
Abstract
Nitric oxide has multiple physiologic roles in the CNS. Inhibiting nitric oxide synthesis might therefore alter functional activity within the brain. We used [14C]-2-deoxyglucose in vivo autoradiography to measure local CMRglc in “knockout” mice lacking the genes for either the endothelial (eNOS) or neuronal (nNOS) isoforms of nitric oxide synthase, and in the progenitor strains (SV129, CS7B1/6). Glucose utilization levels did not significantly differ between nNOS and eNOS knockout mice and C57B1/6 mice in any of the 48 brain regions examined, but were relatively lower in some subcortical regions in SV129 mice.
Keywords
Nitric oxide (NO) is a vital messenger molecule involved in the regulation of numerous physiologic processes within the central, cardiovascular, and peripheral nervous systems (Garthwaite 1991; Bredt and Snyder 1992; Ohno et al., 1993; Dawson and Snyder, 1994). Also, NO may mediate cell death secondary to N-methyl-D-aspartate (NMDA) receptor activation in processes such as ischemia and in neurodegenerative disorders including Alzheimer's disease and Huntington's disease. Nitric oxide is implicated in neurotoxic processes because of its capacity to interact with reactive oxygen intermediates, for example, its reaction with superoxide anion (O2−) to form the powerful oxidant peroxynitrite (ONOO−), which further decomposes to hydroxyl radicals (OH−) and nitrite ions (NO2−).
Nitric oxide is a labile free-radical gas that is not stored within tissues but diffuses freely across membranes. Many of its biological effects are mediated by activation of soluble guanylyl cyclase. It is generated by the calmodulin-dependent oxidation of
Since NO subserves multiple roles in modulating neuronal signal transduction, perturbations in NO synthesis might be expected to alter functional activity within the brain. To gain insight into the contributions of nNOS and eNOS isoforms to integrated functional activity, we used [14C]-2-deoxyglucose in vivo autoradiography to investigate the effects of chronic inhibition of NO production on local cerebral metabolic rates for glucose (1CMRglc) in freely moving nNOS and eNOS knockout mice, compared with levels in the two progenitor mouse strains, SV129 and C57B1/6. An abstract of this work has previously been published (Browne et al., 1997).
MATERIALS AND METHODS
Animals
The NOS knockout mice were generated by targeted disruption of nNOS or eNOS genes in J1 embryonic stem cell lines derived from SV129 mice (Huang et al., 1993, 1995). Embryonic stem cells were injected into blastocysts isolated from C57B1/6 mice, blastocysts reimplanted into pseudopregnant (C57B1/6 x DBA/2) F1 mice, and the resultant chimeric mice back-crossed to C57B1/6 mice. Homozygous eNOS and nNOS knockout (–/–) mice were identified by polymerase chain reaction analysis of genomic DNA isolated from mouse tail. Homozygous wild-type litterrnate control animals were unavailable at the time of this study. We therefore compared glucose use values in knockout mice with levels in the two progenitor strains, SV129 and C57B1/6 (Taconic Farms, Germantown, NY, U.S.A.).
Measurement of 1CMRglc
Adult male mice (20 to 28 g) were anesthetized with 0.5% to 1% halothane in 70%/30% nitrous oxide-oxygen mix, and PE-10 cannulae inserted into one femoral artery and vein. Cannulae were fed under the skin to extrude at the nape of the neck, sutured in place, and lines stoppered. Femoral wounds were sutured closed after applying Xylocaine 2% gel, and mice allowed to recover from anesthesia for 1 hour. The Pa
Glucose use was measured in 22 conscious mice using the fully quantitative [14C]-2-deoxyglucose procedure. Measurement was initiated by a 30-second intravenous injection of 3 µCi [14C]-2-deoxyglucose in 60 µL of sterile heparinized saline, flushed through the cannula with a saline bolus (30 µL). Ten timed arterial blood samples (approximately 30 µL) were taken over 45 minutes sampling times, weighted to reflect 14C-2-deoxyglucose uptake in the period immediately after injection and immediately centrifuged. Plasma 14C and glucose concentrations were measured by liquid scintillation analysis (5 µL) and semiautomated glucose oxidase assay (Beckman; 10 µL). At 45 minutes, the mice were decapitated, and their brains were removed and rapidly frozen in isopentane at −43°C. Brains were cut into 20-µm coronal cryostat sections, triplicate consecutive sections collected onto heated coverslips at 140-µm intervals throughout the brain, and exposed to 14C-sensitive film (Kodak Biomax-MR, Glucose Analyzer II, Arlington Heights, IL, U.S.A.) for 7 to 9 days with precalibrated 14C standards (Amersham, U.K.). Isotope concentrations in 48 brain regions were measured by densitometric analysis (MCID, Imaging Research, St. Catherines, Ontario, Canada), relative to the 14C standards. The 1CMRglc was calculated using Sokoloff's operational equation using the lumped constant for the rat (Sokoloff et al., 1977) in the absence of calculated rate constants for mouse brain.
Statistical analysis
Comparisons between 1CMRglc values for each structure and physiologic variables were made by analysis of variance followed by post hoc Fisher's protected least signficant difference test with Bonferroni correction for multiple comparisons.
RESULTS
Physiologic variables
Basal Pa
Physiologic variables in NOS knockout mice
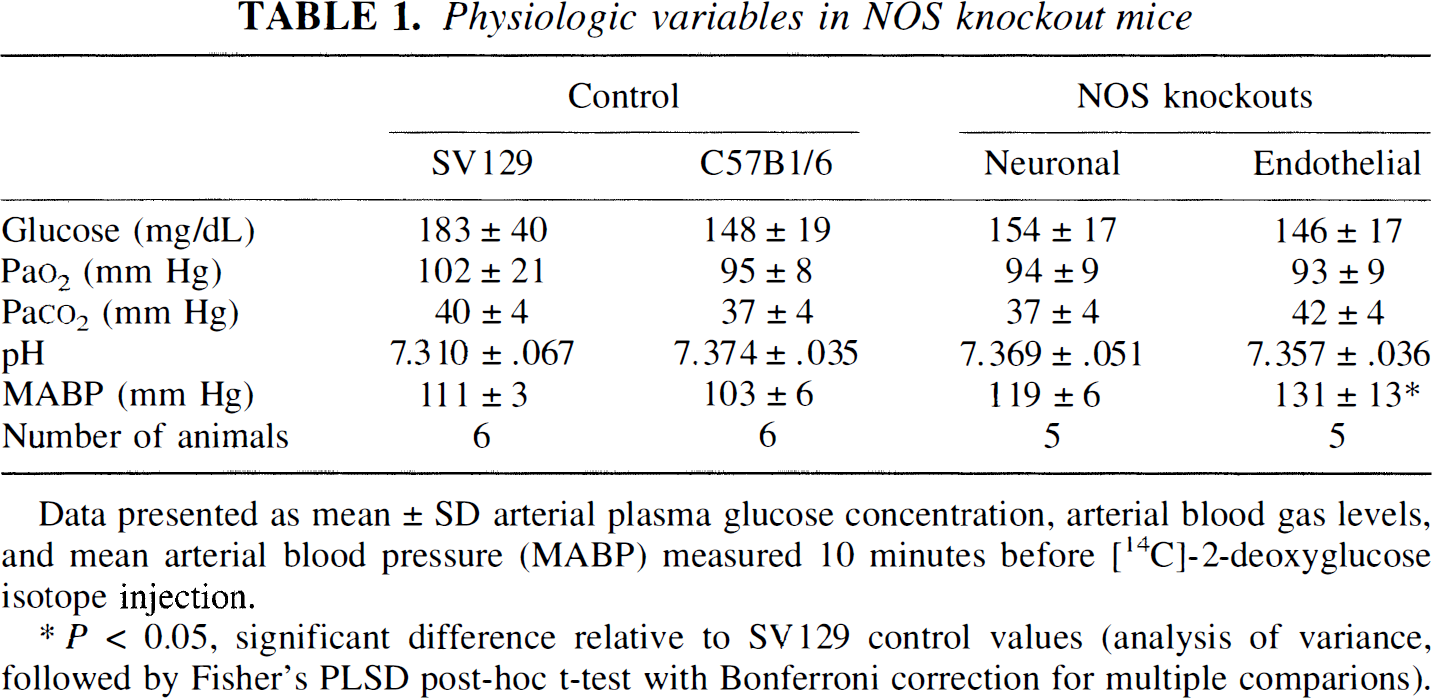
Data presented as mean ± SD arterial plasma glucose concentration, arterial blood gas levels, and mean arterial blood pressure (MABP) measured 10 minutes before [14C]-2-deoxyglucose isotope injection.
P < 0.05, significant difference relative to SV129 control values (analysis of variance, followed by Fisher's PLSD post-hoc t-test with Bonferroni correction for multiple comparions).
Glucose utilization
The 1CMRglc values are presented in Table 2. There were no statistically significant differences in 1CMRglc between eNOS and nNOS knockout mice in any of the 48 brain regions examined. The 1CMRglc did not significantly differ between NOS knockout mice and C57B1/6 mice. Glucose use values in SV129 mice generally were lower than levels in the corresponding regions of both C57B1/6 and NOS knockout mice. Significant reductions in 1CMRglc values were evident in the inferior colliculus and globus pallidus of SV129 mice relative to eNOS mice, and in hippocampus CA1, medial septum, and internal capsule relative to nNOS levels.
Cerebral glucose utilization in NOS-knockout mice
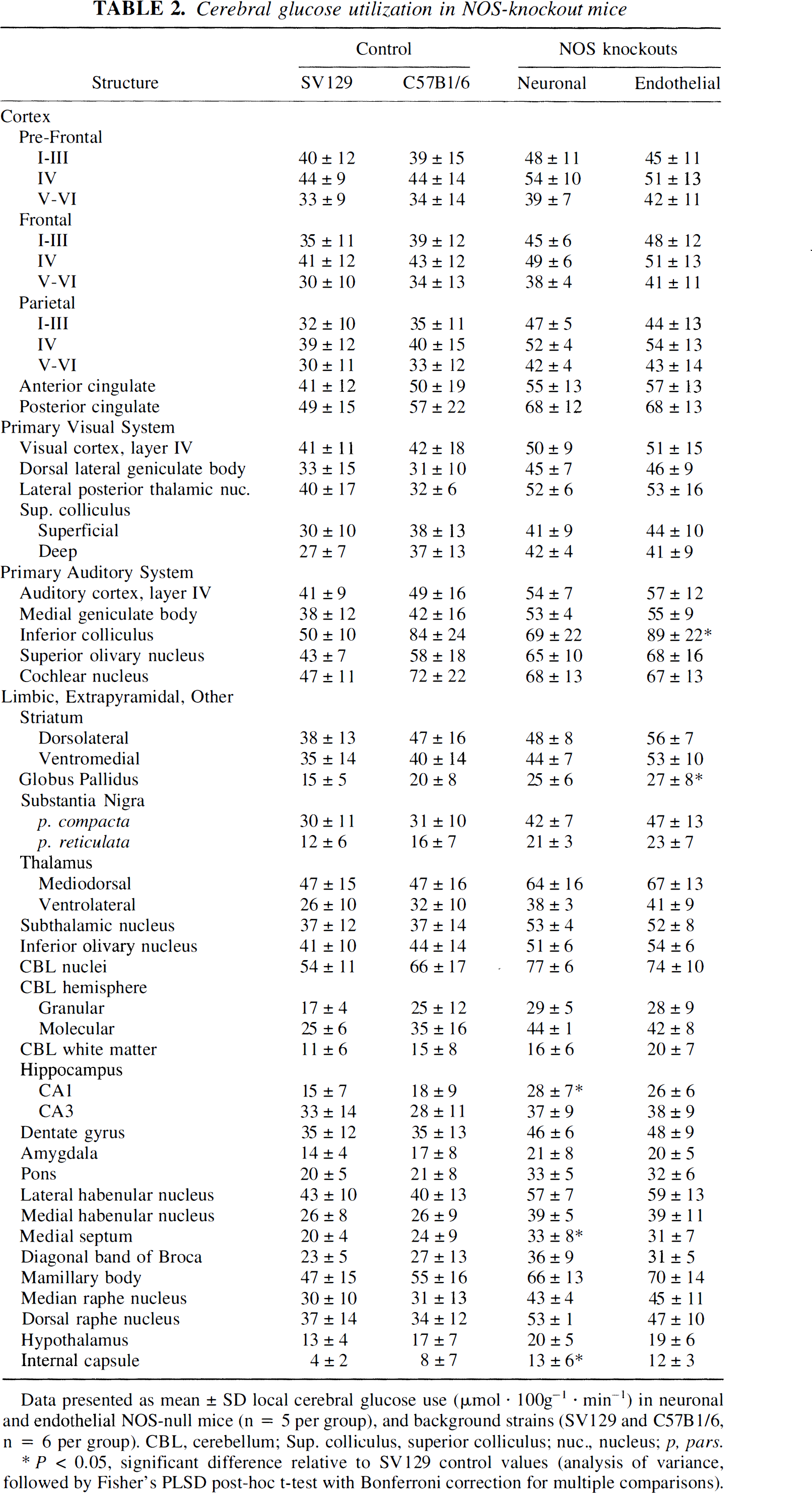
Data presented as mean ± SD local cerebral glucose use (µmol · 100g−1 · min−1) in neuronal and endothelial NOS-null mice (n = 5 per group), and background strains (SV129 and C57B1/6, n = 6 per group). CBL, cerebellum; Sup. colliculus, superior colliculus; nuc., nucleus; p, pars.
P < 0.05, significant difference relative to SV129 control values (analysis of variance, followed by Fisher's PLSD post-hoc t-test with Bonferroni correction for multiple comparisons).
DISCUSSION
Since nNOS and eNOS subserve different functions within the CNS, we postulated that inactivation of the genes responsible for synthesis of either isoform might differentially alter the pattern of glucose metabolism within the brain, a marker for functional activity. Despite the widespread distribution of nNOS throughout the brain, blockade of neuronal NO synthesis did not alter energy metabolism in any of the 52 brain regions examined. Furthermore, cerebral glucose use did not differ between eNOS and nNOS knockout mice despite their differential cellular locations and functions. Whereas nNOS is present throughout the CNS, eNOS is predominantly found within vascular endothelium and a small population of neurons. Both NOS isoforms are activated by increased intracellular Ca2+ levels, but with different trigger mechanisms: nNOS after NMDA receptor stimulation, and eNOS by muscarinic or bradykinin receptor-mediated activation of the phosphoinositide cycle. Both eNOS and nNOS null mice respond differently to pathologic stimuli, with nNOS null mice being protected against ischemic and striatal NMDA lesions (Huang et al., 1994; Hara et al., 1996; Ayata et al., 1997). These results implicate nNOS in neurotoxicity after ischemia and excitotoxicity, whereas eNOS appears to be protective. However, a major concern with the use of knockout animals is the presence of developmental abnormalities or physiologic compensation for the deleted gene. The studies reported here show that differences in glucose utilization do not account for the disparate results observed in nNOS and eNOS knockout mice.
The finding that 1CMRglc does not differ between eNOS and nNOS knockout mice suggests either that cerebral glucose utilization is not influenced by NO-mediated events, or more likely, that compensatory mechanisms may ameliorate the impact of NOS gene deletion. Such mechanisms might involve upregulation of activity of the unaffected constitutive NOS isoform, or the inducible isoform, or other messenger pathways that also target guanylyl cyclase, for example, carbon monoxide or arachidonic acid pathways (Dawson and Snyder, 1994). Other examples of physiologic compensation by non-NO, non-cGMP-dependent pathways include the cerebral blood flow response to hypercapnia (Irikura et al., 1995) or whisker stimulation (Ma et al., 1996; Ayata et al., 1996). Our results are in agreement with previous reports that NOS inhibition (using the nonspecific NOS inhibitor NG-nitro-
Glucose use did not differ between NOS knockout and C57B1/6 mice. However, the 1CMRglc values reported in C57B1/6 mice in this study generally are lower than values reported elsewhere (Jay et al., 1985). Possible reasons for this discrepancy include inter-experimenter differences and differences in determination of the arterial input curve, for example, resulting from slow arterial catheter flow rates affecting sample times. The 1CMRglc values in subcortical regions of SV129 mice generally were lower than in the corresponding regions of both C57B1/6 and NOS knockout mice. The SV129 mice also exhibited increased basal arterial plasma glucose levels relative to the other strains (although not statistically significant), raising the possibility that increased stress in SV129 mice might affect measured glucose use values. An alternative hypothesis is that a strain difference contributes to the difference in 1CMRglc values, supported by recent reports that SV129 mice show increased susceptibility to kainate-induced hippocampal cell death compared with both C57B1/6 mice and SV129xC57B1/6 hybrids (Schauwecker and Steward, 1997), and that C57B1/6 mice are more susceptible to cell damage after global ischemia than SV129 mice (Fujii et al., 1997).
In summary, the current study demonstrates that developmental inhibition of synthesis of either endothelial or neuronal isoforms of NOS does not induce overt changes in cerebral functional activity, as reflected by 1CMRglc, in the mature mouse brain. Our findings support previous reports that the different responses of eNOS and nNOS knockout mice to cerebral ischemia and excitotoxicity reflect primary roles for nNOS in neurotoxic mechanisms, and for eNOS in preserving cerebral blood flow, rather than secondary effects of the gene deletion on glucose utilization.