
Abstract
Previous studies have shown that 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) protect the brain against ischemic injury by upregulating endothelial nitric oxide synthase (eNOS). Here, we tested the hypothesis that statins provide additional beneficial effects by also upregulating endogenous tissue plasminogen activator (tPA) and enhancing clot lysis in a mouse model of embolic focal ischemia. Heterologous blood clots (0.2 mm) were injected into the distal internal carotid artery to occlude blood flow in the middle cerebral artery territory after long-term (14 days) simvastatin, atorvastatin or vehicle treatment. Ischemic lesion volume, neurologic deficits, as well as residual blood clots were measured at 22 h. Reverse transcription-polymerase chain reaction assessed mRNA levels of eNOS, tPA, and the endogenous plasminogen activator inhibitor PAI-1. Ischemic lesion volumes and neurologic deficits were significantly reduced in wild-type mice by both simvastatin and atorvastatin. Statins increased eNOS and tPA mRNA levels but did not change mRNA levels of PAI-1. In eNOS knockout mice, atorvastatin reduced the volume of ischemic tissue and improved neurologic outcomes after arterial occlusion by blood clot emboli. In contrast, statins did not have protective effects in tPA knockout mice after embolic focal ischemia, but only in a filament model where focal ischemia was achieved via mechanical occlusion. These results suggest that statins protect against stroke by multiple mechanisms involving both eNOS and tPA. The involvement of each pathway may be revealed depending on the choice of experimental stroke model.
Keywords
Introduction
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase is the rate-limiting enzyme in the biosynthesis of cholesterol. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) are potent reversible inhibitors of this enzyme, and are widely prescribed to treat patients with hypercholesterolemia. Accumulating evidence suggests that statins may possess beneficial effects in addition to their cholesterol-lowering actions. For example, statins reduce cardiovascular mortality even in normocholesterolemic or hypocholesterolemic patients (Sacks et al, 1996). In experimental models of cerebral ischemia, statins protect against tissue injury by upregulating endothelial nitric oxide synthase (eNOS) (Amin-Hanjani et al, 2001; Endres et al, 1998), augmenting cerebral blood flow (Yamada et al, 2000), and probably by suppressing eNOS-dependent mechanisms regulating platelet aggregation and leukocyte adhesion (Gertz et al, 2003; Laufs et al, 2000).
More recently, it was shown that statins increase fibrinolytic mechanisms or decrease prothrombotic mechanisms. In aortic endothelial cells, statins increase mRNA and enzymatic activity of tissue type plasminogen activator (tPA) (Essig et al, 1998), and decrease mRNA and activity of plasminogen activator inhibitor-type 1 (PAI-1) (Bourcier and Libby, 2000). Tissue plasminogen activator is the major physiologic plasminogen activator and is synthesized by the endothelium. PAI-1 is the major endogenous inhibitor of both t-PA and urokinase-type plasminogen activator and therefore plays a dominant role in the control of fibrinolysis; it is also synthesized within the endothelium. These additional antithrombotic and fibrinolytic effects of statins may promote important benefits in cerebral ischemia resulting from embolic arterial occlusion. In this study, we showed that statins upregulated both endogenous eNOS and tPA, and both pathways can contribute towards cerebroprotection in stroke. In eNOS knockout mice subjected to embolic clot ischemia, statins reduced infarct size and improved outcomes. In tPA knockout mice, statins were effective in a mechanical model of arterial occlusion but not in the embolic clot model. These data show that statins possess multiple cerebroprotective actions in stroke involving both eNOS and tPA, and the importance of each pathway may be revealed by the use of different models of experimental stroke.
Materials and methods
Animal Models of Focal Cerebral Ischemia
Male SV-129 mice (25 to 30 g, Taconic Farms) were used in the initial experiment for testing statins in embolic stroke. In the second set of experiments, male C57BL/6 mice (20 to 25 g, Charles River) were used to match the genetic background of our subsequent eNOS and tPA knockout mouse studies. All experiments were performed following an institutionally approved protocol in accordance with the NIH Guide for the Care and Use of Laboratory Animals. An embolic occlusion method was used to induce focal cerebral ischemia. Using a facemask, mice were induced with 2.0% halothane and anesthesia was maintained with 0.5% halothane in 70% N2O and 30% O2. Rectal temperatures were maintained at 37°C±0.5°C by means of a feedback-regulated heating pad (FHC, Brunswick, ME, USA). Arterial blood pressure, pH, and gases were monitored in selected animals via a PE-10 catheter placed in the femoral artery. The method used to prepare and inject a blood embolus was adapted and modified from Zhang et al (1997). One day before ischemic onset, arterial blood was withdrawn from donor mice into PE-50 tubing, stored at room temperature for 2 h, and then kept at 4°C for 22 h. Coagulated blood was subsequently sectioned into 12.5 mm segments, washed with saline and transferred to a modified PE-50 catheter with a tip diameter of 0.2-mm for injection into the internal carotid artery. Under a surgical microscope, the right common, internal and external carotid arteries were exposed by blunt dissection via midline cervical incision. After the ligation of the ptergyopalatine and distal end of the external carotid arteries, the modified PE-50 tubing with the 12.5 mm clot was connected to a 50-μL Hamilton lock syringe, and gently inserted through the external carotid artery and advanced until the catheter tip was positioned just proximal to the origin of the middle cerebral artery. The clot was slowly injected into the internal carotid artery along with a small amount of saline. After 5 min, the catheter was withdrawn from the external carotid artery. Successful occlusion of the middle cerebral artery was verified for 15 to 20 mins by laser Doppler flowmetry. To compare responses in the clot model versus mechanically induced ischemia, the standard intraluminal filament model of focal cerebral ischemia was also used (Huang et al, 1994). Endothelial nitric oxide synthase knockout mice were subjected to embolic clot focal ischemia. tPA knockout mice were subjected to both embolic clot occlusion as well as the standard filament mechanical occlusion.
Drug Treatments
Simvastatin (20 mg/kg), atorvastatin (20 mg/kg), or vehicle were administered as subcutaneous injections daily for 14 days. Simvastatin (1.2 mg/mL) was prepared in a phosphate-buffered saline (PBS) (pH 7.4) solution containing 10% ethanol and chemically activated by alkaline hydrolysis before administration. Atorvastatin (4 mg/mL) was dissolved in a PBS (pH 7.4) solution containing 45% 3-hydroxypropyl-B-cyclodextrin and 10% ethanol. Treatments followed previously established administration protocols (Amin-Hanjani et al, 2001; Endres et al, 1998; Yamada et al, 2000).
Measurement of Neurologic Deficits, Residual Clot Grade, and Infarct Volume
Mice were blindly scored for neurologic deficits on a 5-point scale: from 0 (no deficit) to 4 (inability to move or retain normal upright posture). Neurologic examinations were performed at 2 and 22 h after injection of clot. All mice were killed at 22 h after ischemia and brains were removed. The status of residual clot in the middle cerebral artery territory was assessed by a masked investigator using a simplified grading scale as follows: grade 2 for fully intact clots present at the junction of the internal carotid artery extending into the proximal anterior and middle cerebral arteries, grade 1 for partially dissolved or broken-up clots, grade 0 for complete lysis with no remnant clot visible. Two-millimeter-thick coronal sections of the brain were cut and stained with 2, 3, 5-triphenyltetrazolium chloride (TTC, Sigma, St Louis, MO, USA). Ischemic lesion volumes were quantified using standard computer-assisted image analysis techniques.
Reverse Transcription Polymerase Chain Reaction
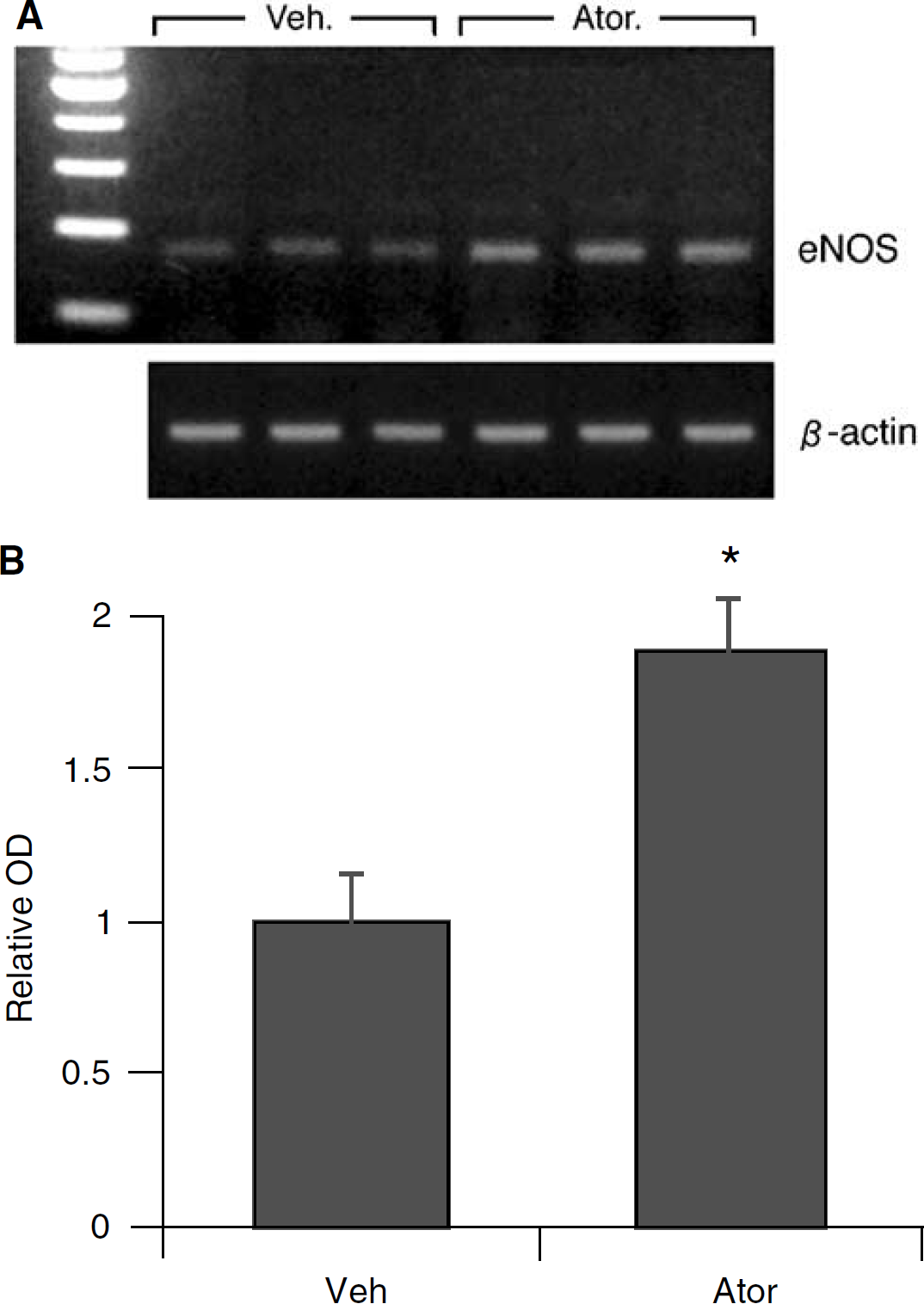
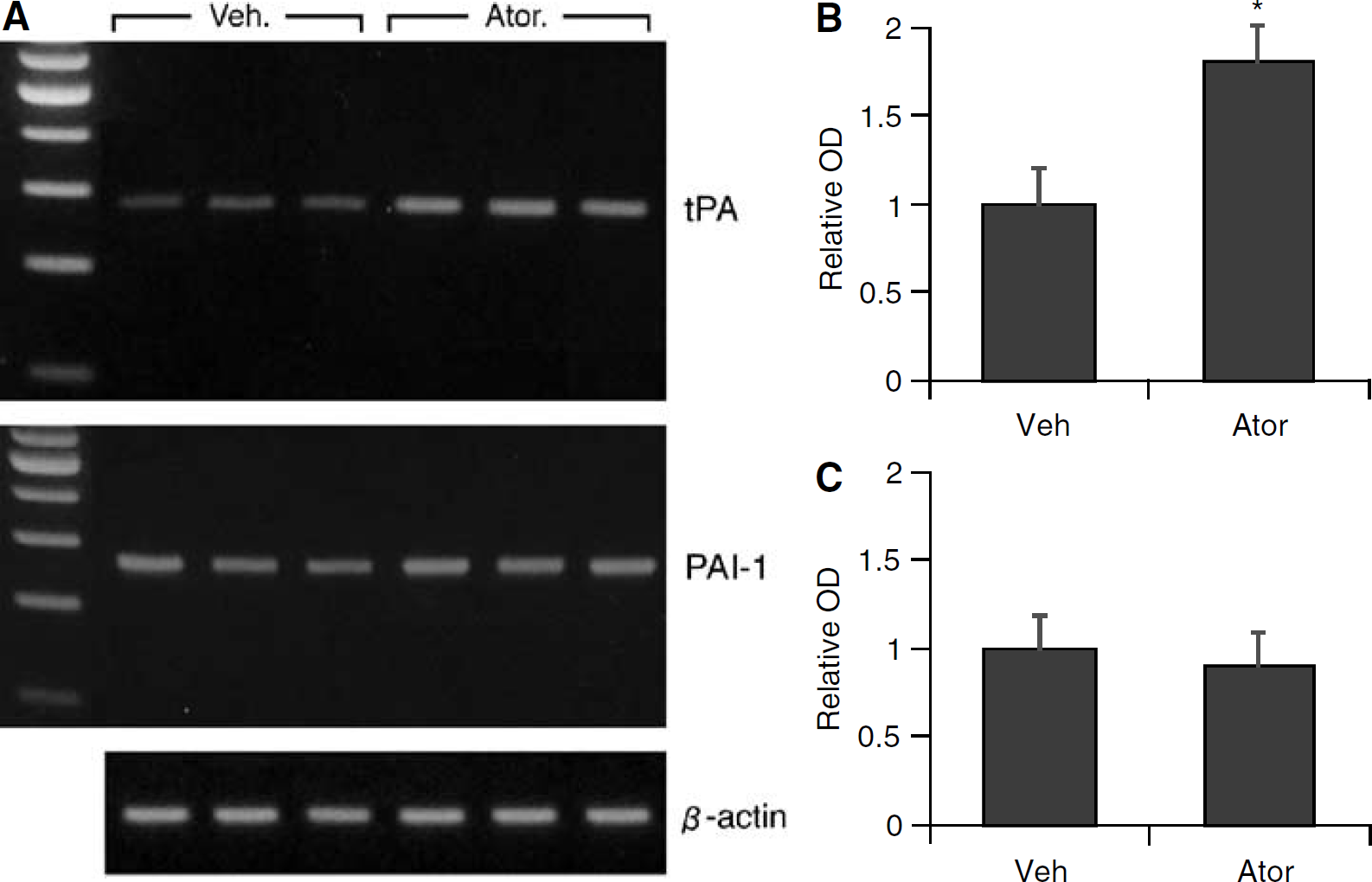
The thoracic-abdominal segments of the aorta were removed and immediately frozen by liquid nitrogen. Total RNA was prepared with RNA isolation reagent (TRIzol Reagent, Life Technologies Inc., Gaitherburg, MD, USA). One microgram of total RNA sample from each animal was subjected to a reverse transcription (RT)-polymerase chain reaction (PCR) analysis for eNOS, tPA, and PAI-1 mRNA. The RT reaction and PCR were performed out using RT-PCR kit (TaKaRa, Co. Ltd., Ohtsu, Japan). The following conditions were used for the RT reaction using oligo-dT primers: 55°C for 20 mins, 99°C for 5 mins, 5°C for 5 mins. The following conditions were used for PCR amplification using specific primers: cDNA products of the RT reaction were denatured for 2 mins at 94°C before cycling at 94°C for 30 secs, 60°C for 30 secs, and 72°C for 90 secs. Cycle numbers were 26, 26, and 32 for eNOS, tPA, and PAI-1, respectively. Primer sequences were as follows: eNOS (5′) primer 5′-TTCCGGCTG-CCACCTGATCCTAA-3′ and (3′) primer 5′-AACATATGTCCTTGC-TCAAGGCA-3′; tPA (5′) primer 5′-CTGAGGTCACAGTCCAAGCAATGT-3′ and (3′) primer 5′-GCTCACGAAGAT-GATGGTGTAAAGA-3′; PAI-1 (5′) primer 5′-TCAGAGCAACAA-GTTCAACTACACTGAG-3′ and (3′) primer 5′-CCCACTGTCAAGGCTCCATCACTT-GCCCCA-3′; beta-actin (5′) primer 5′-TGGAATCCTGTGGCATCCATGAAAC-3′ (3′) primer 5′-TAAAACGCAGCTCAGTAACAG-TCCG-3′. Primer sequences were obtained from previously published studies (Bourcier and Libby, 2000; Endres et al, 1998; Essig et al, 1998). The RT-PCR products were separated on 1.8% agarose gel and visualized with ethidium bromide. Control experiments showed that the amount of final RT-PCR product was proportionally reflected in the amount of input total RNA sample after each specific cycle of amplification.
Statistical Analysis
Quantitative data were expressed as mean+s.e.m. Ischemic lesion volumes and RT-PCR measurements of mRNA levels in various groups were compared using analysis of variance followed by Tukey—Kramer tests. For comparing neurologic deficits and residual clot grades between groups, the nonparametric Kruskal—Wallis tests were used, followed by Mann—Whitney U-tests. Differences with P<0.05 were considered statistically significant.
Results
In all groups of mice (SV-129 and C57BL/6), statin treatments did not alter physiologic parameters including arterial blood pressure, pH, pO2, and pCO2 (Table 1). Laser Doppler flowmetry showed that similar perfusion deficits were achieved in all experimental groups during ischemia (Table 2).
Physiologic variables
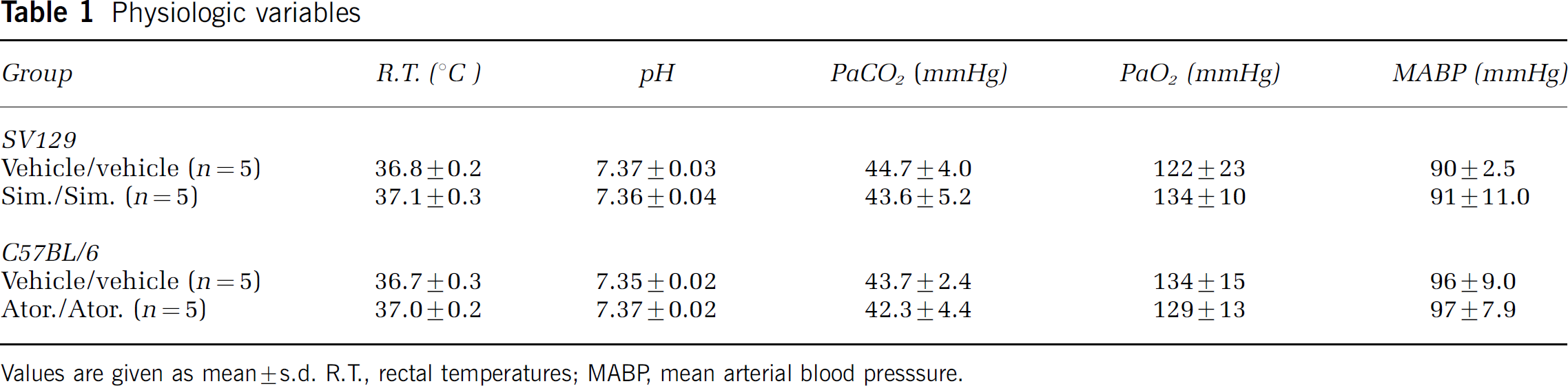
Values are given as mean±s.d. R.T., rectal temperatures; MABP, mean arterial blood presssure.
Regional cerebral blood flow
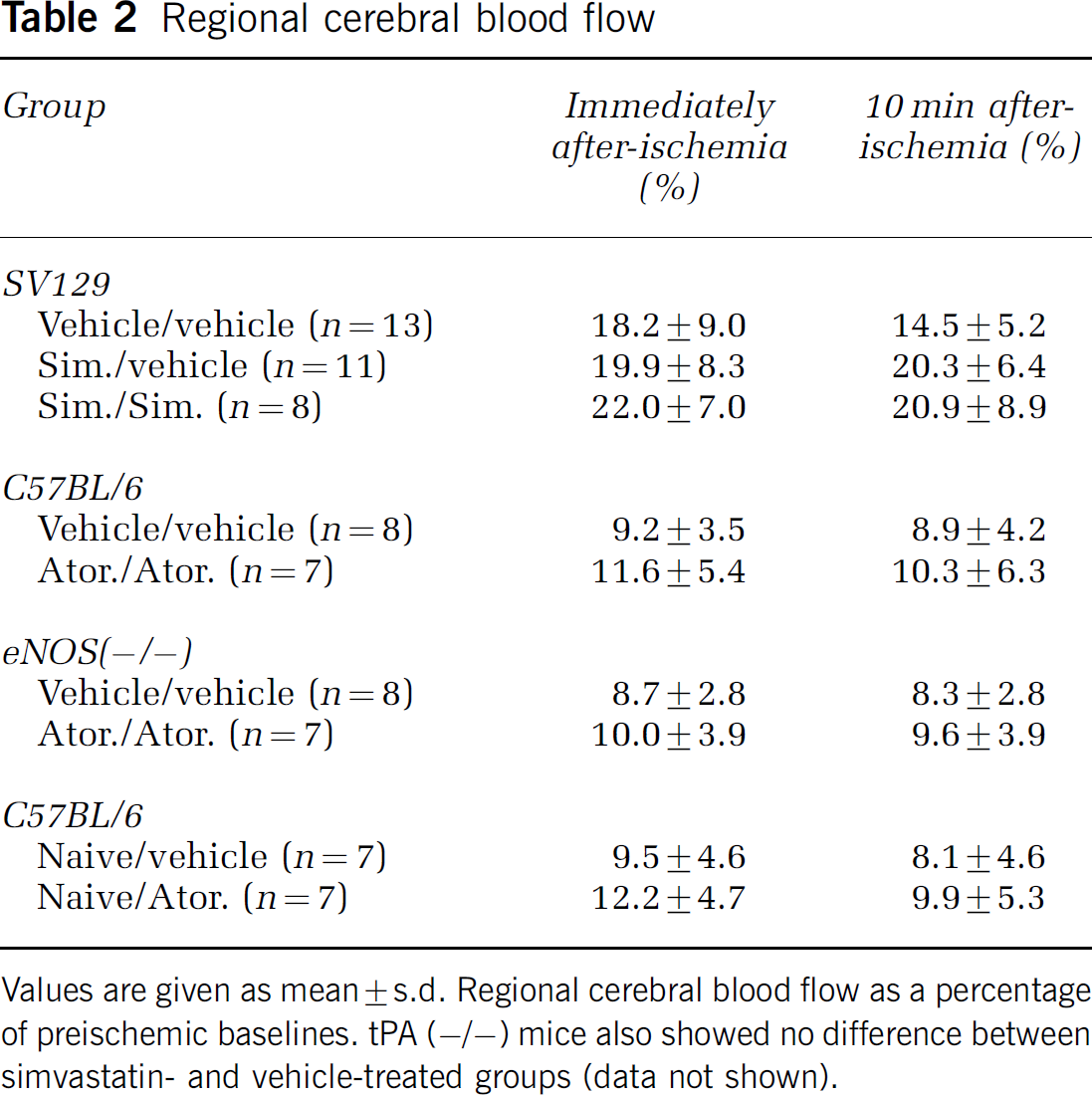
Values are given as mean±s.d. Regional cerebral blood flow as a percentage of preischemic baselines. tPA (−/−) mice also showed no difference between simvastatin- and vehicle-treated groups (data not shown).
Statins Reduced Ischemic Lesion Volumes after Embolic Focal Ischemia
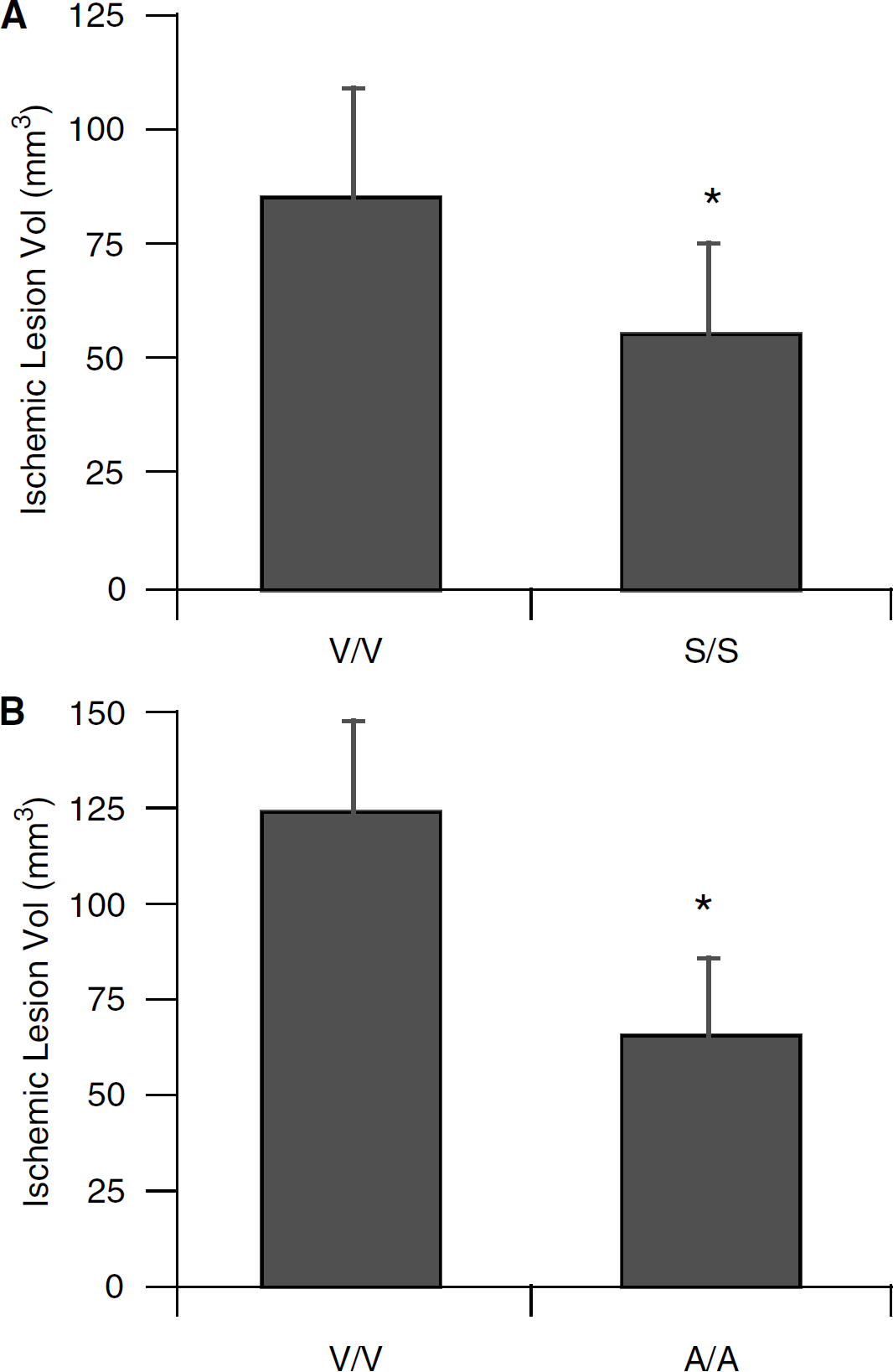
In the embolic clot model, ischemic tissue comprised the middle cerebral artery territory, including striatum and overlying cortex. Simvastatin reduced ischemic lesions in SV-129 mice (Figure 1A), and atorvastatin reduced ischemic lesions in C57BL/6 mice (Figure 1B). The extent of protection was approximately 50% to 60%, consistent with previous reports. Along with this cerebroprotective effect, both statins improved neurologic outcomes after focal ischemia (Table 3).
Neurologic deficit and residual clot grade
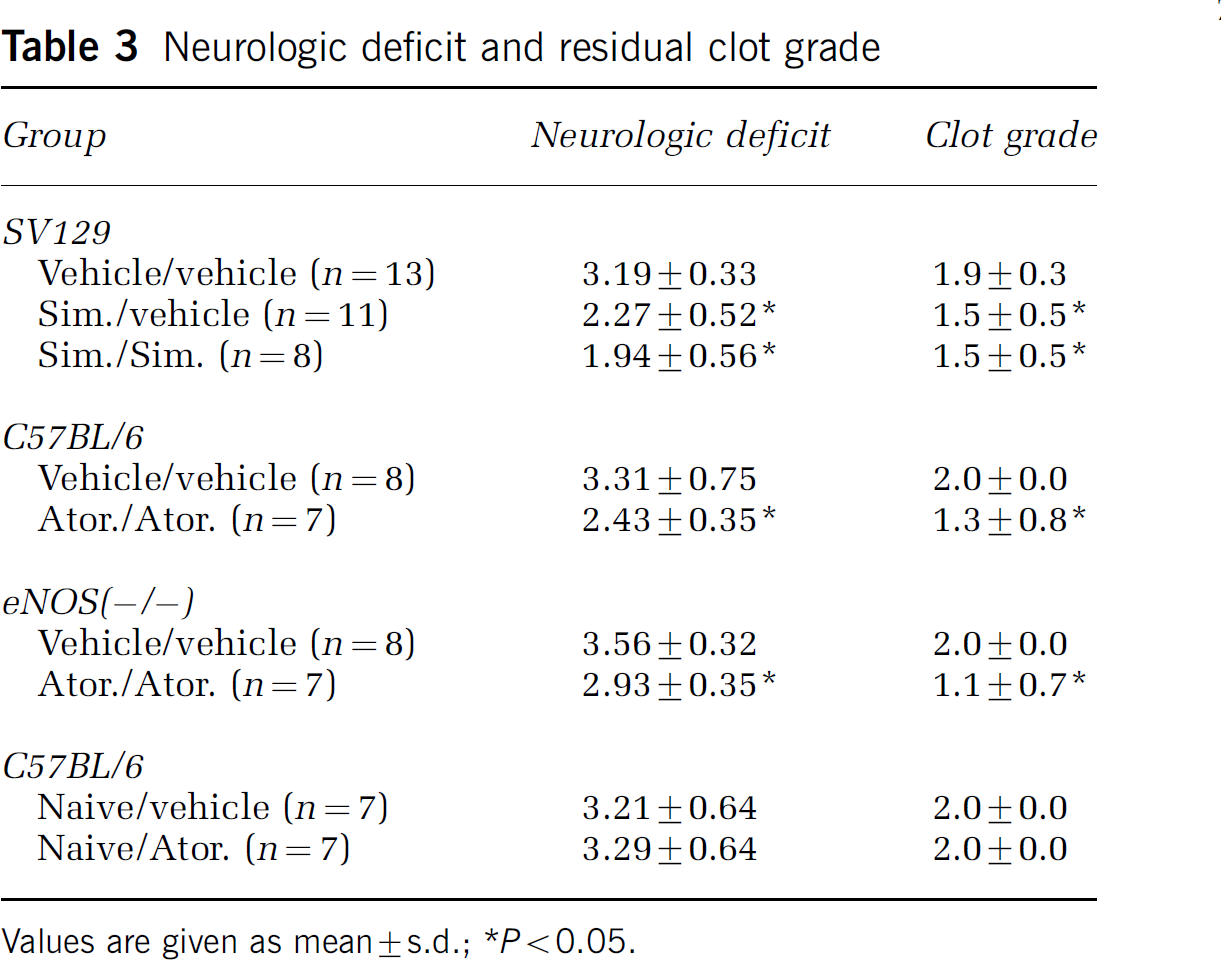
Values are given as mean±s.d.;
P<0.05.
(
To exclude the possibility that the statins affected the integrity of donor blood clots used for embolic stroke, additional experiments were performed to compare stroke outcome when clots originating from vehicle-treated mice were tested and compared with clots prepared from statin-treated mice. Protective effects were not observed when clots derived from statin-treated mice were injected into vehicle-pretreated mice subjected to embolic focal ischemia (Figure 2A). In contrast, regardless of whether the clots were derived from statin or vehicle-treated mice, chronic statin pretreatment significantly reduced ischemic injury (Figures 1A and 2B).
(
Dissolution of Blood Clot Emboli was Enhanced in Statin-Treated Mice
The amount of clot present in cerebral arteries was assessed 22 h after the onset of ischemia. In vehicle-treated controls, residual blood clots typically extended into the distal segments of the middle cerebral arteries (Figure 3). A significant reduction in residual clot was found in both atorvastatin- and simvastatin-treated animals (Figure 3, Table 3). Semiquantitative RT-PCR was used to assess levels of eNOS, tPA, and PAI-1 mRNA in tissue samples from the dissected aorta. Atorvastatin increased eNOS and tPA mRNA levels (Figures 4A, 4B and 5A, 5B) but showed no effects on PAI-1 mRNA after the specified treatment (Figures 5A and 5C).
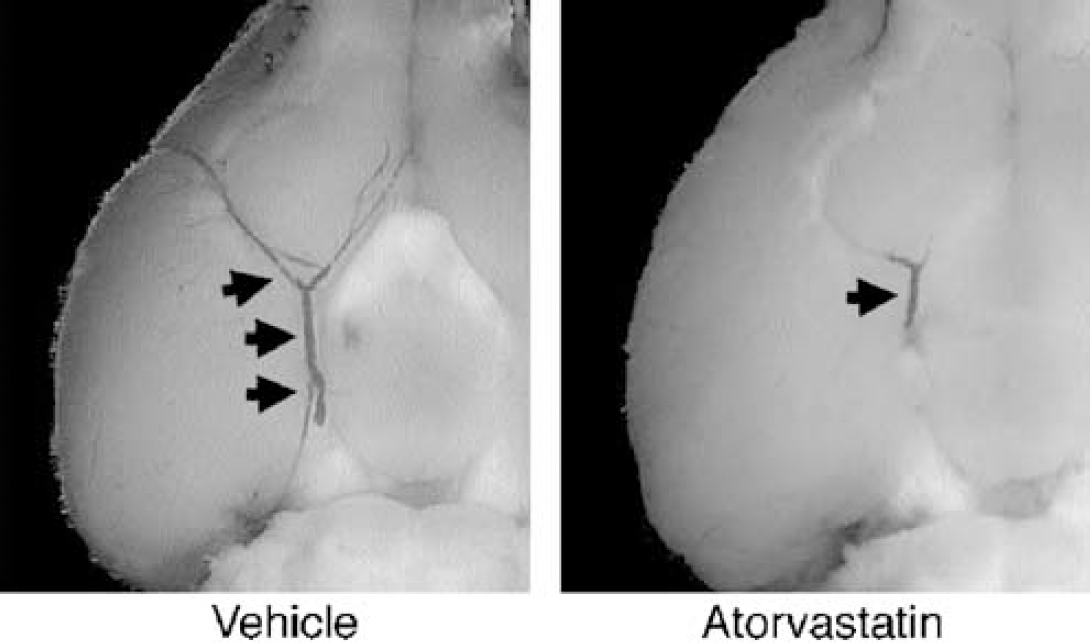
Representative photographs of the residual clots in the middle cerebral arteries of ischemic brains at 24 h. Atorvastatin-treated mice had significantly reduced residual clot compared with vehicle-treated controls (see Table 3 for quantified data).
(
(
Statin Cerebroprotection was Preserved in Endothelial Nitric oxide Synthase Knockout Mice after Embolic Stroke
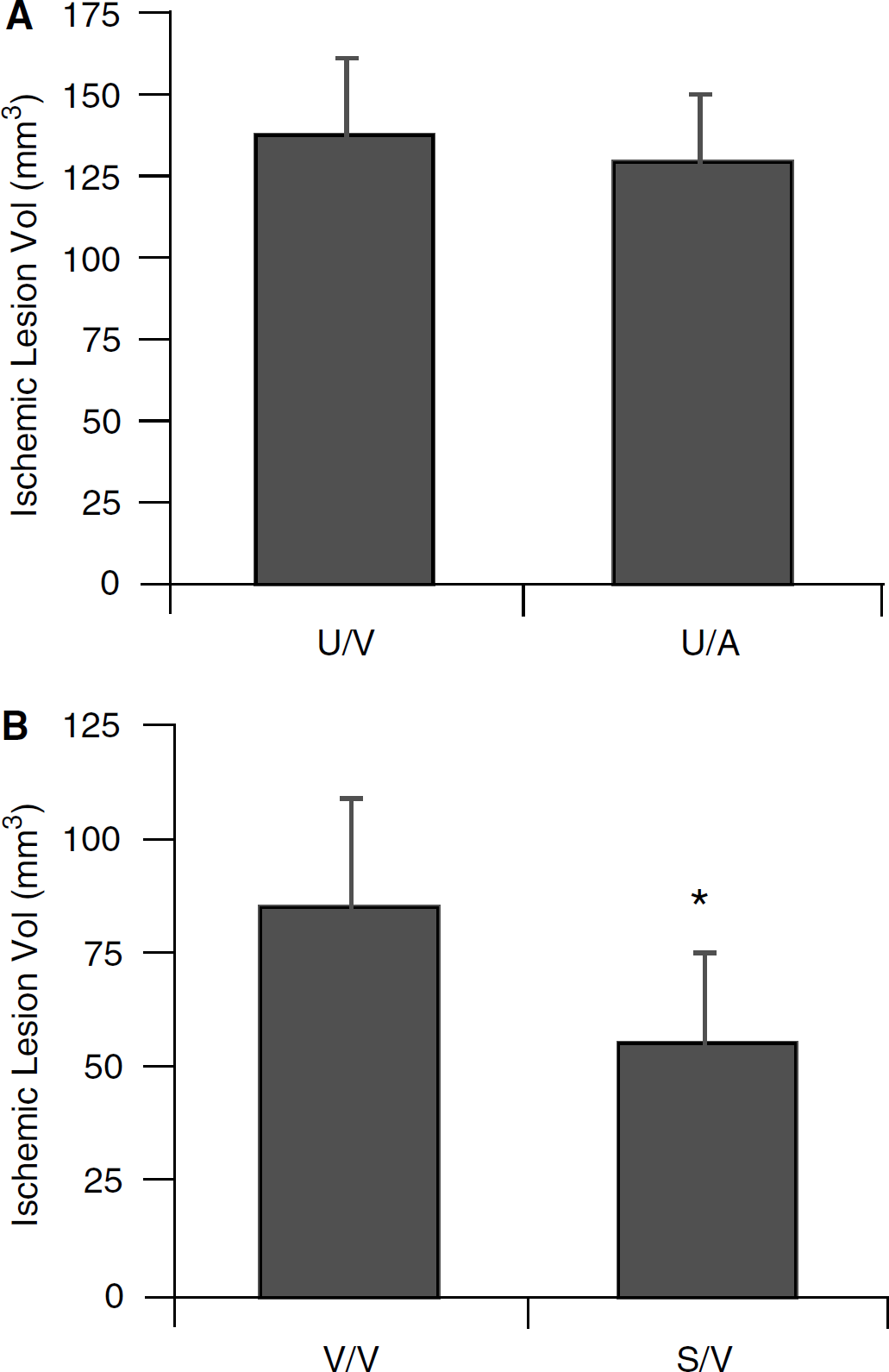
Endothelial nitric oxide synthase-deficient knockout mice were treated with either 20 mg/kg of atorvastatin or vehicle for 14 days, then subjected to embolic focal ischemia. Similar decreases in cerebral perfusion were achieved in statin-treated and control mice when measured by laser Doppler flowmetry (LDF) (Table 1). However, ischemic lesion volume was significantly reduced in atorvastatin-treated eNOS knockout mice compared with vehicle-treated eNOS mutant mice (Figure 6). Correspondingly, neurologic outcomes were also improved by atorvastatin (Table 3).
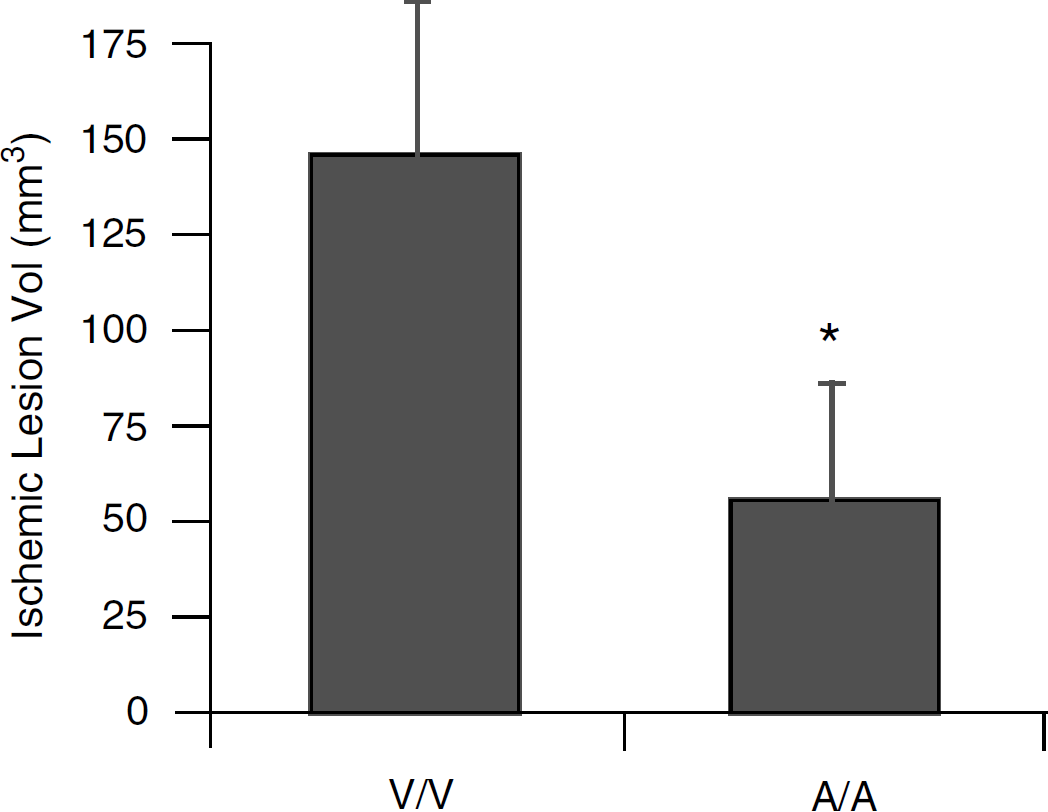
Twenty-four-hour ischemic lesion volumes (mean+s.e.m.) in endothelial nitric oxide synthase (eNOS) knockout mice after embolic focal ischemia were significantly reduced (*P<0.05) in the atorvastatin-treated group (n=10) compared with vehicle-treated controls (n=10). V/V=vehicle-treated eNOS knockouts occluded with clots from vehicle-treated knockout donors. A/A=atorvastatin-treated eNOS knockouts occluded with clots from atorvastatin-treated knockout donors.
Tissue Plasminogen Activator Knockout Mice were not Protected by Statins after Embolic Stroke
Tissue plasminogen activator-deficient knockout mice were treated with either 20 mg/kg of simvastatin or vehicle for 14 days, then subjected to either embolic clot focal ischemia or mechanically induced focal ischemia. Twenty-four-hour ischemic lesion volumes were significantly reduced in simvastatin-treated tPA knockouts compared with vehicle-treated mice after 2-h transient focal cerebral ischemia achieved via filament occlusion (Figure 7). However, simvastatin did not significantly alter ischemic lesion volumes in tPA knockout mice after cerebral ischemia achieved via embolic clot occlusion (Figure 7).
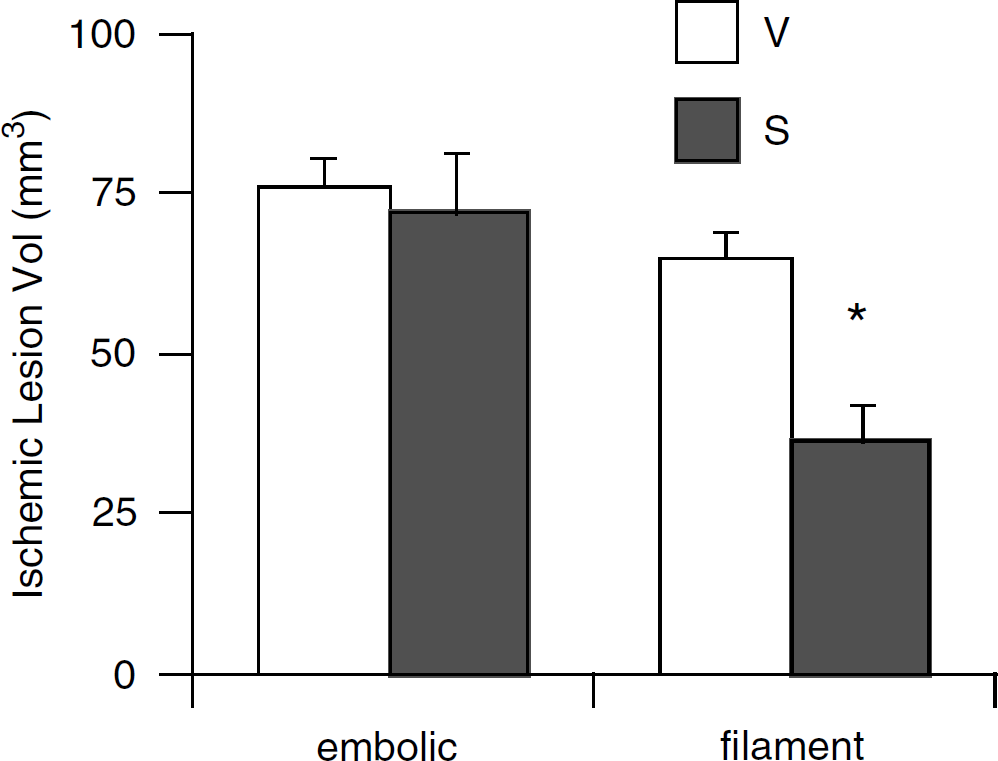
Twenty-four-hour ischemic lesion volumes (mean+s.e.m.) in tissue plasminogen activator (tPA) knockout mice after embolic focal ischemia were significantly reduced (*P<0.05) in the simvastatin-treated group (n=6) compared with vehicle-treated controls (n=5) after filament arterial occlusion. However, simvastatin did not reduce ischemic lesion volumes in tPA knockout mice when focal ischemia was achieved via embolic clot occlusion. V=vehicle-treated mice occluded with clots from vehicle-treated donors. S=simvastatin-treated mice occluded with clots from simvastatin-treated donors. n=6 per group.
Discussion
We previously showed that chronic statin pretreatment protects against ischemic brain injury (Amin-Hanjani et al, 2001; Endres et al, 1998; Yamada et al, 2000). These protective effects depend in part on eNOS upregulation and are shared by drugs that block the enzymatic conversion of HMG-CoA to mevalonic acid (i.e., the statins). Protection by statins was attributed to their ability to elevate absolute cerebral blood flow (Yamada et al, 2000) and to suppress platelet aggregation (Gertz et al, 2003; Laufs et al, 2000) via augmenting eNOS mRNA and protein levels as well as NOS enzymatic activity. Because NO potently dilates vascular smooth muscle and suppresses both platelet aggregation and neutrophil adhesion, and because eNOS deletion or enzymatic inhibition augments infarct size, an important role for NO generated by vascular tissue was hypothesized in cerebral ischemia (Iadecola, 1997; Ignarro, 2002). Strong evidence linking the protective effects of statins to NO generation was provided by experiments in eNOS null mice in which neuroprotection in the presence of statin pretreatment was blocked along with the blood flow enhancing effects (Endres et al, 1998).
It is becoming increasingly recognized that the neuroprotective effects of statins may include an even wider range of actions (Laufs and Liao, 2003; Liao, 2002). For example, acute treatment with statins at the time of stroke onset provides neuroprotection (Sironi et al, 2003); this may be because of allosteric enzymatic NOS activation mediated by PI3 kinase and eNOS phosphorylation by the kinase Akt (Kureishi et al, 2000). In addition, statins upregulate endogenous tPA and downregulate PAI-1 (Bourcier and Libby, 2000; Essig et al, 1998). Many statins including those tested herein reportedly decrease PAI-1 production in cultured human vascular cells under basal conditions and especially after adding pro-inflammatory stimuli such as TNF-alpha and IL-1 alpha (Wiesbauer et al, 2002). Furthermore, statins reportedly improve the fibrinolytic profile in patients with coronary artery disease (Seljeflot et al, 2002), although not all studies agree (Davignon and Laaksonen, 1999; Rosenson and Tangney, 1998). In the present study, we showed that statins were protective in a mouse embolic clot model of focal ischemia. Importantly, these protective effects were preserved in eNOS knockout mice, unlike the findings in a filament model of mechanical arterial occlusion (Endres et al, 1998). Hence, under some circumstances, statins can promote additional actions unrelated to hemodynamic or platelet- and neutrophil-suppressing NO effects that are beneficial for stroke.
We speculate that neuroprotection found after administering statins in this clot model may relate to tPA and its effects on thrombosis. We observed decreased residual clot in the statin-treated group 22 h after embolic stroke. Moreover, the increased tPA mRNA levels we measured could lead to reduced thrombin development. Despite this tPA increase, statins administered long-term to clot donor mice did not apparently alter stroke outcome in vehicle-treated mice rendered ischemic. In other words, outcome did not differ significantly whether the donor animals were pretreated with statin or vehicle. This may indicate that statins do not modify clot formation ex vivo; its effect in the embolic model may relate to suppression of clot stability at sites of local vascular injury after embolic occlusion.
In this study, we showed that levels of tPA mRNA were elevated in statin-treated mice although there were no statistically significant differences for PAI-1. This stands in contrast to reports noted above showing that PAI-1 mRNA levels were downregulated by nearly all statins (Wiesbauer et al, 2002) and might reflect differences in conditions between in vivo and in vitro models. As a second possibility, mRNA levels were not measured in the brain, the most relevant vascular bed, but rather in the aorta. Although it is expected that the effects of statins on PAI-1 transcription would be uniform in all vascular beds, we cannot state this unequivocally, nor can we rule out that statins suppressed PAI-1 mRNA levels only after embolic occlusion and vascular lesion. Nevertheless, the finding that statins enhanced clot dissolution and reduced infarction size supports the notion that the statins increased thrombolysis within the brain.
The pleiotropic effects of statins extend beyond their actions on the fibrinolytic system and NO bioavailability (Takemoto and Liao, 2001). For example, statins exhibit antioxidant and antiinflammatory activity, suppress apoptosis in endothelial cells and promote angiogenesis, among other actions. Isoprenoid intermediates in the cholesterol pathway are key molecules in cell signaling. Inhibition of isoprenoid synthesis appears to modulate eNOS, PA-1, and t-PA mRNA levels. In fact, the mRNA changes are reversed by treatment with mevalonic acid (Laufs and Liao, 2003; Wiesbauer et al, 2002). Briefly, the isoprenoid geranylgeranyl pyrophosphate and subsequent geranylgeranylation of the G-protein signaling molecule Rho is implicated in eNOS, PA-1, and t-PA regulation (Laufs and Liao, 2000). Inhibition of isoprenoid synthesis by statins suppresses Rho GTPase activity and relocates Rho to the cytoplasm, rendering it inactive. Inactivation of Rho GTPase activity disinhibits relevant downstream mechanisms, regulating mRNA stability among other mechanisms still to be clarified.
An important finding in the present study is the dependence of results on the choice of experimental stroke model. Statins were protective in eNOS knockout mice subjected to embolic clot focal ischemia. In contrast, tPA knockout mice were protected by statins only when arterial occlusion was achieved mechanically but not via embolic occlusion. These data suggest that in the embolic clot model, statin upregulation of endogenous tPA may predominate, whereas in the filament occlusion model, eNOS upregulation appears more important. To our knowledge, gene-specific drug effects that are model dependent have not previously been documented in the brain ischemic literature. Taken together with our past experiments, these findings suggest that both eNOS and tPA are key modulators of ischemic damage and can be regulated by statins singly or in combination for the prevention of stroke.
In conclusion, the present study showed that statins might also be cerebroprotective against embolic ischemic stroke via eNOS independent pathways. Statin-induced upregulation of endogenous tPA response may be especially relevant in the context of acute embolic stroke in the clinical realm. More detailed studies are warranted to investigate these pathways and potential interactions between tPA therapy with prior statin use in stroke.