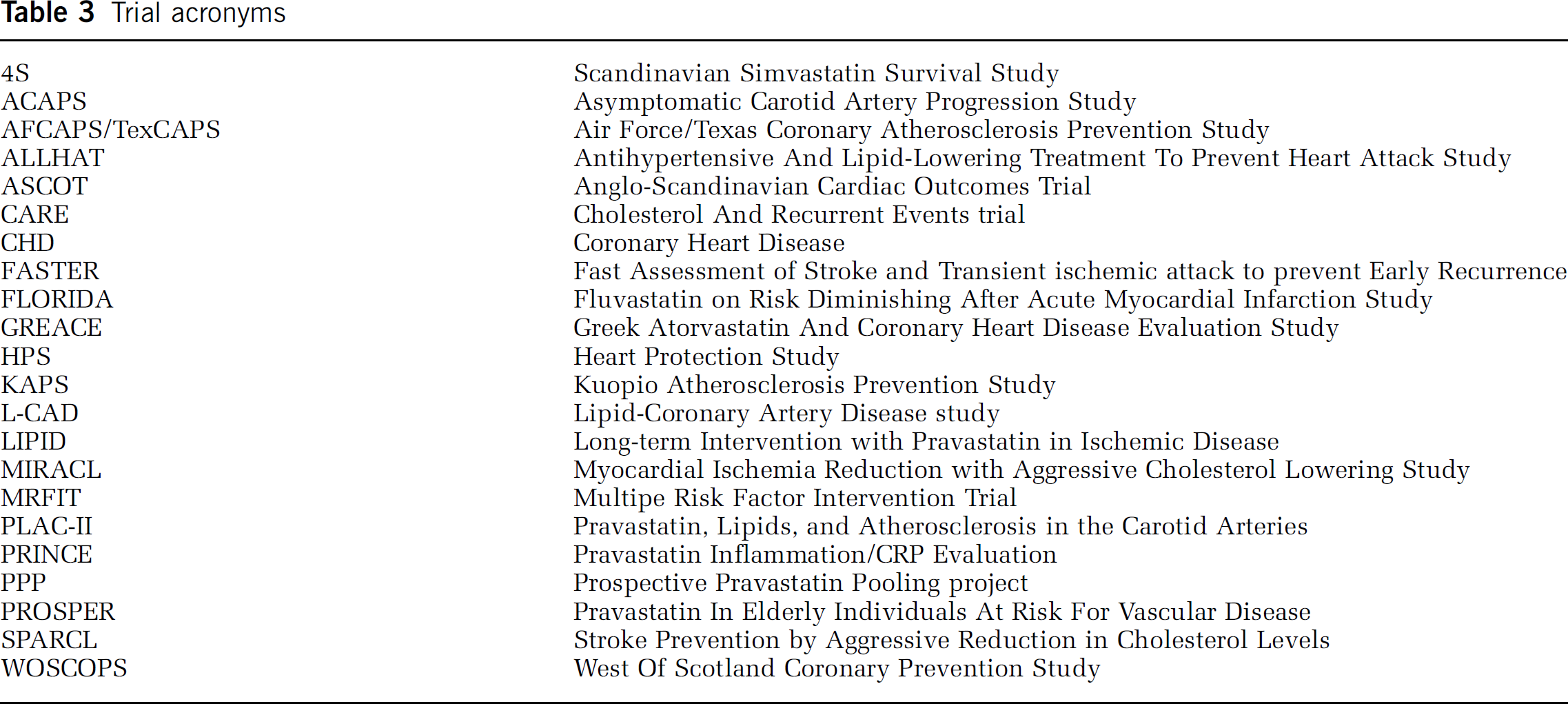
Important statin trials
Modified from Hess et al (2000), Bösel and Endres (2002), Amarenco et al (2004).
pts. = patients; LDL = low-density lipoprotein; ARR = absolute risk reduction (in %); NNT = number needed to treat; RRR = relative risk reduction (in %); ns = nonsignificant.
Secondary prevention: The stroke protective effects of statins were particularly evident in secondary prevention trials of patients with preexisting coronary heart disesase, other vascular disease, or diabetes (Table 4). Absolute stroke risk was relatively low in these study populations (<1% per year) and the resuling reductions in absolute risk were modest. Accordingly, the resultant ‘number needed to treat’ (NNT) to avoid one stroke were very high. Moreover, there is limited evidence regarding the protective effects of statins in the subgroup of patients with previous stroke but without coronary heart disease, peripheral artery disease or diabetes. The HPS trial included a subgroup of 3,280 patients with cerebrovascular disease and 1,822 of them had no established coronary heart disease (Heart Protection Collaborative Disease Group, 2002, 2004). In this subgroup, 40 mg simvastastin reduced the combined vascular end point by about 20% (4.9% absolute risk reduction over 5 years corresponding to an NNT of 102 per year). Surprisingly however, in patients with preexisting cerebrovascular disease there was no apparent reduction in the stroke rate with 22 (100 versus 122) fewer ischemic strokes and twice as many hemorrhages (21 versus 11) in the simvastastin group (Heart Protection Collaborative Disease Group, 2004; Coull, 2004). Taken together, the role for statins in recurrent stroke prevention is not yet defined (Coull, 2004).
Elderly patients: Stroke risk increases with age. However, in the statin trials only a smaller subgroup of study participants were older than 65 years and only a small percentage older than 75 years. The PROSPER (Pravastatin in elderly individuals at risk for vascular disease) trial investigated the effects of 40 mg pravastatin in 5,804 men and women aged 70 to 82 years (Shepherd et al, 2002). The study included individuals with established vascular disease or with a high-risk profile but without previous vascular events. Pravastastin reduced the combined vascular end point by 15% (relative risk) and had no effect on stroke incidence (Shepherd et al, 2002). Interestingly, there was a trend for fewer TIAs in the treatment group. Stroke rate in PROSPER was lower than expected (actual 4.5% versus expected 7% to 8%) and the duration of the trial was relatively short (only 3 versus 5 to 6 years in the other trials). Taken together, however, there is no convincing evidence that statins reduce stroke risk in the elderly.
Primary prevention: The Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) randomized 19,343 individuals with arterial hypertension and >3 additional cardiovascular risk factors. In the lipid-lowering arm of the trial (ASCOT-LLA) individuals received a fixed dose of 10 mg atorvastatin or placebo. This strategy of a fixed statin dose (without titration to target lipid levels) was even termed ‘fire and forget'. The lipid-lowering arm was stopped prematurely after 3.3 years because the primary trial end point was significantly reduced in the treatment group. Stroke risk was significantly reduced by 27% (relative risk) (Sever et al, 2003). In contrast, in the WOSCOPS trials that included middle-aged men with high cholesterol levels but without manifest vascular disease, there was no significant reduction in cerebrovascular events (Shepherd et al, 1995). It is interesting to note that in primary prevention trials, statins reduced stroke risk in hypertensive but not hypercholesterolemic individuals. Other trials that looked at stroke incidence after statin treatment are the ALLHAT (Antihypertensive and Lipid-Lowering Treatment to prevent Heart Attack Study; The ALLHAT officers, 2002) and the GREACE study (Greek Atorvastatin and Coronary Heart Disease Evaluation Study; Athyros et al, 2002).
The current advisory statement of the AHA Stroke Council regarding the use of statins for stroke prevention indicates that (i) the majority of patients with a history of ischemic stroke or transient ischemic attack could benefit from statin use, that (ii) the effects are apparently independent of cholesterol levels; and that (iii) initiation of statin treatment during hospitalisation for first ischemic stroke of atherosclerotic origin is probably justified. (iv) The results of the ongoing SPARCL trial (Stroke Prevention by Aggressive Reduction in Cholesterol Levels) will provide additional information about the role of statins in the minority of patients with prior stroke but no history of other vascular disease where evidence is presently less clear (The Stroke Council, 2004; SPARCL investigators, 2003).
Nonillness mortality, hemorrhagic stroke: In the early 1990s, it was noted that in several cholesterol-lowering intervention trials total mortality was not reduced. In fact, lowering of vascular mortality was apparently counteracted by an increased ‘nonillness' mortality that included death from accidents, suicide and violence (Muldoon et al, 1990). Meta-analyses of the large statin trials, however, did not observe such an increase in nonillness mortality (Muldoon et al, 2001). In fact, several studies even suggest that ‘psychological well-being’ may be improved with statin intake (Young-Xu et al, 2003; Yang et al, 2003). Since a number of studies have shown an increased risk for hemorrhagic strokes in individuals with low cholesterol levels (vide supra) it was suggested that lowering cholesterol levels with statins may increase the risk for cerebral hemorrhages. However, an increased risk of hemorrhagic stroke was not observed in any of the statin trials (Byington et al, 2001; Woo et al, 2004). For example, the Prospective Pravastatin Pooling Project (PPP) that prospectively analyzed data from the WOSCOP, CARE, LIPID trial found 0.5 hemorrhagic strokes per 1,000 patients per year in the pravastatin group versus 0.4 hemorrhagic strokes in the placebo group (thereof 0.4 versus 0.3 non-fatal strokes and 0.1 versus 0.2 fatal strokes, respectively) (Byington et al, 2001).
Experimental Evidence Regarding Statins and Stroke Incidence
In general, there is little evidence from experimental studies regarding therapeutic modulation of stroke risk. In fact, only in spontaneous hypertensive stroke prone rats (SHSPR) can spontaneously occurring cerebrovascular events be studied in an experimental setting (Tagami et al, 1987). Of interest, SHSPR have a decrease in endothelial nitric oxide synthase (eNOS) expression in the brain (Kimoto-Kinoshita et al, 2000). Statins upregulate the expression of eNOS in SHSPR (Kishi et al, 2003). Moreover, long-term oral administration of simvastastin in SHSPR reduces both the incidence and the size of spontaneously occurring strokes although blood pressures and plasma cholesterol levels were not different (Kawashima et al, 2003). Moreover, stroke-associated symptoms and early mortality were markedly reduced in the statin-treated group (Kawashima et al, 2003).
Statins and Stroke Outcome
‘From Prevention to Protection'
The above evidence shows that long-term statin treatment reduces stroke risk. In addition, a number of studies suggest that statins might also be of benefit for stroke outcome. The following clinically relevant questions emerge: (1) Pretreatment: Does long-term statin (pre-) treatment improve outcome and reduce brain injury in the case of a cerebrovascular event? (2) Acute neuroprotection (posttreatment): Does acute statin treatment within minutes or hours after stroke onset improve stroke outcome? (3) Regeneration and secondary prophylaxis: Does early statin administration after acute cerebrovascular (and coronary) syndromes improve morbidity and mortality after several months?
Experimental Evidence
Neuroprotection in focal cerebral ischemia: Animal model experiments show that statin treatment improves stroke outcome and reduces tissue damage after focal brain ischemia. Long-term statin pretreatment increases eNOS expression in the vasculature and in platelets, decreases markers of platelet activation, augments absolute cerebral blood flow (CBF) (Figure 4), reduces lesion volume (Figure 5) and improves neurologic function after middle cerebral artery (MCA) occlusion and reperfusion in the mouse (Endres et al, 1998; Laufs et al, 2000b,
c
; Amin-Hanjani et al, 2001; Laufs et al, 2002; Gertz et al, 2003). Interestingly, increases in CBF were also observed in the nonischemic hemisphere after 14 days of statin pretreatment (Endres et al, 1998). The stroke-protective effects were not at all observed in animals lacking eNOS expression (eNOS knockout mice), therefore some—if not all—of the stroke-protective effects of statin in this rodent mouse model are mediated by the upregulation of endothelial nitric oxide synthase (NOS) (Table 5). The protective effects were dose-dependent and could be observed with different statins, including simvastatin, lovastatin, atorvastatin, mevastatin, and
rosuvastatin (Figure 5). It would be interesting to investigate whether statin (pre)treatment also increases CBF in SHSPR, which have compromised vascular tone. As mentioned earlier, SHSPR have a decrease in eNOS expression in the brain, which is upregulated by statins in this strain (Kishi et al, 2003). Additional intravenous infusion of
Actions of statins on nitric oxide (NO) metabolism

Modified from Laufs (2003).
NO = nitric oxide; Ox-LDL = oxidized low-density lipoprotein; eNOS = endothelial nitric oxide synthase; HSP = heat-shock protein.
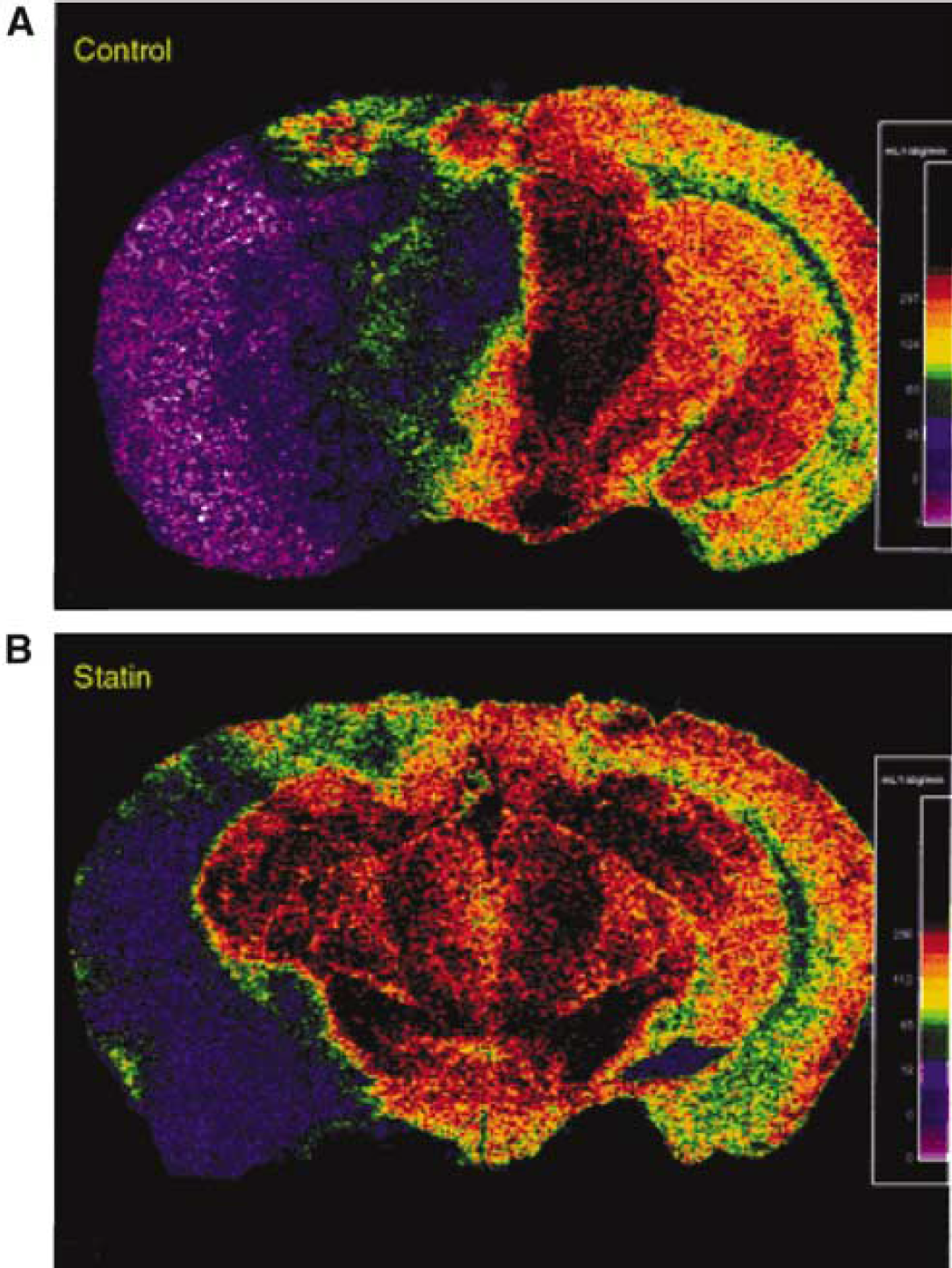
Statins increase cerebral blood flow (CBF) during brain ischemia. This pseudocolor coronal brain section (mid-thalamic level) from [14C]-iodoantipyrine autoradiography shows regional variations in CBF after MCA occlusion in mice treated with vehicle (
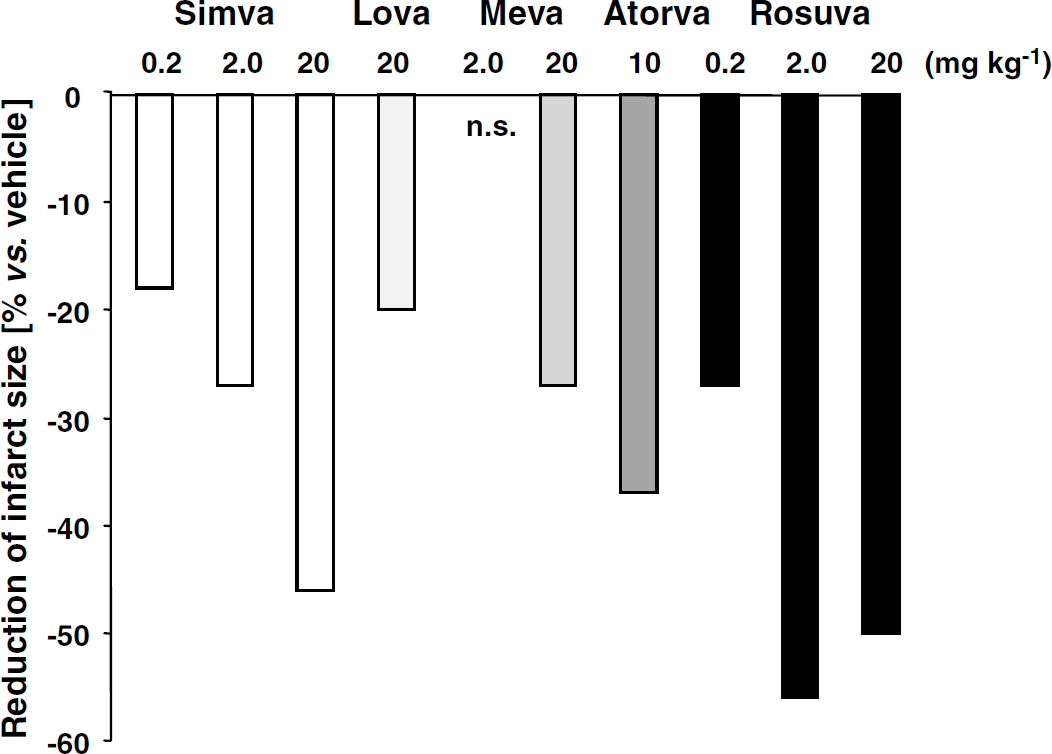
Stroke-protective effects of statins. Efficacy of different statins for infarct reduction (percent reduction of direct lesion size compared with vehicle-treated controls) in a mouse model of transient cerebral ischemia. Animals were pretreated for 14 days with different statins at indicated doses via subcutaneous injection and then exposed to 2-h filamentous MCA occlusion followed by reperfusion (1-h occlusion for atorvastatin). Simva = simvastatin; Lova = lovastatin; Meva = mevastatin; Atorva = atorvastatin; Rosuva = rosuvastatin; n.s. = not significant. Adapted from Endres et al (1998), Laufs et al (2000), Amin-Hanjani et al (2001), Laufs et al (2002), Gertz et al (2003).
Posttreatment: In addition to these protective effects induced by the long-term pretreatment with statins, the therapeutic benefit of statins was found to extend to a 3-h poststroke treatment paradigm (Sironi et al, 2003). In this study, rats were exposed to permament occlusion of the MCA and brain infarct volume was measured by apparent diffusion coefficient maps and T2-weighted images by MRI at 24 and 48 h (Sironi et al, 2003).
Neurorestorative effects: Chopp and co-workers showed that statins also enhance functional outcome and induce brain plasticity with prolonged oral administration of simvastatin or atorvastastin starting 1 day after focal cerebral ischemia in rats. These neurorestorative effects seem to be mediated by synergistic effects on angiogenesis, neurogenesis, and synaptogenesis (Chen et al, 2003). In fact, 1 and 3 mg/kg atoravastatin but not a higher dose increased vascular endothelial growth factor (VEGF) and vessel density in the ischemic brain, enhanced synaptophysin immunoreactivity and increased cell proliferation in the subventricular zone and dentate gyrus (Chen et al, 2003).
Global ischemia: A number of reports indicate that statin pretreatment is also effective in reducing delayed neuronal death in the hippocampal CA1 subfield after global ischemia (Daimon et al, 2004; Kumagai et al, 2004; Kurosaki et al, 2004). Both pravastatin and the novel pitavastastin were effective in reducing neuronal cell death. Protection was associated both with upregulation of eNOS (Kurosaki et al, 2004) and superoxide dismutases (SODs) particularly copper/zinc-SOD (Kumagai et al, 2004). Other mechanisms may involve inhibition of astrocytic activation (Muramatsu et al, 2004).
Neonatal ischemia: In models of neonatal ischemia statins protect against long-lasting behavioral and morphologic consequences of hypoxic/ischemic brain injury. Prophylactic administration of 20 mg/kg simvastatin improved performance in a number of behavioral tests (water maze, radial arm maze, etc.) and reduced histologic damage after ligation of one common carotid artery followed by 3-min hypoxia (Balduini et al, 2001). The protective effects were not observed when simvastatin was administered after the insult and were associated with decreased expression of inflammatory cytokines (Balduini et al, 2003).
Subarachnoid hemorrhage: Statin pretreatment may also ameliorate cerebral vasospasm resulting from subarachnoid hemorrhage via eNOS upregulation (McGirt et al, 2002). In this study, mice were pretreated with 20 mg/kg simvastatin for 14 days and then subjected to endovascular perforation of the right anterior cerebral artery. Several days later neurologic deficits were scored and MCA diameter and eNOS protein expression were measured.
Taken together preclinical evidence shows that statins reduce stroke occurrence (SHSPR), augment CBF and improve acute stroke outcome when administered long-term before the event as well as with posttreatment, and that statins may augment recovery and rehabilitation. The mechanisms may relate to augmentation of NO bio-availability as well as additional pleiotropic effects as discussed below.
Endothelium-function and NO bio-availability: Statins increase eNOS expression and augment NO bioavailability via several cholesterol-dependent and -independent mechanisms (Laufs, 2003; Endres et al, 2004) (Table 5). Endothelial NOS expression is negatively regulated by oxidized LDL cholesterol via two mechanisms. One mechanism relates to caveolin-1, the major constituent of the cell-membrane invaginations called caveolae, the other to the upregulation of lectin-like LDL-1 receptor (Laufs, 2003). Hence, reducing cholesterol also indirectly increases eNOS expression. Cholesterol-independent mechanisms include the direct upregulation of eNOS by statins via inhibiting geranylgeranylpyrophosphate (GGPP) synthesis and increasing the inactive cytosolic state of the small GTPAse Rho (Laufs et al, 1998; Laufs and Liao, 1998; Hernandez-Perera et al, 1998). Rho cycles between a membrane-bound and an cytosolic state and it is the active form anchored to the membrane that negatively regulates eNOS mRNA stability via actin remodeling (Laufs et al, 2000b) (Figure 6). In addition, statins directly activate eNOS via protein kinase Akt, which is an important regulator of a number of cellular processes including metabolism and apoptosis. Activation of phosphatidyl 3-(PI3) kinase provokes the phosphorylation and activation of Akt, and PI3 kinase inhibitors block the effects of statins on Akt (Kureishi et al, 2000). It is the activation of Akt by statins that acutely increases NO production and improves endothelium-dependent vasodilation (Kureishi et al, 2000). Indeed, improvement of endothelium-dependent vasodilation is one of the earliest clinically recognizable effects of statins and this clearly proceeds cholesterol reduction (O'Driscoll et al, 1997).
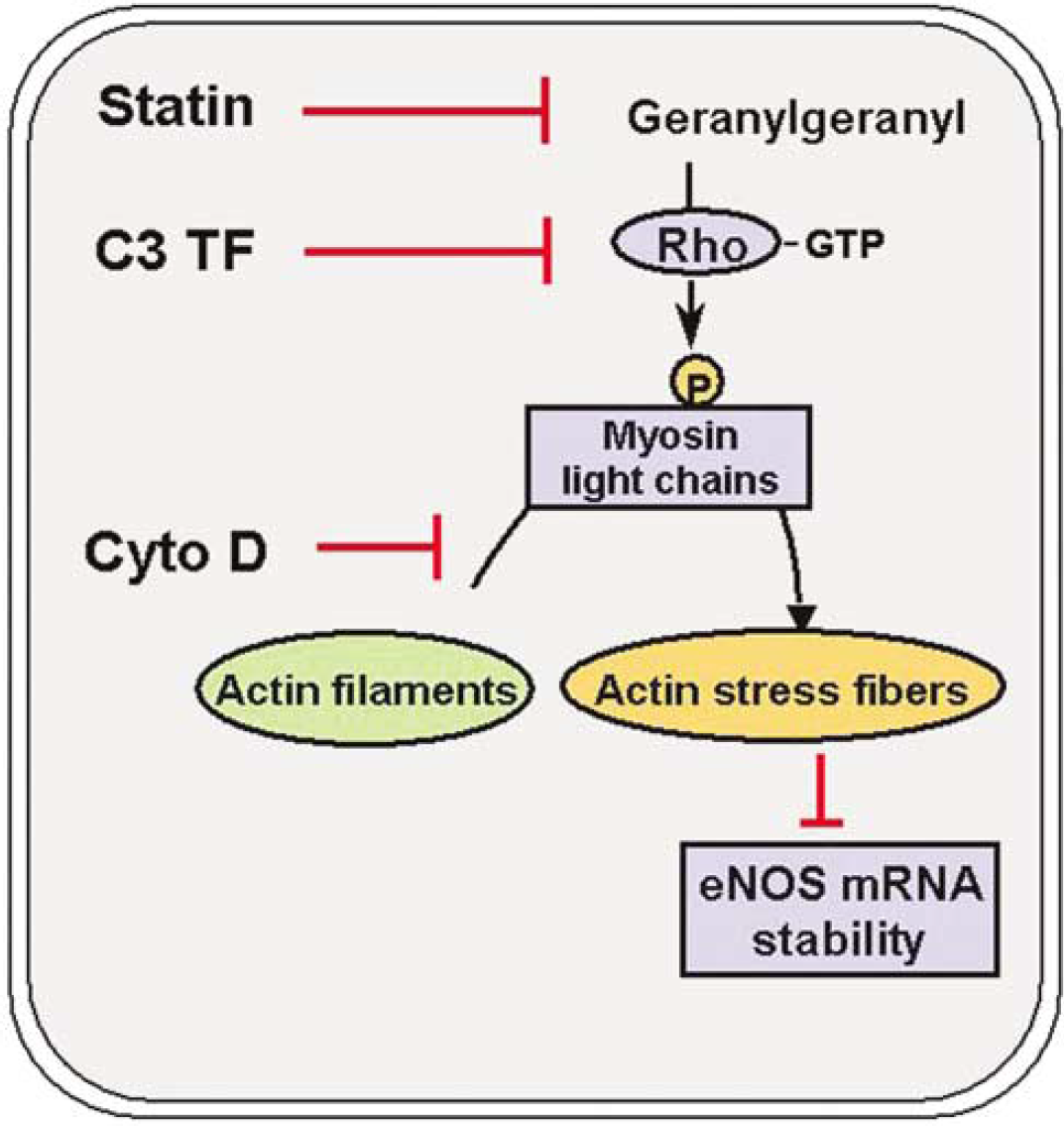
Regulation of eNOS by the actin cytoskeleton. Inhibition of Rho GTPase acitivity leads to eNOS upregulation via effects on the actin cytoskeleton. Rho and Rho kinase confer phosphorylation of myosin light chain, which in turn induces the formation of actin stress fibers. eNOS mRNA abundance is regulated negatively by actin stress fiber formation. Statins (via inhibition of geranylgeranylation), C3 transferase from clostridium botulinum (3CTF; via inhibition of Rho GTPase activity) and the fungal toxin cytochalasin D (via direct disruption of microfilaments) all mediate the upregulation of eNOS mRNA and protein (modified from Endres and Laufs, 2004).
Antithrombotic effects: Thrombosis superimposed on atherosclerosis (‘atherothrombosis') plays an important role during brain infarctions. In animal experiments, statins reduce indirect markers of platelet activation and thrombus formation at high dose, which—at least in part—might be mediated by eNOS and platelet inhibitory effects of NO (Laufs et al, 2000c). In addition, statins regulate fibrinolytic balance as they upregulate tissue plasminogen activator (tPA) and simultaneously inhibit plasminogen activator inhibitor (PAI) (Bourcier and Libby, 2000). Interestingly, stroke-protective effects of statins were also demonstrated in a mouse model of embolic stroke and were apparently independent of eNOS upregulation (Asahi et al, 2000).
Antioxidative effects: Another important pleiotropic mechanism of statins is an antioxidative effect via inhibition of Rac1 and NADPH oxidase (Figure 1) (Endres and Laufs, 2004). There are several sources of superoxide radicals (ROS) within vascular cells, including NAD(P)H-oxidase, xanthine oxidase and NOS. When the availability of its cofactor tetrahydrobiopterin is limited, eNOS can uncouple and generate superoxide anions (Vasquez-Vivar et al, 1998; Landmesser et al, 2003). Interestingly, statins have been shown to potentiate the synthesis of tetrahydrobiopterin in rat smooth muscle cells in culture (Hattori et al, 2002), which may shift the balance away from NOS-generated superoxide production to the generation of NO. The majority of ROS, however, occurring in the vascular wall seems to originate from membrane-bound NAD(P)H oxidase (Gorlach et al, 2000). The latter is an enzymatic system composed of several subunits, including p22phox, gp91phox/mox1, p40phox, p47phox, and p67phox (Griendling and Ushio-Fukai, 1998). In vascular cells, activation of the angiotensin AT1 receptor by angiotensin II is one of the most prominent mechanisms of ROS production. HMG-CoA reductase inhibition confers a reduction of angiotensin II-induced release of free radicals from vascular smooth muscle cells, which is mediated by two important mechanisms. First, statins down-regulate AT-1 receptor gene expression mediated by destabilization of AT-1 mRNA (Wassmann et al, 2001). Moreover, angiotensin II activates the small GTP-biding protein rac1, which is critically involved in the activation of NAD(P)H-oxidase (Wagner et al, 2000); statins inhibit the activation of rac1 by angiotensin II by preventing the geranylgeranyl-dependent translocation of rac1 from the cytosol to the cell membrane (Maack et al, 2003) (Figure 1). In fact, statins induce downregulation of vascular AT1 receptor and reduction of vascular ROS production also in spontaneously hypertensive rats in vivo (Wassmann et al, 2001).
Antiinflammatory and immunomodulatory effects: Over the last decades the importance of imflammation for atherosclerosis and also acute neurodegeneration has become increasingly appreciated. There is compelling evidence that statins lower a number of markers of inflammation, including C-reactive protein (CRP), soluble intercellular adhesion protein or interleukin-6 (Albert et al, 2001; Ridker et al, 2001). Certain statins interfere with the interaction of LFA-1 with intercellular adhesion molecule 1, a mechanism that is independent of HMG-CoA reductase inhibition (Weitz-Schmidt et al, 2001). In the CNS, statins may inhibit the induction of inducible NOS and cytokines (Pahan et al, 1997). In addition, statins have direct effects on the immune system as they inhibit the interferon-γ-induced expression of MHC class II on B-cells and macrophages (Kwak et al, 2000), suggesting a role for statins in immunomodulation (Palinski, 2000). These immunomodulatory effects may reduce organ rejection after transplantation (Kobashiagawa et al, 1995) or improve outcome from immune-mediated disease such as rheumatoid arthritis or multiple sclerosis (Aktas et al, 2003; Youssef et al, 2002).
Regulation of stem and progenitor cells: Statins modulate stem cell regulation and increase the number of progenitor cells. For example, statins increase the number of circulating endothelial progenitor cells (EPCs) by activating the pathway involving phosphoinositide 3-kinase (PI3K) and PKB/Akt (Dimmeler et al, 2001; Llevadot et al, 2001; Werner et al, 2002). This was also shown in patients with coronary heart disease (Vasa et al, 2001). Mobilization of these bone-marrow-derived cells may enhance reendothelialization after vascular injury. In the brain, EPCs contribute to neovascularization after ischemic stroke (Zhang et al., 2002). Therefore, the formation of new blood vessels in the adult brain after stroke is not restricted to the migration and proliferation of local endothelial cells, but may also involve the engraftment of circulating EPCs. Hence, statins may promote both angio- and vasculogenesis in the postischemic brain. In addition, statins induce proliferation of neuronal progenitor cells in the brain, which may contribute to neurogenesis and synaptogenesis after stroke (Chen et al, 2003).
Direct neuroprotective effects: Statins may exert direct effects in the brain parenchyma. It should be noted that BBB permeability differs among statins and correlates in part with their respective lipo***versus hydrophilicity (simvastatin is an example of an BBB permeable statin while pravastatin or rosuvastatin hardly penetrate the CNS). In addition, even statins that are not BBB permeable may enter the brain after BBB disruption, for example, after onset of cerebral ischemia. Zacco et al (2003) recently demonstrated that statins protect cortical neurons from excitotoxicity in culture. Neuroprotective efficacy between different statins was similar to the rank order of potency for the inhibition of HMG-CoA reductase enzyme and neuroprotection was dependent on HMG-CoA reductase inhibition as cotreatment with mevalonate or cholesterol bypassed the effect. It was suggested that the neuroprotective effects were dependent on intracellular depletion of cholesterol. Resistance of cultures to excitotoxicity developed only after days of statin exposure and was associated with reduced N-methyl-
Clinical evidence that statins improve stroke outcome: A number of clinical reports indicate that individuals on statin medication have improved outcome from a number of different diseases including sepsis (Almog et al, 2004), cardiac transplantation (Kobashiagawa et al, 1995), vascular events after vascular surgery (Durazzo et al, 2004), in-hospital mortality after major noncardiac surgery (Lindenauer et al, 2004), or vascular outcome in patients with peripheral artery disease (Schillinger et al, 2004).
A number of smaller, retrospective clinical trials have analyzed whether patients that were on statin medication at stroke onset had better outcomes compared with patients that were not treated with a statin. An earlier, retrospective pilot case-referent study investigated the influence of statin pretreatment on stroke outcome but the results—albeit in favor of a benefit—were not significant (Jonsson and Asplund, 2001). Greisenegger et al (2004) describe in a cross-sectional study within a prospective cohort of ischemic stroke patients that statin use is negatively associated with stroke severity: of 1,691 patients, 152 were taking statins. Patients with a modified Rankin score of 5 or 6 (which corresponds to severe disability or death) at 1 week after stroke were significantly less frequent in the statin group (Greisenegger et al, 2004). Of note, particularly patients with diabetes benefitted from statin use. Another small prospective trial in which consecutive patients with acute ischemic stroke were included that favorable outcome at 3 months (modified Rankin score) was independently associated with prior statin use in addition to age and National Institutes of Health Stroke Scale (NIHSS) score at admission (Marti-Fabregas et al, 2004). Similarly, in an observational study, Yoon et al (2004) found that patients with acute ischemic stroke that were taking statins at the time of admission had good outome 51% in compared with only 38% of patients not taking statins. The Heart and Estrogen–Progestin Replacement Study (HERS) showed a trend for a reduced number of fatal strokes with statin use (hazard ratio 0.52, 95% confidence interval 0.23 to 1.18; Bushnell et al, 2004). Taken together, a number of smaller studies suggest that statins may provide benefits for long-term functional outcome when administered before the onset of cerebral ischemia. However, randomized controlled trials will be required to further substantiate the validity of these preliminary findings.
Four randomized trials have examined the role of statins for acute coronary syndromes: the Myocardial Ischemia Reduction With Aggressive Cholesterol Lowering (MIRACL) study, the Pravastatin Turkish Trial, the Fluvastatin on Risk Diminishing After Acute Myocardial Infarction (FLORIDA) study, and the Lipid-Coronary artery Disease (L-CAD) study (Wright et al, 2002). Three trials showed benefit of statin use and analysis of stroke as a predefined secondary outcome parameter in the MIRACL study showed that early treatment of patients with acute coronary syndromes reduced also the incidence of stroke (which was a predefined secondary endpoint) by 50% (Schwartz et al, 2001; Waters et al, 2002). Currently, the hypothesis that early treatment with statins after a cerebrovascular event reduces morbidity and mortality after several months is being tested in the prospective FASTER trial.
Are the Noncholesterol Effects of Statins as Shown in Experimental Studies Responsible for the Stroke-Protective Effects in Patients?
Most experimental work is performed in healthy, young, male laboratory animals while statins exert most clinical benefits in patients with coronary heart disease and patients at high vascular risk because of multiple risk factors. (i) The reduction in cardiovascular morbidity and mortality with decreases in left ventricular thrombi may account for a reduction in cardioembolic strokes (Waters et al, 2002; Amarenco et al, 2004). (ii) In addition, as noted by Wallis, the MR-FIT trial shows that in patients with coronary heart disease stroke risk increases with high cholesterol levels. It is therefore plausible that in the study populations that were included in the statin trials cholesterol may play an underestimated role for stroke pathogenesis (Wallis et al, 1998; Iso et al, 1989). (iii) Lowering the severity of atherosclerosis and plaque stabilization particularly in the carotid and vertebral arteries is another important mechanism. A number of trials including ACAPS, LIPID Atherosclerosis Substudy, KAPS and PLAC-II have shown that statins reduce carotid atherosclerosis (Adams et al, 1995; Furberg et al, 1994; Crouse et al, 1995; Salonen et al, 1995; Hodis et al, 1996; MacMahon et al, 1998). (iv) Lastly, statins may lower blood pressure in individuals with hypertension (Wilkinson and Cockrift, 2000). Given the fact that hypertension is the single most important stroke risk factor and that lowering of diastolic blood pressure by 5 mm Hg reduces stroke incidence by >40% even subtle effects of statins on blood pressure may have biologically significant effects on stroke incidence. Indeed, in a crossover study, monotherapy with pravastatin lowered mean systolic and diastolic BP by 8 and 5 mm Hg respectively (Glorioso et al, 1999). This study was confirmed by at least two others (Borghi et al, 2000; Sposito et al, 1999). In support of this hypothesis, statins reduce stroke risk in hypertensive but not in hypercholesterolemic individuals without manifest vascular disease (ASCOT versus WOSCOPS trial). Indeed, statins reduce the expression of angiotensin II type 1 (AT1) receptors and patients pretreated with statins show less blood pressure increase on infusion with angiotensin II (Nickenig et al, 1999). Clinical data support the idea that statins may act synergistically to lower blood pressure particularly when coadministered with angiotensin-converting enzyme (ACE) inhibitors or AT1 receptor blockers (Sposito et al, 1999; Borghi et al, 2000).
Effects of Statin Withdrawal
A number of cardiovascular drugs like β-blockers or nitrates exert negative vascular effects when treatment is suddenly withdrawn. Data from clinical and experimental studies suggest that abrupt discontinuation of statin treatment may also confer adverse vascular effects.
Experimental Evidence
In endothelial cells in culture eNOS is upregulated by long-term statin treatment via decreased geranylgeranylation of rho GTPase (Figure 1; Table 5) (vide supra, Laufs and Liao, 1998). Withdrawal of statin treatment, however, results in a transient increase of rho activity, causing a suppression of endothelial NO production indicative of a withdrawal phenomenon (Laufs et al, 2000a). The underlying mechanism is a negative feedback regulation of rho gene transcription mediated by the actin cytoskeleton: statins inhibit isoprenoid-dependent rho membrane translocation and GTP-binding activity, at the same time, however, they upregulate the expression of rho GTPase in the cytosol. Rho gene expression but not mRNA half-life is controlled by the actin cytoskeleton via a negative feedback mechanism. After withdrawal of statin treatment GGPP becomes available leading to membrane anchoring of isoprenylated rho and resultant overshoot activation of rho GTPase activity. This results in downregulation of endothelial NO production below baseline levels (Laufs et al, 2000a). In animals, sudden withdrawal of statin treatment resulted in massive eNOS downregulation in the brain and aorta (Figure 7) (Gertz et al, 2003). Vice versa, markers of platelet activation were downregulated by statin treatment but withdrawal of statins resulted in upregulation after 2 days. Moreover, the stroke-protective effects of long-term statin treatment were completely abrogated after withdrawal of statin treatment (Figure 7).
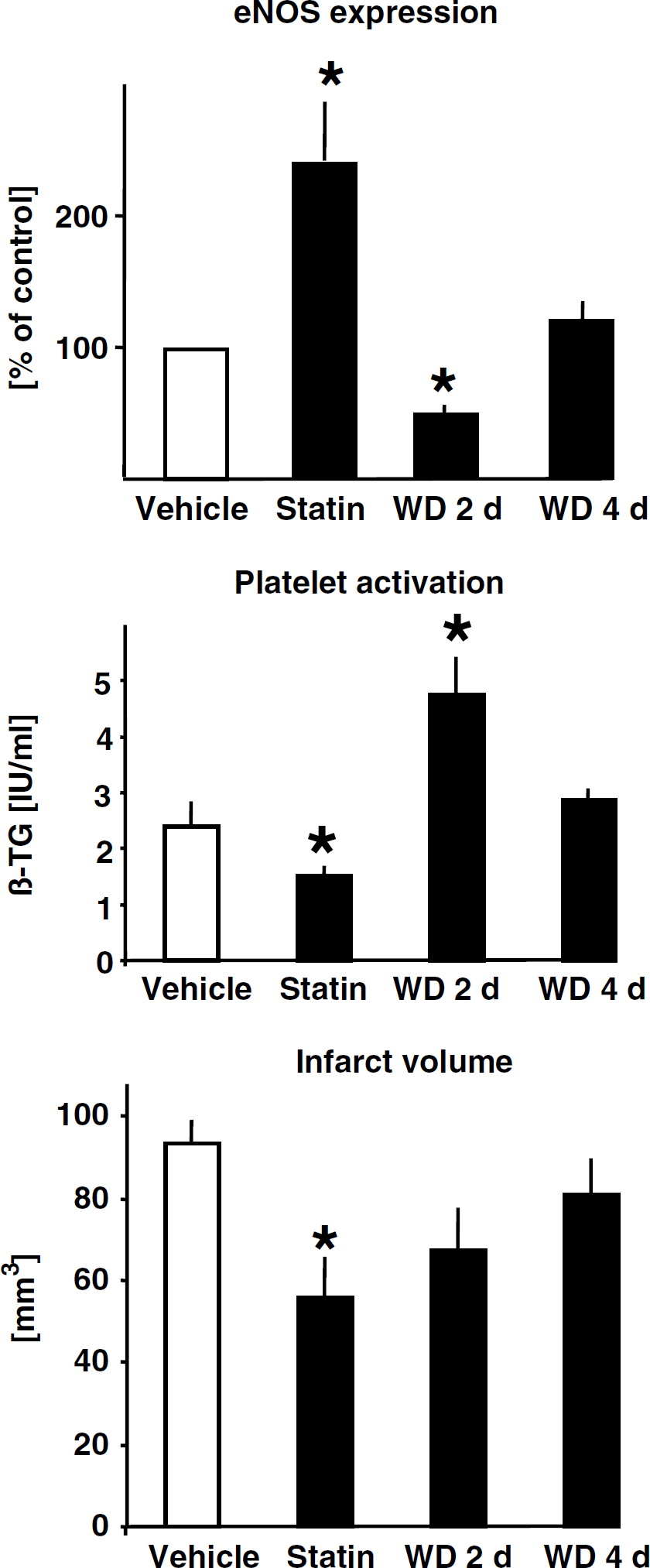
Effects after withdrawal (WD) of statin treatment. Wild-type mice were chronically treated with a statin or vehicle. Middle cerebral artery occlusion (for 1 h plus 23 h reperfusion) was induced in mice chronically treated with statins or 2 or 4 days (WD 2 d, WD 4 d) after withdrawal of treatment. Expression of eNOS mRNA was determined in aortas and markers of platelet activation (i.e., β-TG = beta thromboglobulin) were measured in the serum. Brain infarct volume was determined by computer-assisted volumetry. *P<0.05; ANOVA plus Tukey's post hoc test (modified from Gertz et al, 2003).
Overshoot activation of rho GTPase and resultant downregulation of eNOS expression may not be the only mechanisms by which statin withdrawal impairs vascular function. Abrupt discontinuation of statin treatment is also associated with increased production of reactive oxygen radicals mediated by NAD(P)H-oxidase, increases in monocytes chemoattractant protein 1 and tissue factor. Similar to the overshoot activation of rho also rac GTPase is anchored to the membrane once GGPP is available which confers NAD(P)H-oxidase activation (Vecchione and Brandes, 2002; Brandes et al, 2003).
Clinical Evidence
In 1998, a report from New Zealand noted a threefold increase in thrombotic vascular events after the treatment of patients with simvastatin was stopped and then continued with relatively lower doses of fluvastatin because of reference pricing for statins (Thomas and Mann, 1998). In healthy young individuals 80 mg atorvastastin improves endothelium function but 24 h after withdrawal vascular function was impaired without accompanying changes in cholesterol levels or inflammatory markers (Laufs et al, 2001). The notion that abrupt withdrawal of statin medication may increase hospital morbidity and mortality after vascular events is supported by a retrospective analysis of the Platelet Receptor Inhibition in Ischemic Syndrome Management (PRISM) trial, which showed that statin pretreatment was associated with improved outcome, which however was completely lost when statins were discontinued after the onset of symptoms (Heeschen et al, 2002, 2003). Spencer et al (2004a) obtained similar findings in the Global Registry of Acute Coronary Events (GRACE): Patients who were pretreated with and continued to take a statin during hospitalization were significantly less likely to die in hospital than those who had never received statin therapy. Conversely, individuals in whom statin therapy was discontinued at the time of admission had a slightly higher risk of death than those never administered statin treatment (Spencer et al, 2004a). Moreover, the National Registry of Myocardial Infarction showed that withdrawal of statin therapy in the first 24 h of hospitalization for non-ST segment elevation myocardial infarction is associated with worse hospital outcome (Spencer et al, 2004b). In contrast, the Treating to New Target (TNT) Study Group found no evidence for an increase in acute coronary syndromes after short-term abrupt discontinuation of statins in stable cardiac patients (McGowan et al, 2004). Presently, there are no data available regarding statin withdrawal in stroke patients. Nevertheless, statin therapy should be continued during hospitalization for stroke and myocardial infarction unless strongly contraindicated.
Concluding Remarks
In addition to their potent cholesterol-lowering effect statins exert numerous pleiotropic vasculo- and cerebroprotective actions. Compelling clinical data show that statins reduce stroke risk although the role of cholesterol as a stroke risk factor is less clear. In the next few years ongoing trials will unequivocally answer the question whether statins reduce stroke recurrence in patients with a previous stroke (even in the absence of additional vascular disease). Animal research strongly supports the notion that statins administered as pre- or posttreatment provide neuroprotection and enhance regeneration after cerebral ischemia. Hence, statin treatment may combine prevention (i.e., reduction of stroke risk) with prophylactic treatment (i.e., improvement of outcome after an insult). For example, in individuals at high risk for stroke such as in patients undergoing cardiac or vascular surgery high-dose statins could be administered as short-term pretreatment. Moreover, in the next years clinical trials will answer the question whether acutely administered statins may improve stroke morbidity and mortality. To do so, intravenous formulations of statins may need to be developed for rapid administration and also for patients that are unable to swallow.
Footnotes
Acknowledgements
The author thanks Michael A Moskowitz, James K Liao and Ulrich Laufs for advice and Golo Kronenberg for critically reading the manuscript.