
Abstract
Secondary brain injury due to ischemia includes the infiltration of leukocytes into the brain parenchyma mediated by activation of nuclear factor-κB (NF-κB), which is activated by proteasome degradation. Neuroprotection with the proteasome inhibitor MLN519 has previously been reported to decrease ischemic brain injury in rats. The authors used higher doses of MLN519 to evaluate the neuroprotection therapeutic window after 24 hours of brain injury in rats as correlated to proteasome levels, activated NF-κB immunoreactivity, and leukocyte infiltration. Male Sprague-Dawley rats were subjected to 2-hour middle cerebral artery occlusion (MCAO) and recovery. MLN519 or vehicle was administered after injury with a single injection given in delayed increments of 2 hours (i.e., 4, 6, or 8 hours after MCAO). Treatment with MLN519 up to 6 hours after MCAO (4 hours after reperfusion) effectively reduced neuronal and astrocytic degeneration, decreased cortical infarct volume, and increased neurologic recovery. These effects were related to >80% reductions in blood proteasome levels, reduced neutrophil infiltration, and a decrease in activated NF-κB immunoreactivity. This improved neuroprotection profile and antiinflammatory effect of MLN519 provides an exciting avenue for potential treatment of focal ischemic brain injury in humans.
Ischemic brain injury due to insults such as stroke is a leading cause of death in the United States. Such injury is also a crucial player in brain damage due to blunt head trauma and is responsible for 500,000 hospitalizations per year (Torbati, 1995). Several therapeutic interventions against ischemic brain injury in the past involved targeting ionic homeostasis with the use of voltage- or ligand-gated ion channel blockers (Muir and Lees, 1995). Unfortunately, several of these strategies have failed in clinical trials, most likely because of their short therapeutic window for intervention after the ischemic insult (De Keyser et al., 1999). Thus, a search for novel pharmacological agents has been initiated targeting downstream or delayed injury processes (Heiss et al., 1993). Such targets include inhibition of apoptosis and reduction of the acute inflammatory response (Barinaga, 1998; Lucchesi, 1993).
Recent neuroprotective strategies include targeting inflammatory mediators or immunomodulation as a whole. Targeting individual inflammatory mediators has proved difficult due to redundancy in function. For example, alternate inflammatory pathways may circumvent the suppression of a single targeted mediator (Stanimirovic and Satoh, 2000). Targeting inflammatory cascades as a whole is another approach, although treatment with glucocorticoids has shown no benefit (Jean et al., 1998). Nonsteroidal antiinflammatory drugs have been reported to be neuroprotective in focal ischemic brain injury models, but the neuroprotective effect has been explained by their ability to suppress mitochondrial calcium transition rather than antiinflammatory effects (Uchino et al., 1995). Novel approaches have now focused on alteration of inflammatory transcriptional factors to simultaneously interfere with the upregulation of multiple inflammatory genes. Nuclear factor-κB (NF-κB) is one such transcriptional factor involved in inflammation, and it can be blocked with agents such as proteasome inhibitors. In particular, MLN519 (previously named PS519) is a selective proteasome inhibitor and has little activity against other enzymes (Soucy et al., 1999). Previous data from our laboratory have, in fact, showed that treatment with MLN519 after experimental stroke in rats reduced brain infarction and leukocyte infiltration into injured brain tissue (Phillips et al., 2000). Recently, a successful phase 1 clinical safety trial was completed for MLN519 (Elliott et al., 2000). In the present study, we evaluated higher doses of MLN519 to extend the therapeutic window of neuroprotective treatment beyond initiation of MCAO in rats and further investigated the mechanism of action of this clinical development drug.
MATERIALS AND METHODS
Surgical procedures
Male Sprague-Dawley rats (270 to 330 g; Charles River Laboratories, Raleigh, VA, U.S.A.) were used in all procedures. Anesthesia was induced by 5% halothane and maintained with 2% halothane delivered in oxygen. Body temperature was maintained normothermic (37°C ± 1°C) throughout all surgical procedures by means of a homeothermic heating system (Harvard Apparatus, South Natick, MA, U.S.A.). Food and water were provided ad libitum before and after surgery, and the animals were individually housed under a 12-hour light/dark cycle. Intravenous (i.v.) polyethylene catheters (PE-50) were chronically implanted into the right jugular veins of all animals. Femoral artery catheters were placed into the right femoral artery using MRE-25 tubing (Braintree Sci., Braintree, MA, U.S.A.). The femoral catheter was attached to PE-50 tubing and tunneled subcutaneously to exit the back for attachment to a fluid swivel system (Instech, Plymouth Meeting, PA, U.S.A.) for continuous measurement of blood pressure and heart rate. Two cortical electrodes (epidural stainless steel screw electrodes, 0–80 × 1/8 in) were permanently implanted and fixed to the skull using dental acrylate cement (Tortella et al., 1997).
Twenty-four hours after the surgical procedures, the rats were reanesthetized and prepared for temporary focal ischemia using the filament method of MCAO and reperfusion as described elsewhere (Britton et al., 1997). Briefly, the right external carotid artery was isolated and its branches coagulated. A 3–0 uncoated monofilament nylon suture (Ethilon, Somerville, NJ, U.S.A.) with heat-blunted tip was introduced into the internal carotid artery via the external carotid artery and advanced (approximately 22 mm from the carotid bifurcation) until a slight resistance was observed, thus occluding the origin of the middle cerebral artery (MCA).
Once the filament was in place, a drop in amplitude of the cortical electroencephalographic (EEG) recording was used to indicate a successful occlusion. The endovascular suture remained in place for 2 hours and then was retracted to allow reperfusion of blood to the MCA. After MCAO surgery, animals were placed in recovery cages in which an ambient temperature of 22°C was maintained. At the conclusion of the experiment (24 hours after MCAO) rats were anesthetized, killed by decapitation, and brains were removed for quantification of infarction and histologic processing.
Infarct analysis
From each rat brain, analysis of ischemic damage included core infarct volume from both cortical and subcortical brain regions. Infarct volume quantitation was achieved using 2,3,5-triphenyl tetrazolium chloride (TTC) staining. Brain sections were taken from the region beginning 1 mm from the frontal pole and ending just rostral to the corticocerebellar junction. Computer-assisted image analysis was used to calculate infarct volumes from seven coronal brain sections (2-mm thick) as described in detail elsewhere (Tortella et al., 1999). Briefly, the posterior surface of each TTC-stained forebrain section was digitally imaged (Loats Associates, Westminster, MD, U.S.A.) and quantified for areas (mm2) of ischemic damage. Core injury was defined as brain tissue completely lacking TTC staining as compared with the contralateral, uninjured hemisphere. Sequential integration of the respective areas yielded core infarct volume (mm3). Similarly, ipsilateral and contralateral hemispheric volumes were measured, where hemispheric swelling (edema) was expressed as the percent increase in size of the ipsilateral (occluded) hemisphere over the contralateral (uninjured) hemisphere.
Physiology experiments
Mean arterial blood pressure (MABP) and heart rate (HR) were monitored in a separate group of awake freely moving rats from the femoral artery catheter before MCAO, continuously for 6 hours after injury, and again at 24 hours using a DigiMed blood pressure analyzer (MicroMed, Louisville, KY, U.S.A.). Vehicle (50% polypropylene glycol in saline, i.v., n = 7) or MLN519 (1.0 mg/kg, i.v., n = 8) was administered 2 hours after MCAO. Blood samples (100 μL) were taken before and 0.5, 2, 3, 4, 6, and 24 hours after MCAO to measure P***o2, P***co2, and pH using an ABL5 blood gas system (Radiometer A/S, Copenhagen, Denmark). Rectal temperatures were recorded in all rats before and 0.5, 2, 6, and 24 hours after MCAO.
Neurologic examination
A neurologic examination was performed on each rat immediately before MCAO and again at 2 and 24 hours after injury. Neurologic scores (NS) were derived using a 10-point sliding scale. Each animal was examined for reduced resistance to lateral push (score = 4), open field circling (score = 3), and shoulder adduction (score = 2) or contralateral forelimb flexion (score = 1) when held by the tail (modified from Bederson et al., 1986). Rats extending both forelimbs toward the floor and not showing any other signs of neurologic impairment were scored 0. Using this procedure, maximal neurologic severity was measured as an NS of 10. In the present study, all rats subjected to MCAO either exhibited a neurologic score of 10 when examined 2 hours after ischemia or immediately before reperfusion, or were excluded from the study.
Electroencephalographic recovery
Brain EEG activity was recorded in anesthetized rats before MCAO and at 2 and 24 hours after injury. Computer-analyzed spectral analysis was used to calculate the percent drop in EEG power after occlusion of the MCA. Animals not exhibiting an 80% drop in EEG power over the injured cortex were excluded from the study. Electroencephalographic activity was recorded and analyzed using QND 3.0 software and amplifiers (Neuro-data Inc., Pasadena, CA, U.S.A.)
20S proteasome assay
Arterial blood samples (0.5 mL) were collected in 10 μL heparin (1,000 U/mL) for proteasome analysis. Samples were taken before MCAO (baseline), at both 1 hour before and 1 hour after MLN519 treatment, and again at 24 hours after occlusion. Proteasome levels were compared with a matched vehicle-treated group at each time point. Proteasome peptidase activity was measured using a modified method described by (Dick et al., 1997). Samples were combined with 20 mmol/L HEPES, 0.035% (w/v) SDS, and 10 μmol/L succinyl-Leu-Leu-Val-Tyr 7-amino-4-methylcoumarin (s-AMC). Substrate hydrolysis of s-AMC was measured by continuously monitoring the fluorescence of the AMC using a Hitachi F2000 fluorescence spectrophotometer. After 3 or 4 minutes, 20.3 μL lactacystin (1 mmol/L) was added to reach a final concentration of 10 μmol/L lactacystin, and substrate hydrolysis was monitored for an additional 6 to 8 minutes. Because this lactacystin treatment is sufficient to inhibit the peptidase activity of the 20S proteasome, the final fluorescence recordings represent background activity of various other peptidases (Dick et al., 1997) and were subtracted from the initial 3- or 4-minute recordings. Enzyme activity was reported as moles s-AMC per milligram protein.
Histology
The TTC-stained brain slices were fixed in 10% formalin and embedded in paraffin (Phillips et al., 2000). Briefly, the tissue was dehydrated with gradient alcohol concentrations, cleared in xylene and embedded with paraffin wax with the use of an automatic tissue processor (Miles Tissue Tek; Sakura Finetek Inc., Torrance, CA, U.S.A.). Slices (6 μm) were cut from the second, third, and fourth coronal brain sections. Standard tissue dehydration and clearing for paraffin embedding removed the TTC stain. Brain sections were stained with hematoxylin and eosin (H&E) with sequential slides prepared for immunohistochemistry. Twelve 40x microscope fields from both striatal and cortical brain regions were used to count infiltrating leukocytes (neutrophils and macrophages) from each of the three brain regions. Extent and location of the lesion, cell morphology, presence of microhemmorhages, and evidence of changes in meninges or ventricles were noted for each brain section. Control experiments included comparisons between both normal uninjured and sham-injured animals. The ischemic injury in the ipsilateral hemisphere of each animal was also directly compared with the contralateral, uninjured hemisphere. All slides were analyzed on an Olympus AX-70 microscope and images captured on a Magnafire digital camera (Opelco Optical, Dulles, VA, U.S.A.).
Degeneration of neurons and axons was analyzed using the anionic fluorochrome FluoroJade. Slides were dewaxed, washed in deionized water, and placed in 0.06% potassium permanganate for 15 minutes. Slides were then washed in deionized water and incubated in 0.001% FluoroJade (Histo-Chem Inc., Jefferson, AZ, U.S.A.) for 30 minutes, rinsed, and coverslipped.
Immunohistochemistry
To study NF-κB activation, we used a monoclonal antibody (anti-NF-κB, p65 subunit; Chemicon International, Inc., Temecula, CA, U.S.A.) that specifically recognizes an epitope on the p65 subunit, which is normally masked by bound IκB-α. This antibody exclusively detects activated NF-κB because it recognizes p65 only in the absence of inhibitor κB-α (Brand et al., 1996; Hickenbottom et al., 1999; Nonaka et al., 1999). Brain sections were dewaxed and incubated for 1 hour with 4% normal horse serum in phosphate-buffered saline (PBS) to block nonspecific antibody binding. The primary monoclonal anti-NF-κB antibody was diluted 1:150 in the solution used to block nonspecific binding. Tissue sections were covered with diluted primary antibody and incubated overnight in a humid chamber with gentle rotary agitation. Sections were washed three times in PBS, and the bound antibody was visualized by the avidin-biotin complex method (Vectastain Elite; Vector Labs, Burlingame, CA, U.S.A.) with peroxidase as the marker enzyme. Biotinylated secondary antibody and avidin-peroxidase solutions were incubated on the sections for 90 minutes each, with three washes of PBS after each step. The immunohistochemical signal was developed using a metal-enhanced diaminobenzidine substrate system (Pierce Chemical Company, Rockford, IL, U.S.A.). In control experiments, primary antibodies were omitted to verify the absence of uncontrolled secondary antibody binding.
Quantitation of NF-κB immunoreactivity was done on cellular nuclei using 40x digital images from infarcted cortical brain tissue. Three cell types were defined: endothelial cells (oblong nuclei surrounding vascular openings), leukocytes (intact round nuclei in and around blood vessels), or perivascular neurons and glial cell types (polygonal degenerate nuclei). Optical density (OD) measurements were measured from the nuclei of NF-κB-positive cells using image analysis software (Inquiry; Loats Associates). Background OD levels from each slide were then subtracted from the OD of each cell nuclei and the mean values calculated for vehicle and MLN519 treatment groups for comparison.
Astrogliosis was assessed using glial fibrillary acidic protein (GFAP) immunostaining (1:1500; Dako, Copenhagen, Denmark) on an Optimax Plus automated tissue stainer (Biogenex, San Ramone, CA, U.S.A.). Slides were dewaxed and incubated for 20 minutes in 3% hydrogen peroxide to block endogenous peroxide followed by incubation in Biogenex Universal Blocking Reagent for 20 minutes to block nonspecific antibody binding. Tissue sections were covered with diluted primary antibody and incubated for 60 minutes. Slides were then incubated with Biognenex prediluted antirabbit link for 20 minutes followed by Biogenex horseradish-peroxidase-labeled streptavidin for 30 minutes, with three washes of PBS after each step. The immunohistochemical signal was developed using AEC substrate (Biogenex) for 5 minutes followed by counterstain with hematoxylin for 1 minute to identify cellular structures. In control experiments, primary antibodies were omitted to verify absence of uncontrolled secondary antibody binding. GFAP-positive cells were counted in a 40x field of the ipsilateral corpus callosum and the periinfarct region of the striatum from each animal, and comparisons were made between vehicle- and MLN519-treated animals. Only strongly reactive, intact cells possessing two or more dendritic ramifications were counted.
Data analysis
Data are presented as the mean ± SD. Statistical analysis of infarct volume, neurologic scores, leukocyte infiltration, IHC quantitation, and changes in physiologic parameters were evaluated by independent t-tests between each matched vehicle- and corresponding MLN519-treated group. P < 0.05 was considered significant.
Compound
MLN519 (Millenium Pharmaceuticals, Cambridge, MA, U.S.A.) was dissolved in a solution of 50% polypropylene glycol (PPG) in physiologic saline immediately before injection and was administered intravenously without handling or disturbing normal animal behavior. A single injection of MLN519 (1.0 mg/kg) or vehicle was delivered intravenously 4, 6, or 8 hours after MCAO (i.e., 2, 4, or 6 hours after reperfusion).
RESULTS
Brain pathology after 2-hour middle cerebral artery occlusion and 24-hour recovery
Control, vehicle-treated rats subjected to 2 hours of MCAO followed by 22 hours of reperfusion exhibited striatal and cortical infarction in the right brain hemisphere, as indicated by TTC-stained tissue sections (Fig. 1). Infarction was evident throughout the basal ganglia in all animals. In particular, ischemic regions included the middle and posterior portions of the caudoputamen as well as the internal capsule and occasionally parts of the anterior thalamus. Widespread GFAP staining was also present throughout the infarcted region, predominately in vehicle-treated animals (Fig. 1).
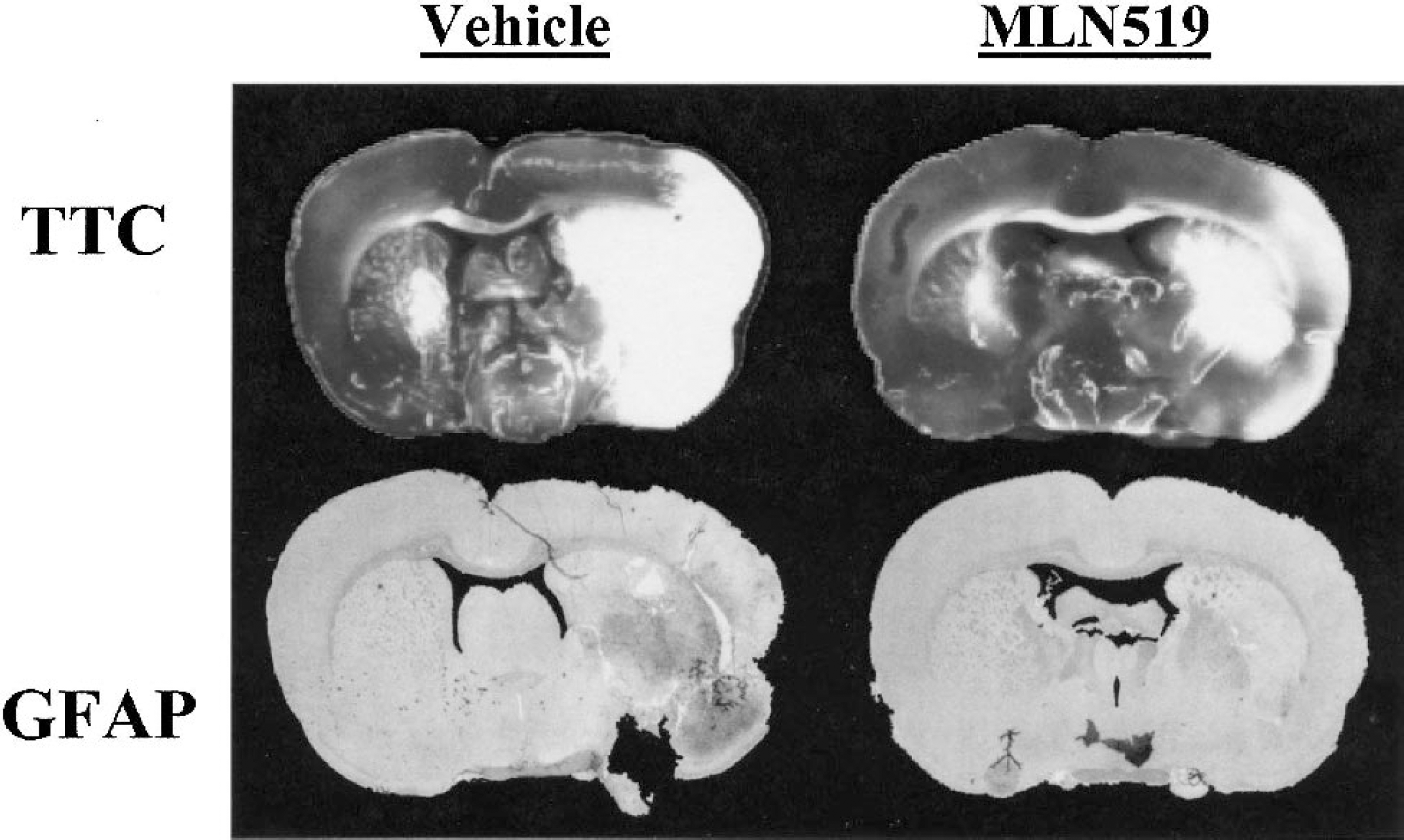
Representative coronal brain sections from vehicle- and MLN519-treated animals at 24 hours after middle cerebral artery occlusion. A direct comparison is presented between 2,3,5-triphenyl tetrazolium chloride (TTC) and glial fibrillary acidic protein (GFAP) staining from sequential brain sections of the same animal. Note the visible GFAP reactivity (dark areas) in the core of the infarct, which corresponds to necrotic debris, presumably from degenerating astrocytes.
Brain infarct volume
At the 24-hour endpoint, average infarct volumes for each of the vehicle-treated groups ranged from 165 to 203 mm3 (cortical) and 66 to 70 mm3 (subcortical) as reported in Table 1. MCAO injury also induced edemic increases in brain volume of 9% to 13% in the vehicle-treated groups (Table 1). MLN519-treated groups exhibited significantly lower cortical infarct volumes with delayed injections of MLN519 4 hours (42% reduction) and 6 hours (48% reduction), but not 8 hours, after MCAO (Table 1). No significant differences were measured in subcortical brain regions between treatment groups. We also measured a significant reduction in edemic brain swelling, but only with the 4-hour post-MCAO delayed injection of MLN519 (Table 1).
Effects of delayed treatment of MLN519 after middle cerebral artery occlusions on infarct volume, percent edema, and neurologic score
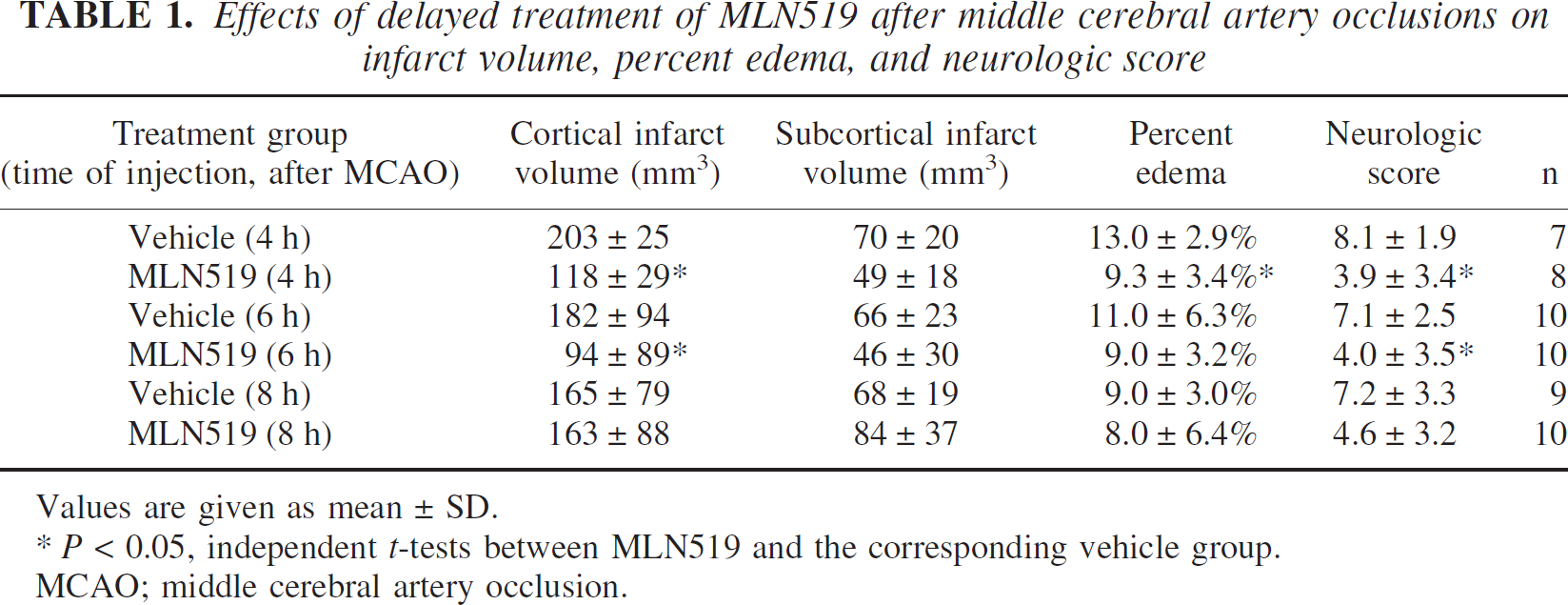
Values are given as mean ± SD.
P < 0.05, independent t-tests between MLN519 and the corresponding vehicle group.
MCAO; middle cerebral artery occlusion.
Neurologic recovery
Significant neurologic impairment was observed at 24 hours in vehicle-treated groups as revealed by average neurologic scores ranging from 7.1 to 8.1 (out of a maximum score of 10, Table 1). As with the reduction in infarct volume, we measured a statistically significant improvement in neurologic recovery 4 or 6 hours after MCAO treatment with MLN519.
Neuronal degeneration
Loss of neuronal integrity was verified with FluoroJade-stained sections (Figs. 2 and 3). Normal neuronal morphology was present in the contralateral (uninjured) parietal cortex of both vehicle- and MLN519-treated rats, as indicated in cortical layers II and III (Figs. 2A and 2C), and was comparable to both normal and sham-injured animals (data not shown). Severe neuronal degeneration was present in the ipsilateral parietal cortex of vehicle-treated animals, as indicated by the strong fluorescence labeling of neurons and loss of axonal processes (Fig. 2B), which was largely ameliorated in MLN519-treated animals (Fig. 2D). Presence of FluoroJade-positive neurons were verified in sequential H&E slides as hypereosinophilic, polygonal, shrunken cells present in the pale staining neuropil of the ischemic parietal cortex of vehicle-treated rats (Fig. 2F). Normal cellular morphology was observed in the contralateral hemisphere of all animals (Figs. 2E and 2G) as well as the ipsilateral parietal cortex of MLN519 treated animals (Fig. 2H).
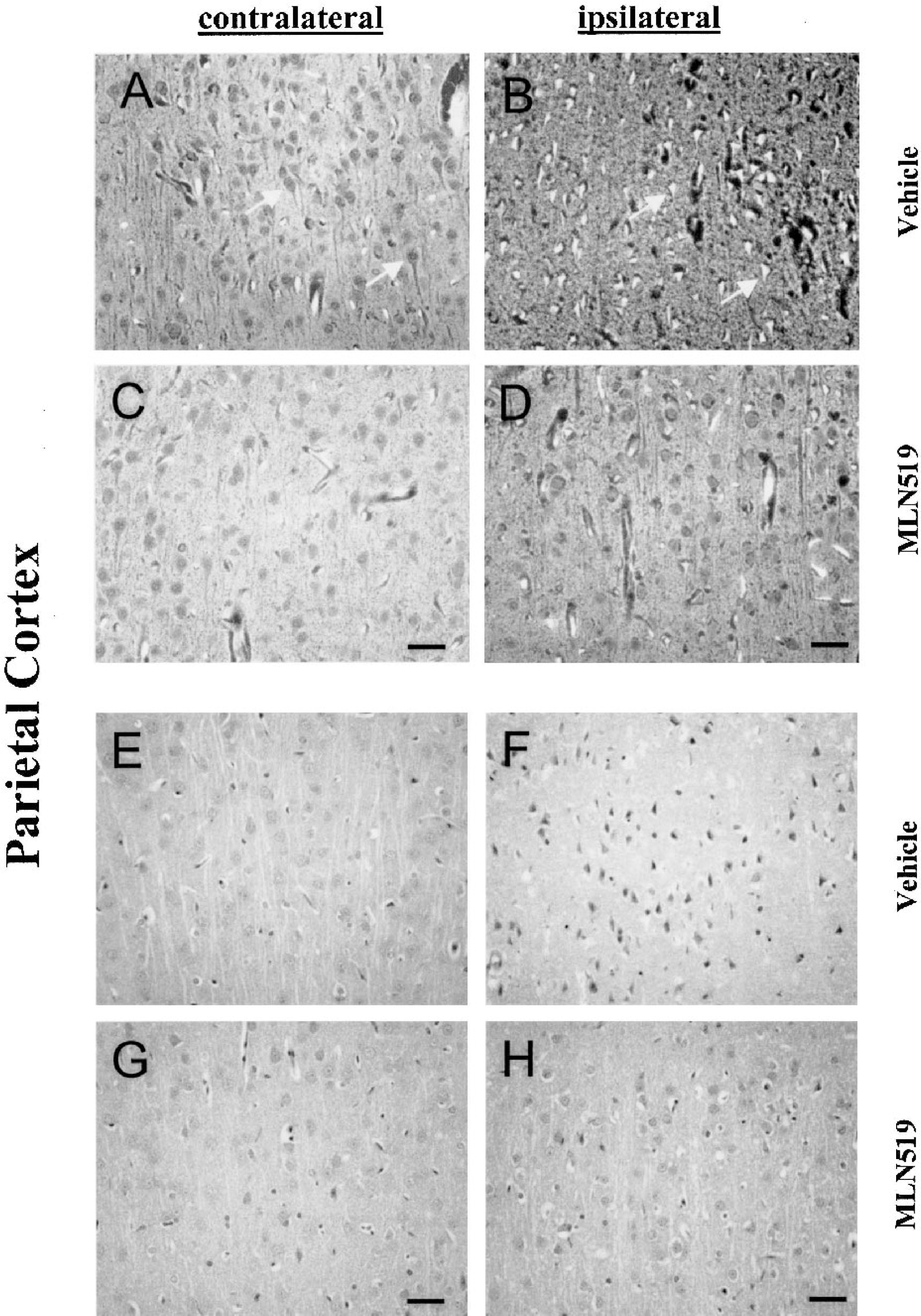
Representative parietal cortex sections stained with FluoroJade
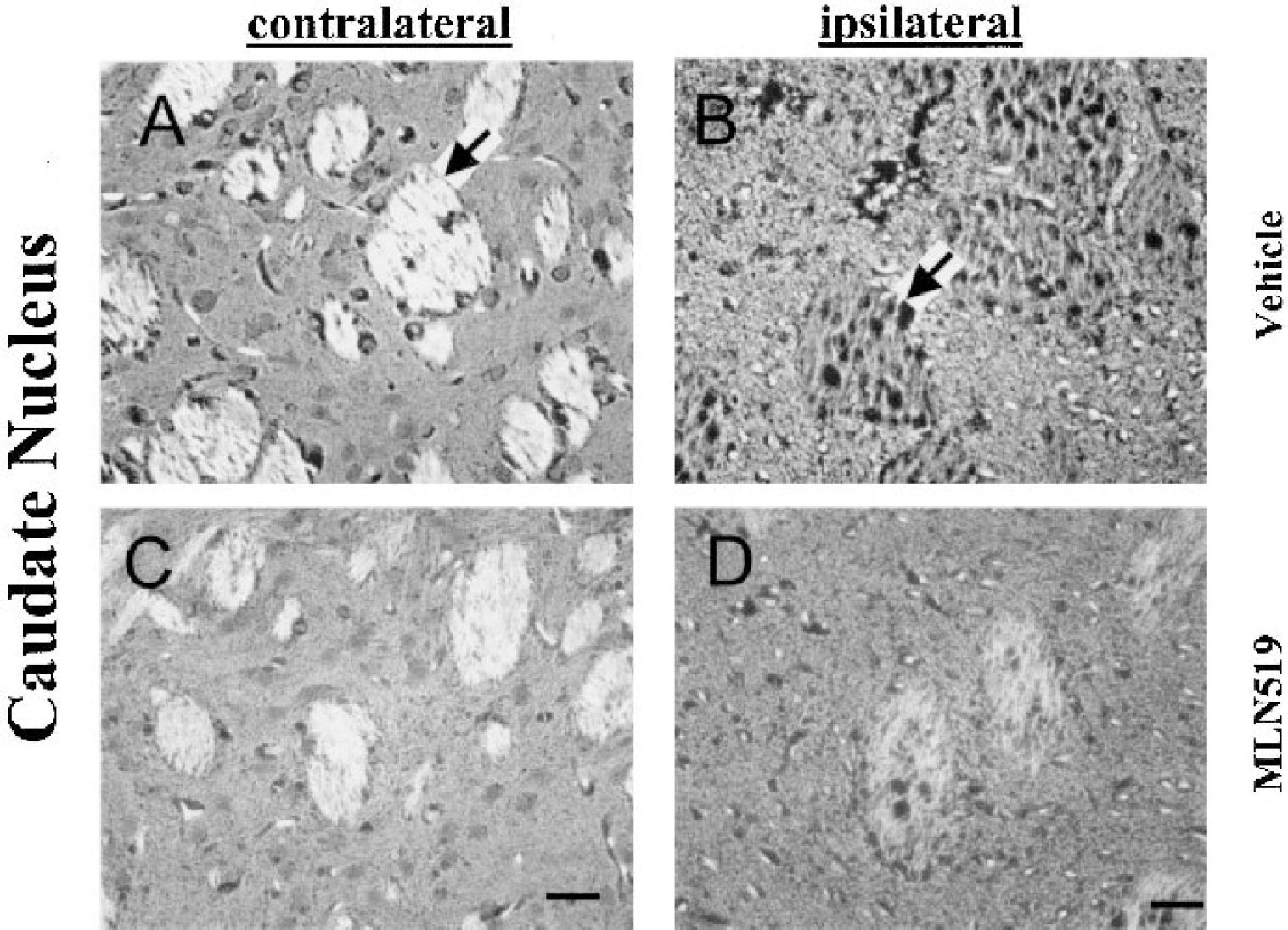
Representative FluoroJade stains of caudate nucleus sections from animals treated with vehicle (50% polypropylene glycol, intravenously [i.v.]) and MLN519 (1.0 mg/kg, i.v.), delivered 6 and 24 hours after middle cerebral artery occlusion. Normal neuronal morphology in the contralateral caudate nucleus
Similarly, normal brain morphology was present in the contralateral caudate nucleus of both vehicle- and MLN519-treated animals as indicated from FluoroJade stained sections (Figs. 3A and 3C). In the ipsilateral caudate of vehicle-treated animals severe neuronal degeneration was present along with a degeneration of axonal bundles (Fig. 3B). Widespread, although reduced, neuronal degeneration was also present in the caudate of MLN519-treated animals but with a striking maintenance of axonal bundle integrity (Fig. 2D).
Astrogliosis
Astrogliosis, as indicated by increased GFAP immunofluorescence, was notable in the ischemic brain regions. Normal or sham-treated animals exhibited little or no GFAP reactivity, similar to the contralateral hemisphere of all MCAO-injured animals (data not shown). The core of the injury contained only scattered GFAP staining due to cellular deterioration (Fig. 1), while the periinfarct region contained GFAP-positive, highly ramified astrocytes in both vehicle- and MLN519-treated animals (Figs. 4A and 4B). These large multipolar cells were strongly reactive for GFAP (Fig. 4C).
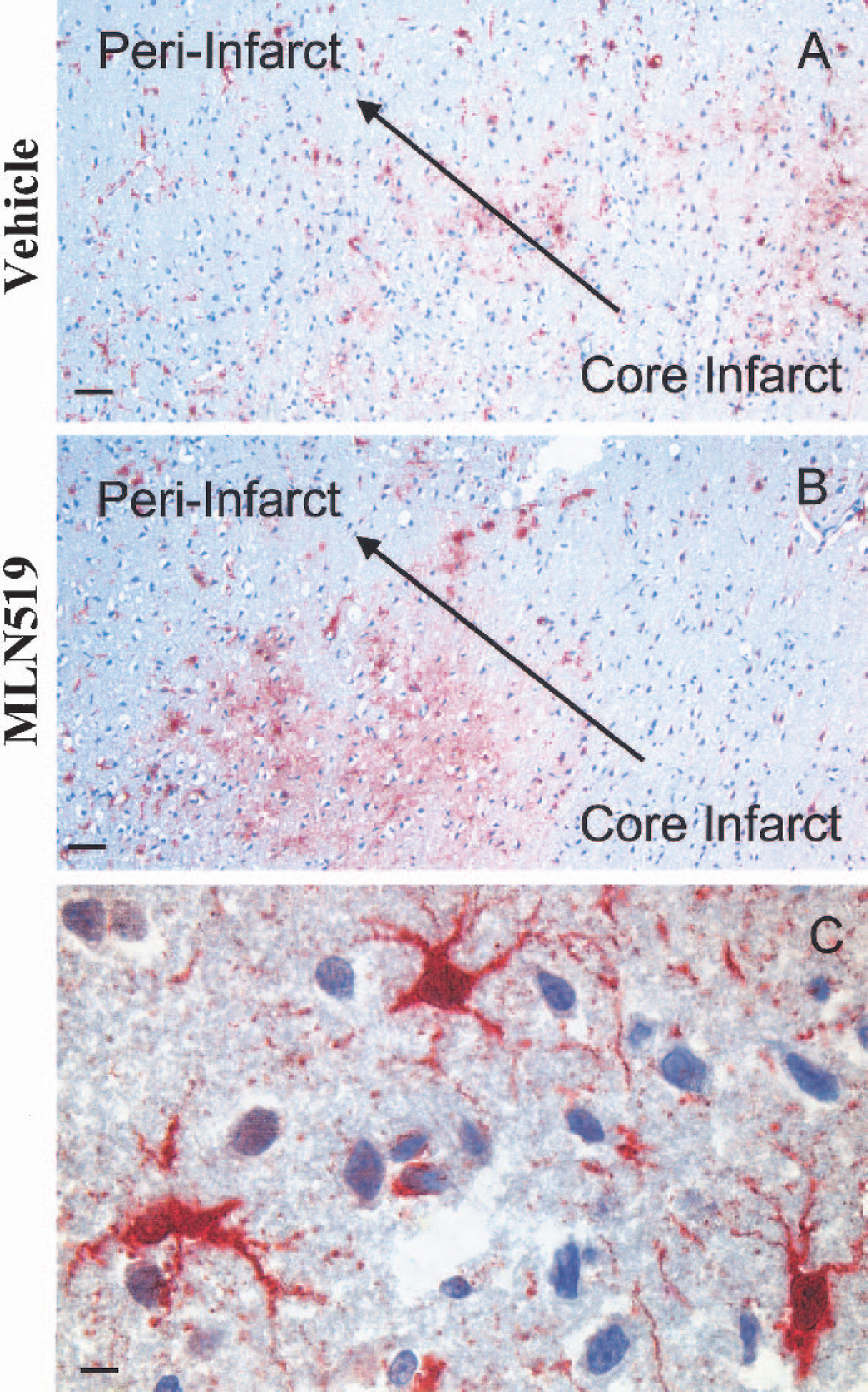
Glial fibrillary acidic protein (GFAP; brown/red = GFAP, blue = hematoxylin counterstain) immunoreactivity in the periinfarct regions of vehicle-
Glial fibrillary acidic protein reactivity was present throughout the corpus callosum in both brain hemispheres, with average cellular densities of 54 ± 14 and 43 ± 12 cells per 40x field in animals treated with vehicle and MLN519 (6 hours after MCAO treatment), respectively. GFAP-reactive cell densities in the periinfarct region of the striatum were 20 ± 5 and 15 ± 4 cells per 40x field in the vehicle- and MLN519-treated groups, respectively. Cortical regions also contained widespread GFAP staining, most notably in the periinfarct regions. GFAP staining was present throughout cortical layer 1 in 70% (7 of 10) of vehicle-treated and 50% (5 of 10) of MLN519-treated (6 hours after MCAO treatment) animals, generally in those animals exhibiting larger infarct volumes. Although MLN519-treated animals exhibited reduced gliosis as determined from GFAP-positive cell counts, the values were not statistically different from vehicle-treated animals.
Leukocyte infiltration
Infiltrating leukocytes, predominately neutrophils, were evident in the ischemic brain region of injured animals at 24 hours, as indicated in H&E slides, with only occasional detection in either normal, sham, or the contralateral hemisphere of MCAO-injured animals. Infiltrating leukocytes were not distributed evenly throughout the ischemic region but were concentrated at the borders of the lesion or around blood vessels. In most cases, larger lesions exhibiting a complete loss of tissue architecture in core regions were associated with the highest number of leukocytes. Average neutrophil counts for vehicle-treated animals ranged from 145 to 178 cells per injured hemisphere, with significant reductions of 36% and 38% after 4- or 6-hour (post-MCAO) delayed treatment with MLN519, with no significant differences measured with the 8-hour delay (Fig. 5).
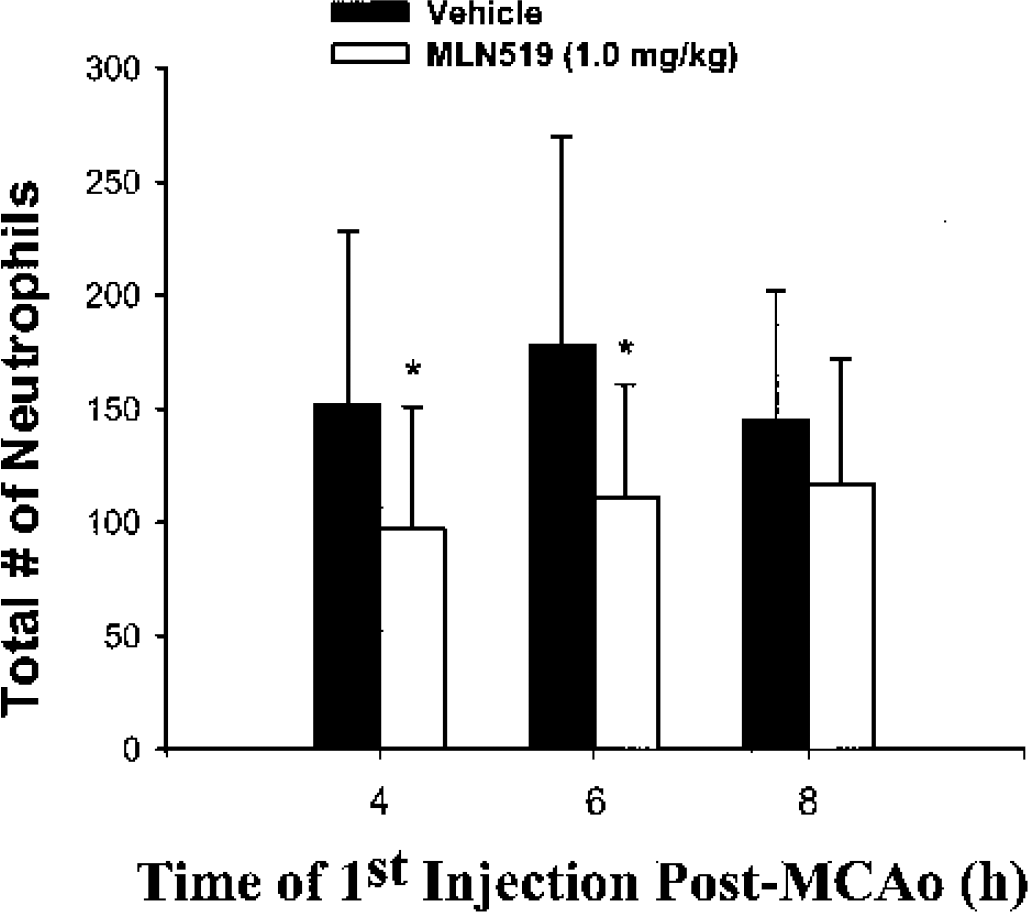
Neutrophil counts from the injured brain hemisphere at 24 hours after middle cerebral artery occlusion (MCAO) in vehicle- and MLN519-treated rats. Values are given as mean ± SD. * P < 0.05 compared with corresponding vehicle control (independent t-tests).
The presence of brain macrophages was also observed 24 hours after MCAO, although in lower numbers. Average macrophage counts ranged from 22 to 45 in the injured hemisphere, although no significant differences were measured between vehicle- and MLN519-treated rats (P > 0.05, independent t-tests).
Proteasome levels
Arterial proteasome levels remained steady throughout the course of MCAO injury, but immediately dropped by 87% to 89% 1 hour after MLN519 injection, regardless of whether the injection was given 4, 6, or 8 hours after MCAO (Fig. 6). Proteasome levels then returned to the normal, preinjury range by 24 hours (Fig. 6).
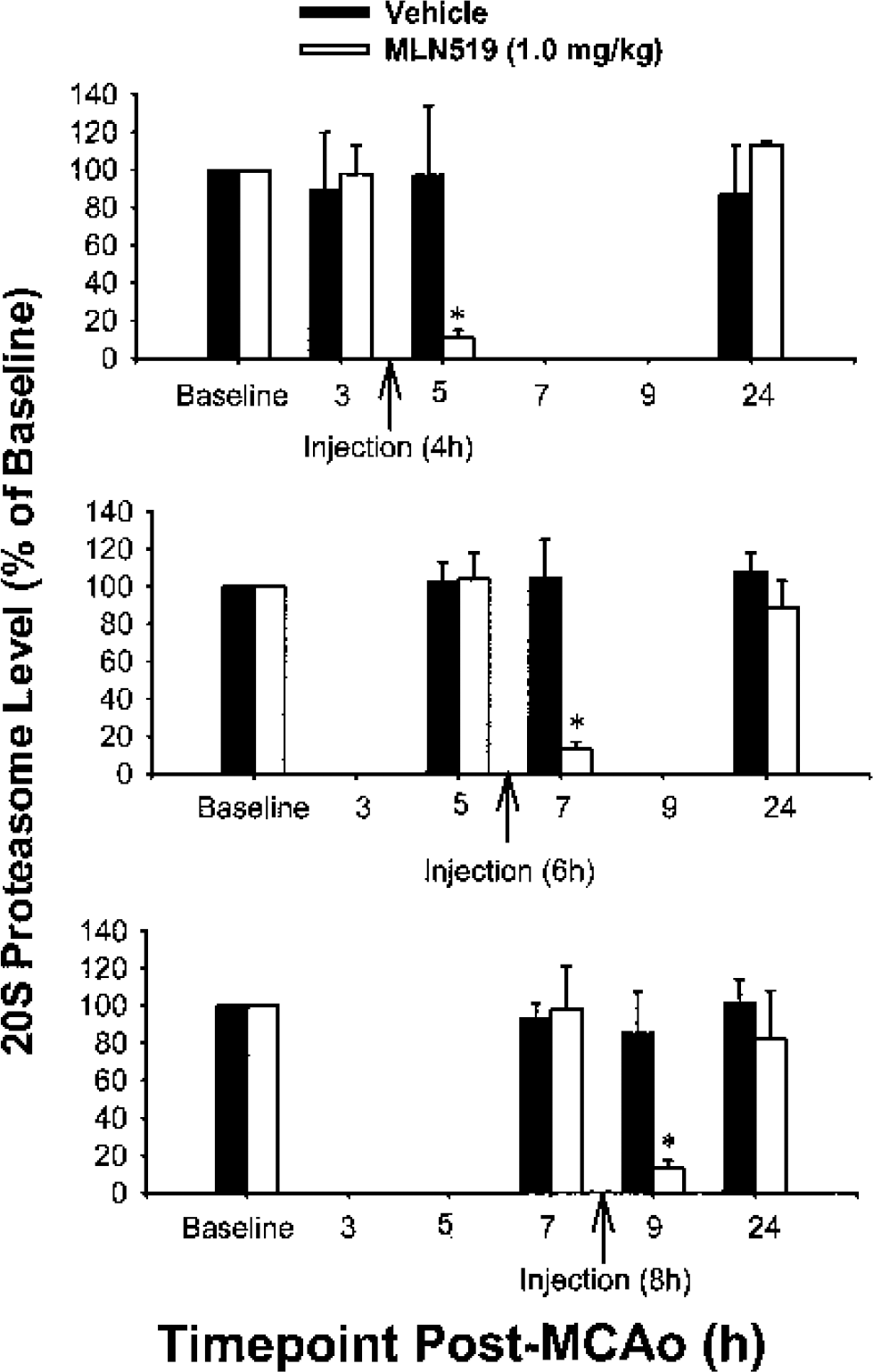
Proteasome blood levels remained steady after middle cerebral artery occlusion (MCAO) injury with significant reductions after MLN519 (1.0 mg/kg, intravenously) treatment and eventual recovery by 24 hours. Values are given as mean ± SD. * P < 0.01 compared with corresponding vehicle control (independent t-tests).
Nuclear factor-κB activation
Figure 7 demonstrates the immunoreactivity observed for activated NF-κB after 24-hour MCAO. Nuclear factor-κB immunoreactivity was barely detectable in the contralateral brain hemisphere, similar to sham or normal rat brain. Intense NF-κB staining was measured in the injured brain, including cortex and striatum coincident with regions of infarction as well as the surrounding periinfarct regions (data not shown). Table 3 compares OD measurements from three groups of cell types, which exhibited NF-κB staining. Nuclear factor-κB immunoreactivity was highest in leukocytes and endothelial cells as compared with the resident brain cells (i.e., neurons and glial cells). Although all cell types exhibited significantly less immunoreactivity after treatment with MLN519, the greatest reduction of NF-κB activation was measured in endothelial cells (47%) and leukocytes (45%).
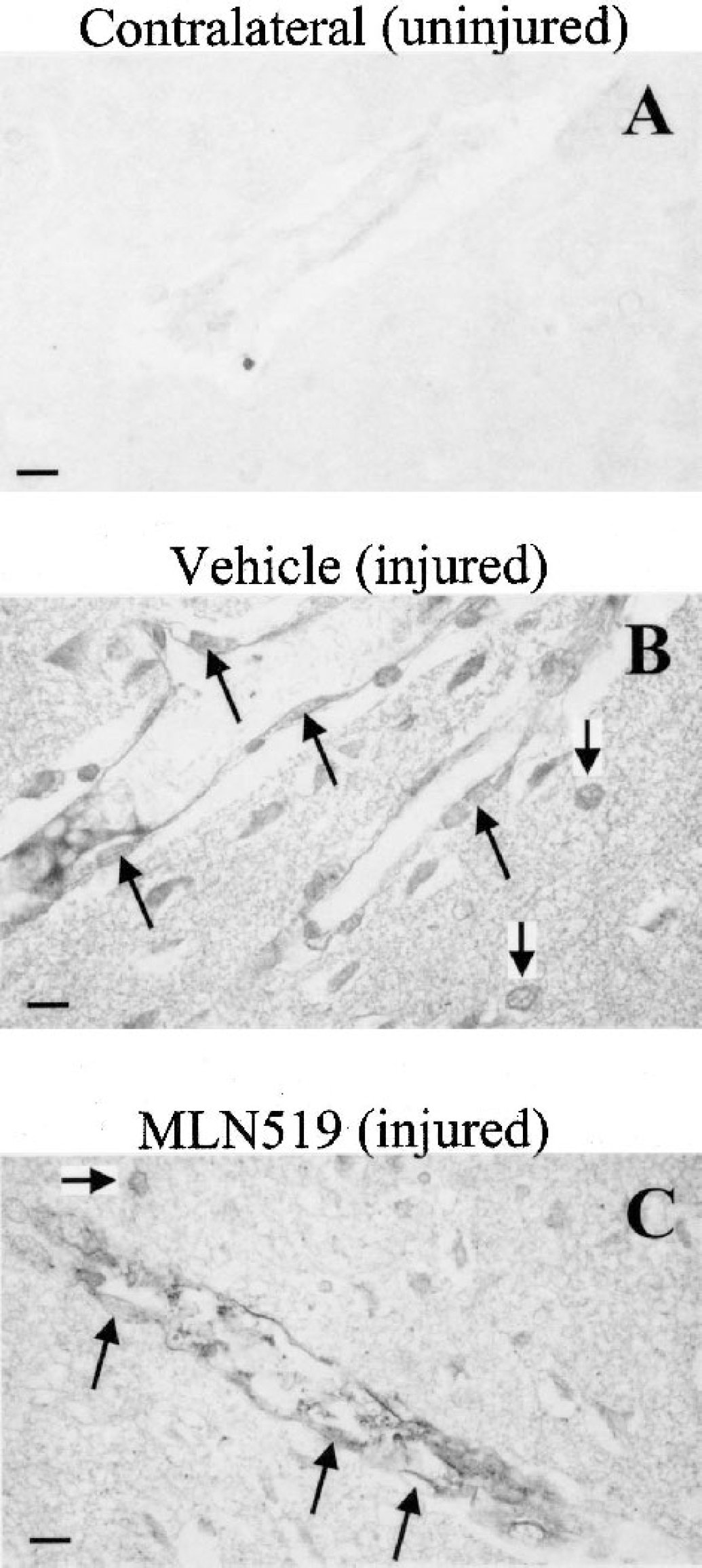
Immunoreactivity of activated nuclear factor-κB (NF-κB) from middle cerebral artery occlusion-injured rats (24 hours after injury). Diffuse staining was present in the uninjured hemisphere
Physiologic parameters
All MCAO-treated animals lost approximately 12% to 14% of their body weight over the 24-hour recovery period, regardless of treatment group, with no significant differences in body weight loss between groups. In the vehicle-treated group, MCAO caused a transient and mild hyperthermia (increase of 1.0–1.4°C), but temperatures returned to within-normal ranges (36.7–37.4°C) by 4 hours after occlusion, similar to our previous findings regarding changes in temperature that are observed with this model (Phillips et al., 2000; Tortella et al., 1999; Williams and Tortella, 2000). At all doses and time points, temperature measurements from MLN519-treated animals were not significantly different (P > 0.05, ANOVA) from the corresponding vehicle-treated animals.
Middle cerebral artery occlusion induced increases in both MABP and HR during the 0.5- to 6-hour postinjury period (Table 2), but both measures returned to within-normal preinjury levels by 24 hours. Partial O2 tension was transiently decreased 0.5 to 2 hours after injury and returned to within-normal preinjury levels by 4 hours after injury. No significant changes were measured between time points for either Pco2 or pH levels. Importantly, no significant changes were measured between vehicle- and MLN519-treated groups for any of the physiologic variables measured (Table 2, P > 0.05, ANOVA).
Time-course changes in physiologic parameters after middle cerebral artery occlusion
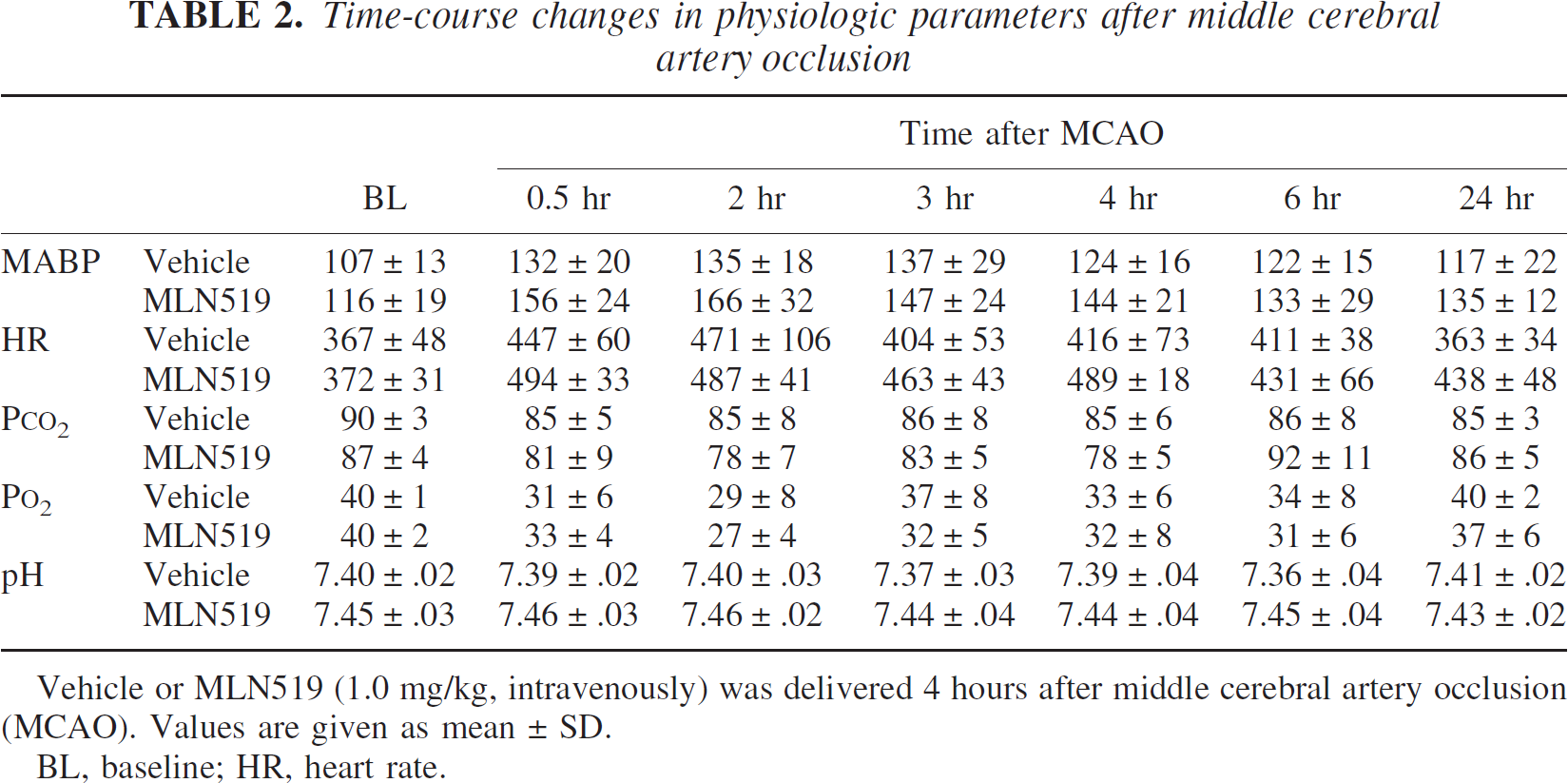
Vehicle or MLN519 (1.0 mg/kg, intravenously) was delivered 4 hours after middle cerebral artery occlusion (MCAO). Values are given as mean ± SD.
BL, baseline; HR, heart rate.
Optical density measurements from nuclear factor-κB-stained brain sections, 24 hours after middle cerebral artery occlusion
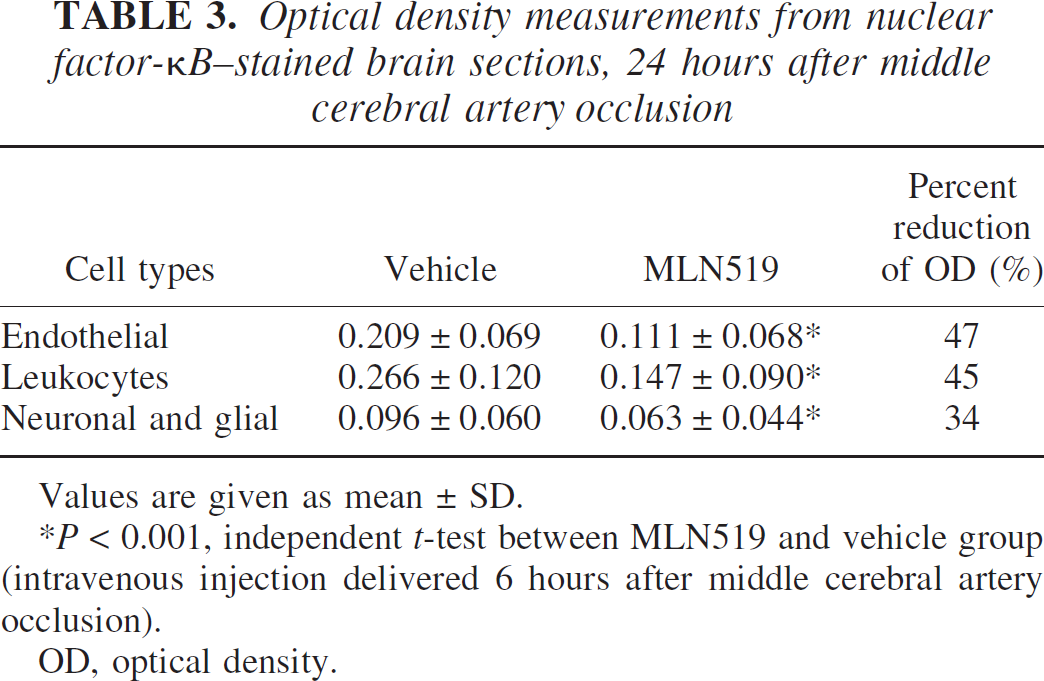
Values are given as mean ± SD.
P < 0.001, independent t-test between MLN519 and vehicle group (intravenous injection delivered 6 hours after middle cerebral artery occlusion).
OD, optical density.
DISCUSSION
Brain ischemia is a pathologic process involving a complex interaction between various perpetrators of cellular death. Loss of blood flow to the brain leads to inhibition of oxidative phosphorylation and results in a maelstrom of cytotoxic activity. At the cellular level, once ionic homeostasis is lost a host of damaging cell-signaling messengers are activated, which activate proteases, phospholipases, and free radical formation. The end result is initiation of cell death defined either by necrotic cell damage due to excitotoxic overload or by activation of delayed apoptotic cascades (Lipton, 1999; Martin et al., 1998). Initially only a small area, the core infarct, is irreversibly damaged after an ischemic attack. The surrounding tissue, known as the periinfarct region, is subject to further damage through secondary death cascades. Recent studies indicate that acute inflammation may be a principal mediator of these secondary cell death responses (Stanimirovic and Satoh, 2000), whereas other investigators suggest that infiltrating leukocytes may only be passive bystanders to the injury (Emerich et al., 2002).
To evaluate the potential in vivo neuroprotective effects of the proteasome inhibitor MLN519, we used a model of focal cerebral ischemia to produce a 2-hour temporary MCAO in the rat, followed by reperfusion of blood to the ischemic region. This model closely resembles clinical stroke, which is also due to predominately acute focal ischemic attacks (Kaufmann et al., 1999; Ringelstein et al., 1992). Reperfusion has been shown to enhance injury due to production of reactive oxygen species and increased edema (Kuroiwa et al., 1988; Lipton and Rosenberg, 1994) but also allows for restoration of metabolic activity to the compromised areas (Hallenbeck and Dutka, 1990; Halsey et al., 1991).
Critically, this model is well established and been shown to be highly sensitive to pharmacological intervention with neuroprotective drugs as a means to reduce the resulting brain infarction (Britton et al., 1997; Clemens and Panetta, 1994; Kawasaki-Yatsugi et al., 1998; Tatlisumak et al., 1998; Tortella et al., 1999; Williams et al., 2000). Previous studies with MLN519 have reported neuroprotection in this same model of MCAO with a dose of 0.1 mg/kg delivered 4 hours after MCAO (Phillips et al., 2000), as well as in combination with tissue plasminogen activator in a model of embolic stroke in rats (Zhang et al., 2001). The present study presents a comprehensive histopathologic analysis of treatment with delayed injections of MLN519 to rescue ischemic brain tissue from injury as well as improve neurologic recovery. Furthermore, we have correlated these neuroprotective effects to reductions in blood proteasome levels, neutrophil infiltration, and NF-κB immunoreactivity.
As confirmed by infarct volume analysis 24 hours after injury, a single injection of MLN519 significantly decreased cortical brain lesion size by 48% when the treatment was delayed for 6 hours after MCAO (4 hours after reperfusion). Brain regions proximal to the origin of the MCA (subcortical) were not significantly affected. Thus, periinfarct brain regions, defined as the ischemic territory most distal to the MCA, seem to be most sensitive to delayed treatments with MLN519. This neuroprotective reduction of infarction also correlated to a significant improvement in neurologic recovery. Although the infarct volume at 24 hours after injury has not fully progressed to maturation, it is >80% of the size acquired by 72 hours after injury in this model of brain injury (Phillips et al., 2000; Tortella et al., 1999; Williams et al., 2000). Beyond 72 hours, the lesion begins to cavitate as the necrotic tissue is cleared, leaving behind a connective tissue matrix (Clark et al., 1993). We have previously shown that lower doses of MLN519 are neuroprotective when evaluated at 72 hours after injury (Phillips et al., 2000), and additional studies are currently underway to evaluate the full therapeutic window and cellular response after longer recovery periods.
FluoroJade staining is a relatively new procedure that is similar to silver staining techniques and offers a definitive and sensitive measurement for neuronal degeneration (Schmued et al., 1997). In this study, FluoroJade reactivity correlated with the cellular pathology determined from sequential H&E-stained sections. The presence of neuronal degeneration throughout the ischemic lesion (FluoroJade stains) was significantly reduced after MLN519 treatment in accordance with the reduction of infarct volume (TTC and H&E stains). In conjunction with the preservation of neuronal morphology, axonal degeneration was also reduced in cortical regions of MLN519-treated animals. Importantly, preservation of axonal bundles was observed in the striatum of these same animals. This maintenance of axonal integrity may be responsible for the attenuation of the neurologic dysfunction as measured here and in other studies (Phillips et al., 2000; Zhang et al., 2001). In particular, protection of parietal neurons in the motor cortex as well as striatal axons may preserve the signaling pathways necessary for normal motor function.
Astroglial activation, as determined from GFAP staining, was present in both vehicle- and MLN519-treated animals. The highest density of activated astrocytes was seen in periinfarct regions and throughout the corpus callosum, which is typical of this type of injury (Clark et al., 1994). GFAP staining was also present throughout the core of the infarct but was diffuse, most likely owing to degenerate astrocytes activated early in the injury, but was no longer intact by 24 hours. Astrocytes play a vital role in the maintenance of normal function of neuronal activity, and may contribute to neuronal survival after ischemic injury (Louw et al., 1998). Reports have indicated that GFAP-null mice have a higher susceptibility to ischemic injury induced by MCAO or global ischemic injury (Nawashiro et al., 2000) and that ischemic neuronal injury is ameliorated by astrocyte activation (Louw et al., 1998). It has also been suggested that activated astrocytes may play a protective role after injury owing to their ability to metabolize excitatory neurotransmitters, release neurotrophic compounds, and buffer extracellular potassium in ischemic brain regions (Louw et al., 1998). In our study the periinfarct regions of both treatment groups included strong reactivity for GFAP, but, in concordance with a reduction of infarct volume after MLN519 treatment, we observed less astrocytic “debris” present throughout the core infarcted tissue.
Focal cerebral ischemia, where reperfusion injury occurs, causes an acute inflammatory response characterized by the infiltration of neutrophils followed at later time points by macrophages into the ischemic region (Clark et al., 1994). Transient MCAO induces a more rapid induction of neutrophil infiltration, as early as 1 hour after reperfusion (Clark et al., 1994; Phillips et al., 2000) as compared with a permanent occlusion of the MCA. Activated leukocytes produce several neurodegenerative mediators including the expression and release of inflammatory cytokines, formation of reactive oxygen species, and activation of arachidonic acid cascades (Stanimirovic and Satoh, 2000). Antineutrophil treatment after 1-hour temporary MCAO in rats has been shown to completely inhibit the increase in extracellular ascorbyl radical (a marker of oxygen radical presence), suggesting that neutrophils are a major source of oxygen radicals during reperfusion (Matsuo et al., 1995). In support of these findings and consistent with the reduction of infarction and neuronal degeneration, treatment with MLN519 was associated with a nearly 40% reduction of infiltrating neutrophils into the perivascular regions of the brain as evaluated 24 hours after injury.
At the nuclear level, one control mechanism promoting the inflammatory response involves the transcription factor NF-κB under control of the ubiquitin-proteasome pathway. Nuclear factor-κB is constitutively present in cells in an inactivated state bound by I-κB. Inflammatory signals, such as IL-1β and TNF-α, promote NF-κB release through the activation of I-κB kinases leading to phosphorylation of I-κB. Once phosphorylated, I-κB is a target for 20S proteasome degradation through the ubiquitin-proteasome pathway, releasing the p50/p65 heterodimer of NF-κB. Nuclear factor-κB then translocates to the nucleus and is involved in promoting the expression of a variety of inflammatory mediators including cytokines, cell adhesion molecules, and chemokines (Carroll et al., 2000). Cytokine upregulation, including formation of IL-1β and TNF-α, induces further NF-κB activation in a feed-forward and rapid upregulation of the inflammatory response. In this study we show that, after MCAO and treatment with MLN519, the measured reduction in neutrophil infiltration was directly associated with both a reduction in circulating 20S proteasome levels and activated NF-κB immunoreactivity in the vascular compartments of the injured brain. In particular, MLN519-treated animals experienced >80% reduction in blood proteasome levels and a 34% to 47% reduction in NF-κB immunoreactivity, depending on the cell type, in necrotic brain regions. This evidence supports the role of proteasome inhibition as an active mediator of inflammatory cell infiltration into ischemic brain tissue. Initial studies with MLN519, however, indicated that poor penetration into ischemic brain regions was achieved when the drug was delivered intravenously (Phillips et al., 2000). This would indicate that the effects of MLN519 treatment are most likely nonneuronal and that the major site of action of MLN519 may, in fact, be on endothelial cells to block the infiltration of leukocytes into the brain.
Sustained increases in NF-κB activation have been shown to occur in a variety of in vivo central nervous system injury models including spinal cord injury, traumatic brain injury, global ischemia, and transient focal ischemia (Berti et al., 2002a; Bethea et al., 1998; Clemens et al., 1998; Nonaka et al., 1999; Schneider et al., 1999; Stephenson et al., 2000). Interestingly, the activation of NF-κB can have dual effects on neuronal survival after injury. For example, the induced upregulation of inflammatory cytokines can initiate an inflammatory response linked to increased necrotic demise of ischemic tissue. Evidence for this is supported by several studies describing effects of reduced NF-κB activation mediating neuronal survival or antiinflammatory mechanisms (Clemens et al., 1998; Lockyer et al., 1998). Furthermore, our results, as well as other data (Phillips et al., 2000; Zhang et al., 2001), suggest that delayed postinjury treatment with the proteasome inhibitor MLN519 reduces brain infarction and increases neuronal viability after temporary focal ischemia in the rat. Alternatively, NF-κB activation has been shown to upregulate anti-apoptotic genes including manganese superoxide dismutase and Bcl-2 (Toyoda et al., 1997), whereas others (Hill et al., 2001) have reported that complete inhibition of NF-κB (as evaluated using electrophoretic mobility shift assay) after direct intracranial injection of the NF-κB inhibitor diethyldithiocarbamate increased cell death induced by transient MCAO in rats. Thus, complete inactivation of NF-κB seems to be neurotoxic after injury, but moderate inhibition or inhibition targeted to endothelial cells and leukocytes likely interferes with the inflammatory diapedesis of leukocytes into the brain and is neuroprotective. The major neuroprotective role of MLN519 treatment, therefore, may be to reduce the adhesion molecule-initiated infiltration of inflammatory blood cells into ischemic brain tissue (Berti et al., 2002a; Lockyer et al., 1998). Currently, we are evaluating the effects of MLN519 treatment to alter the expression of a variety of inflammatory genes under the control of NF-κB (Berti et al., 2002b). Furthermore, these results would suggest that neurodegeneration associated with NF-κB activation is a delayed response allowing an extended window of intervention with agents such as MLN519 to interfere with the development of ischemic brain injury.
In conclusion, MLN519 treatment provided an excellent therapeutic window for the treatment of ischemia-reperfusion brain injury in rats. The neuroprotective effects included a reduction in core infarct volume, increased neuronal survival and axonal viability, as well as an improved neurologic recovery after a 6-hour (post-MCAO) delayed injection of MLN519 after injury. This coincided with a drop in blood proteasome levels and a reduction in both neutrophil infiltration and activated NF-κB immunoreactivity. No significant changes in physiologic parameters were observed between vehicle-and MLN519-treated rats. Currently available management protocols for clinical stroke may therefore benefit from antiinflammatory treatment with compounds such as MLN519 that target the neuropathology associated with reperfusion injury.