
Abstract
The cytokine interleukin-1 (IL-1) has been implicated in the exacerbation of ischemic damage in the brains of rodents. This study has ascertained the cellular localization and chronologic and topographic distribution of pro/mature interleukin-1β (IL-1β) protein 0.5, 1, 2, 6, 24, and 48 hours after ischemia by subjecting rats to permanent unilateral occlusion of the middle cerebral artery. Interleukin-1β was localized immunocytochemically in vibratome sections of perfusion-fixed brains. The cells that expressed IL-1β had the morphologic features of microglia and macrophages. Interleukin-1β was first detected 1 hour after occlusion in ipsilateral meningeal macrophage-like cells. By 6 hours, pro/mature IL-1β-immunoreactive (IL-1βir) putative microglia were present in the ischemic cerebral cortex, corpus callosum, caudoputamen, and surrounding tissue. By 24 and 48 hours after ischemia, the number and spread of IL-1βir cells increased greatly, including those resembling activated microglia and macrophages, as the core of the infarct became infiltrated. Interleukin-1βir cells also were present in apparently undamaged tissue, adjacent to the lesion ipsilaterally, and contralaterally in the cerebral cortex, dorsal corpus callosum, dorsal caudoputamen, and hippocampus. These results support the functional role of IL-1 in ischemic brain damage and reveal a distinct temporal and spatial expression of IL-1β protein in cells believed to be microglia and macrophages.
There is mounting evidence that the cytokine interleukin-1 (IL-1) is involved in the pathogenesis of neuronal loss caused by an ischemic insult to the brain. It has been shown that interleukin-1β (IL-1β) messenger ribonucleic acid (mRNA) is produced rapidly in rat brain after experimental ischemia (Buttini et al., 1994; Liu et al., 1993; Minami et al., 1992). In addition, assays on tissues from the cerebral cortex of the rat reveal increased levels of bioactive IL-1β protein after head trauma (Taupin et al., 1993) and raised levels of IL-1β, measured by immunoassay, induced by occlusion of the middle cerebral artery (MCAO) (Iannotti et al., 1993). Besides the induction of IL-1β mRNA and protein after brain injury, administration of recombinant IL-1β produces marked exacerbation of ischemic brain damage in the rat (Loddick and Rothwell, 1996; Yamasaki et al., 1995). Early involvement of IL-1 in the neurodegenerative process has been substantiated by the effects of experimental administration of its naturally occurring receptor antagonist. Thus, either intracerebroventricular (Relton and Rothwell, 1992; Loddick and Rothwell, 1996) or peripheral (Garcia et al., 1995b; Relton et al., 1996) injection of recombinant human IL-1 receptor antagonist (rhIL-1ra) markedly diminishes the size of the infarct, edema, and the neurologic deficit caused by permanent MCAO in the rat. Consequently, the number of necrotic neurons within the territory of the MCAO is reduced significantly in rats treated with rhIL-1ra (Garcia et al., 1995b). Intracerebroventricular injection of rhIL-1ra also has been shown to decrease the volume of the infarct and thus to inhibit neuronal death caused by fluid percussion injury to the rat brain (Toulmond and Rothwell, 1995). Using a different approach, Betz and others (1995) attenuated the infarct volume after MCAO by adenoviral-induced over-expression of IL-1ra in the rat brain. Furthermore, inhibition of IL-1β activity by central administration of an IL-1β neutralizing antibody after transient MCAO (Yamasaki et al., 1995), or injection of interleukin-1β converting enzyme (ICE) inhibitor after permanent MCAO (Loddick et al., 1996), significantly reduces ischemic damage in the rat.
These data strongly implicate IL-1β in ischemic brain damage. However, it is not known how the chronologic and topographic distribution of IL-1β protein relates to the morphologic changes that take place in cells as the infarct evolves. Nor has it been ascertained which cells in the brain produce endogenous IL-1β protein after ischemia. Evidence from in vitro studies suggests that microglia and macrophages are a major source of IL-1 protein (Giulian et al., 1986; Hetier et al., 1988), although IL-1 expression also has been detected in astrocytes (Fontana et al., 1982; Nieto-Sampedro and Berman, 1987). Interleukin-1β-immunoreactive neurons have been reported in the hypothalamus and hippocampus of normal human and rat brain (Breder et al., 1988; Lechan et al., 1990; Van Dam et al., 1992) and also in the hippocampus of perinatal rats after excitotoxic damage (Hagan et al., 1996). In contrast, in experimental allergic encephalomyelitis (Bauer et al., 1993) and after peripheral administration of endotoxin to rats (Van Dam et al., 1992, 1995), microglia and macrophages, but not neurons, expressed immunoreactive IL-1β (IL-1βir).
The current study relates the temporal and topographic distribution of IL-1β expression to that of ischemic neuronal damage using permanent MCAO in the rat as the ischemic insult. Cellular IL-1β expression was assessed by immunocytochemical study at 0.5, 1, 2, 6, 24, and 48 hours after permanent MCAO.
MATERIALS AND METHODS
Surgical procedure
Permanent, unilateral occlusion of the middle cerebral artery (MCA) was performed on male Sprague Dawley rats weighing 230 to 270 g (Charles River UK, Ltd., Margate, U.K.) according to a modification of the method of Tamura and colleagues (1981) as described by Loddick and Rothwell (1996). The rats were anesthetized by inhalation of 4% halothane in oxygen and maintained by 2% to 3% halothane during surgery. After exposure of the subtemporal fossa, craniotomy was performed using a dental drill, the dura was pierced, and the left MCA exposed and occluded, proximally, below the lenticulostriate branch and at the level of the inferior cerebral vein, using a bipolar coagulator (Aesculap, Sheffield, U.K.). The artery then was cut between these two points to prevent recanalization. For sham operations, the MCA was exposed in the same way but not occluded, and a small amount of tissue was cauterized to mimic physical damage caused by artery occlusion. After surgery, soft tissues were replaced and the wound stitched. Animals were maintained on a heated mat until they regained consciousness.
Tissue preparation
Experimental and sham-operated animals were killed 0.5, 1, 2, 6, 24, and 48 hours after surgery. At the 0.5-, 1-, and 2-hour time points, three experimental and two sham rats were used, whereas at 6, 24, and 48 hours, four experimental and two sham rats were used. After anesthesia by administration of intraperitoneal sodium pentobarbitol, the rats were perfused transcardially with phosphate-buffered saline (PBS; 0.01 mol/L, pH 7.4) followed by 4% formaldehyde in PBS. The brains were left intact inside the skull for 24 hours at 4°C to reduce the possibility of morphologic damage, then were removed and washed several times in PBS and then sectioned coronally at 80 μm on a vibratome (Series 1000, The General Scientific Co., Ltd, Redhill, U.K.). Sections were collected in PBS from the infarcted area or equivalent in shams (i.e., from the level of the lateral septal nuclei and fundus striati through to the temporal cortex and the ventromedial hypothalamic nucleus) (Bregma 1.7 mm to Bregma −3.6 mm; Paxinos and Watson, 1986).
Immunocytochemistry
Vibratome sections of perfusion-fixed brains were used for this study because they produce the best combination of good morphologic features and immunostaining. Such tissue has a better morphologic appearance than either frozen or paraffin wax-embedded material. Furthermore, immunostaining by free-floating is a more sensitive method than staining mounted sections because more of the antigen is accessible to the antibody.
The IL-1βir cells were detected using an affinity purified polyclonal antibody raised against recombinant rat IL-1β in sheep (S328B4, dilution 1:750, National Institute for Biological Standards and Control [NIBSC], Potters Bar, U.K.), which recognizes both the inactive pro and active mature forms of IL-1β (S. Toulmond and S. Poole, unpublished data). To help confirm the identification of the IL-1βir cells, a monoclonal mouse anti-rat antibody specific for ramified and activated microglia as well as macrophages (OX-42, dilution 1:500, MCA275G, Serotec, Oxford, U.K.), or one specific for macrophages, including phagocytic microglia (ED-1, dilution 1:500, MCA341, Serotec, Oxford, UK) were used.
Serial sections were divided rostrocaudally into four sampling areas. Two sections from each area were labeled with the antibodies as follows. The sections were stained free-floating using an avidin-biotin-peroxidase method. All primary and biotinylated secondary antibodies were diluted with 0.05 mol/L TRIS buffered saline (pH 7.6) containing 0.1% bovine serum albumin (TBS/BSA). First, endogenous peroxidase activity was blocked by treating the sections with 0.3% hydrogen peroxide (vol/vol) in 0.05 mol/L TBS for 30 minutes, followed by normal goat or donkey serum diluted 1:5 in TBS/BSA. After overnight incubation in the primary antibodies at 4°C, sections were incubated with the appropriate biotinylated secondary antibodies (dilution 1:2000 to 1:3000) followed by avidin-peroxidase (2.5 μg/mL), both for 60 minutes at room temperature. The immunoreactive product was visualized with 0.025% (wt/vol) 3,3′-diaminobenzidine tetrahydrochloride dihydrate containing 0.015% (vol/vol) hydrogen peroxide. One of each pair of immunostained sections was counterstained with toluidine blue to aid evaluation of the ischemic damage.
To confirm definitively the identification of the IL-1β immunopositive cells, an additional brain was perfusion-fixed as described earlier, 24 hours after MCAO, and embedded in paraffin wax. Alternate consecutive sections (7 μm) were stained with the IL-1β antibody (dilution 1:200) or with the biotinylated lectin Griffonia simplicifolia isoform B4 (GSA I-B4, dilution 10 μg/mL; L-2104, Sigma Chemical Co., U.K.), a specific marker for microglia, and macrophages. An avidin-peroxidase protocol similar to that described earlier was used. Also, double labeling with the IL-1β antibody and GSA I-B4 was performed on paraffin wax sections. 3,3′-diaminobenzidine tetrahydrochloride dihydrate was used to visualize the IL-1β immunostaining, and a peroxidase substrate SG kit (SK-4700, Vector Laboratories, Peterborough, U.K.), the GSA I-B4 positive staining.
Controls were performed by replacing the primary or secondary antisera with TBS/BSA and by preabsorption of the IL-1β antibody with recombinant rat IL-1β protein at a range of concentrations from 0.1 nmol/L to 1.0 mmol/L.
Analysis
For each brain, the area of ischemic damage was outlined on maps of the four coronal sampling areas throughout the lesion by microscopic examination. The topography of the IL-1βir microglia was located on the sections and then marked onto the brain maps. The results are represented pictorially in Fig. 1, in which each brain map shows the pattern of IL-1β staining averaged out for the group of rats at each time point after MCAO. The IL-1βir macrophages were not included, since they were too numerous to resolve, especially 48 hours after MCAO. It was not the aim of the study to formally quantify the number of IL-1βir cells, since thick vibratome sections are not suitable for that purpose. In such sections, the fragile infarcted tissue tends to fragment during sectioning, resulting in the loss of small, variable amounts of tissue. This makes it difficult to systematically sample the sections in exactly the same topographic regions at each time point. Since the tissue in the infarct is not homogeneous, variations in section thickness can occur that dictate the number of cells present. In addition, changes in the size and shape of the cells with time require the use of the optical dissector counting method. This technique would be suitable only if the antibody penetrated the whole of the section evenly.
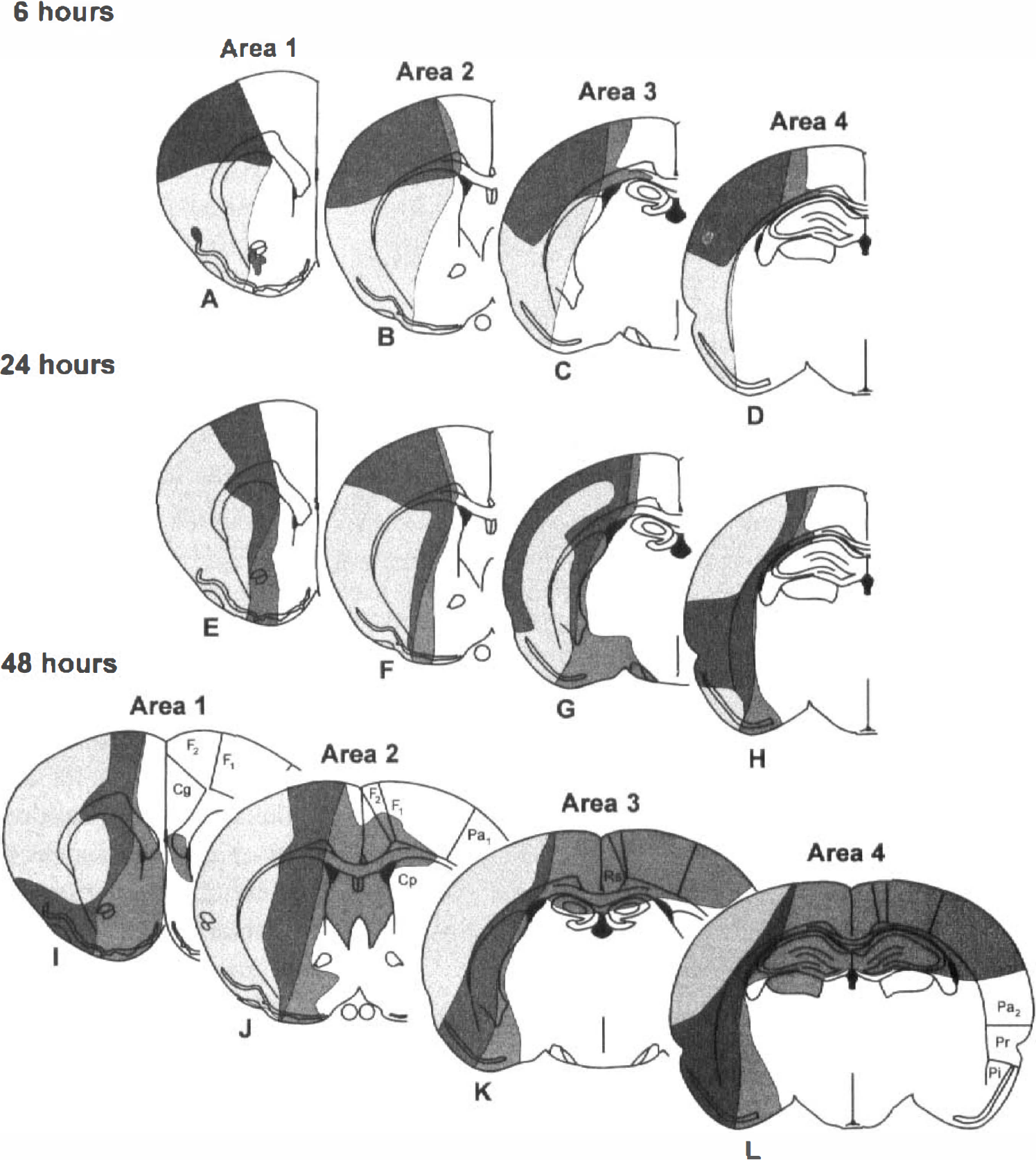
Schematic representation of the topography of ischemic damage (light shaded area) and pro/mature interleukin-1β (IL-1β)-immunopositive putative microglia (dark shaded area) at four coronal levels and various times after middle cerebral artery occlusion (MCAO):
RESULTS
Gross effects of middle cerebral artery occlusion
The ischemic zone in the MCAO rats was consistently identified from 6 hours onward in the cortex and lateral caudoputamen of the left cerebral hemisphere as a distinct pale-stained area (Fig. 1). The topographic extent of the ischemic damage was consistent among animals at the 6-, 24-, and 48-hour time points. Earlier damage (0.5, 1, and 2 hours) was more scattered and is not represented diagrammatically. No such ischemic zone was seen in the sham-operated animals. Although no specific behavioral analysis was performed, it was observed that all rats subjected to MCAO manifested typical behavioral effects such as splaying of the contralateral extremities and circling. Some animals also exhibited “barrel rolling” (seizure activity). Core body temperatures have been observed previously to rise slightly (0.5°C) and transiently (up to 4 hours) after surgery, but there was no significant difference between the temperature of sham-operated and MCAO rats up to 24 hours after surgery (Loddick, 1996).
Identification of pro/mature interleukin-1β-immunopositive cells
Immunolabeling with the IL-1β antibody was abolished by preabsorption with recombinant rat IL-1β protein at a concentration of 0.1 μmol/L (Fig. 2).
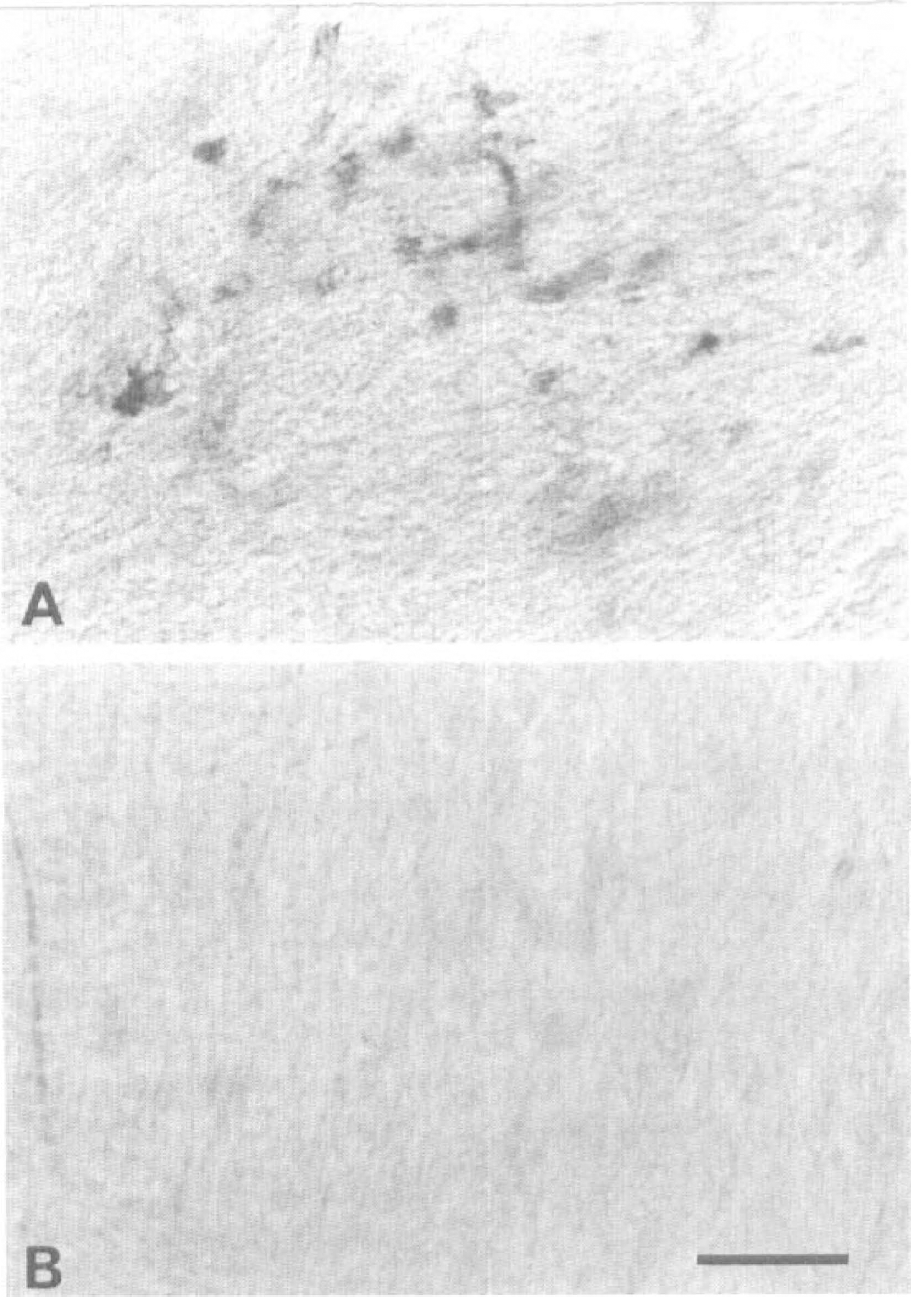
Preabsorption of the sheep anti-rat IL-1β antibody with recombinant rat IL-1β protein in vibratome sections (80 μm) 24 hours after MCAO.
Consecutive paraffin wax sections labeled with IL-1β antibody or biotinylated GSA I-B4, a specific marker for microglia and macrophages, showed that by using blood vessels as landmarks, equivalent areas containing GSA I-B4-positive microglia/macrophages coincided in paired sections with IL-1βir cells (Fig. 3A and B). Double labeling also indicated that the IL-1βir cells had morphologic features consistent with those of microglia and macrophages (data not shown).
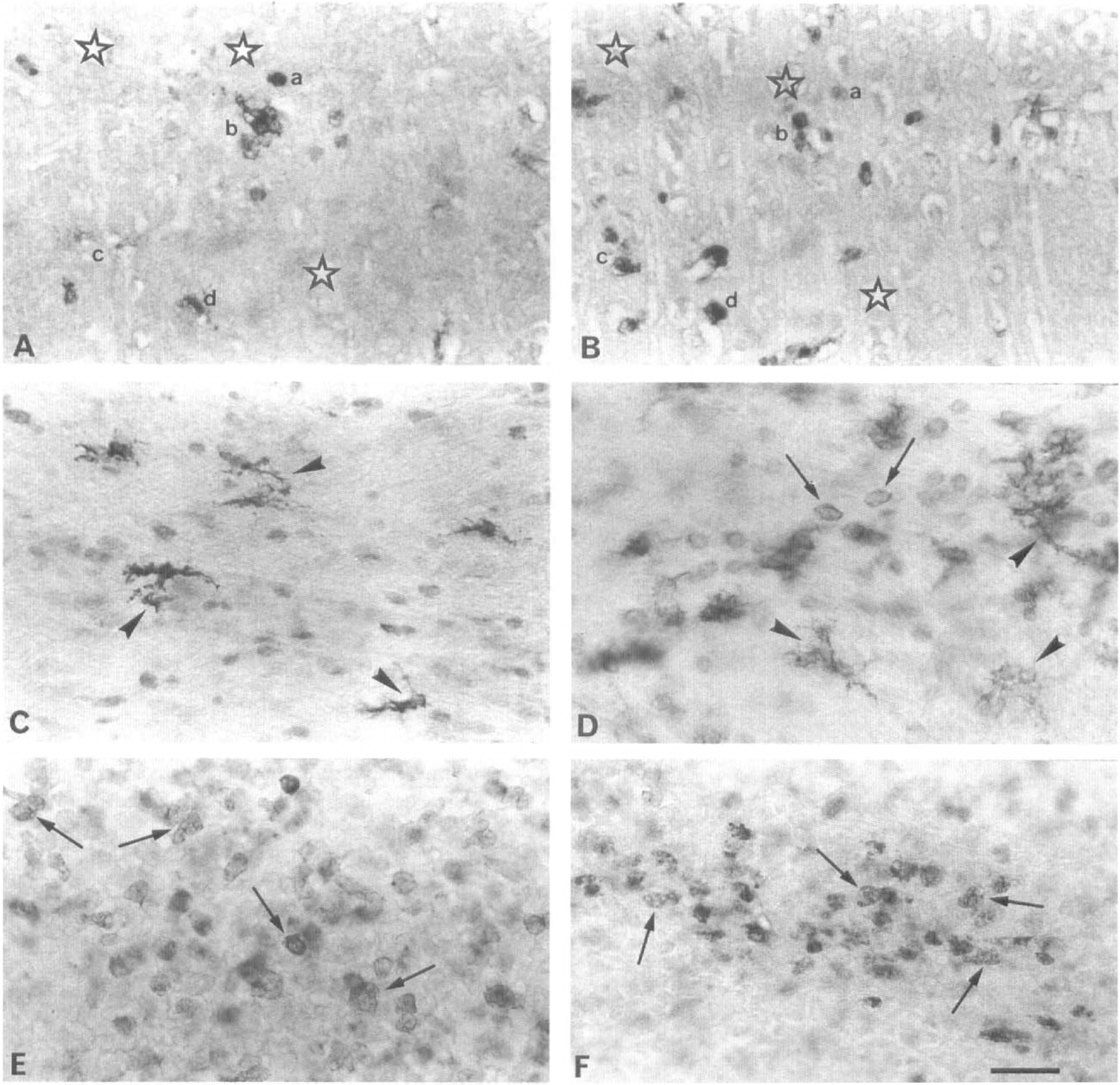
Labeling of consecutive paraffin wax sections (7 μm) from the ipsilateral frontal cortex 24 hours after MCAO, with the antibody to IL-1β
In addition, immunolabeling with OX-42, ED-1, and the antibody to IL-1β revealed that the IL-1βir cells, which had small cell bodies and ramified thin processes, resembled microglia (compare Fig. 3C and D), as well as macrophages within the infiltrated core of the lesion (compare Fig. 3E and F) and in the meninges (compare Fig. 4A through C). No IL-1βir neurons were observed in MCAO or sham-operated rats.
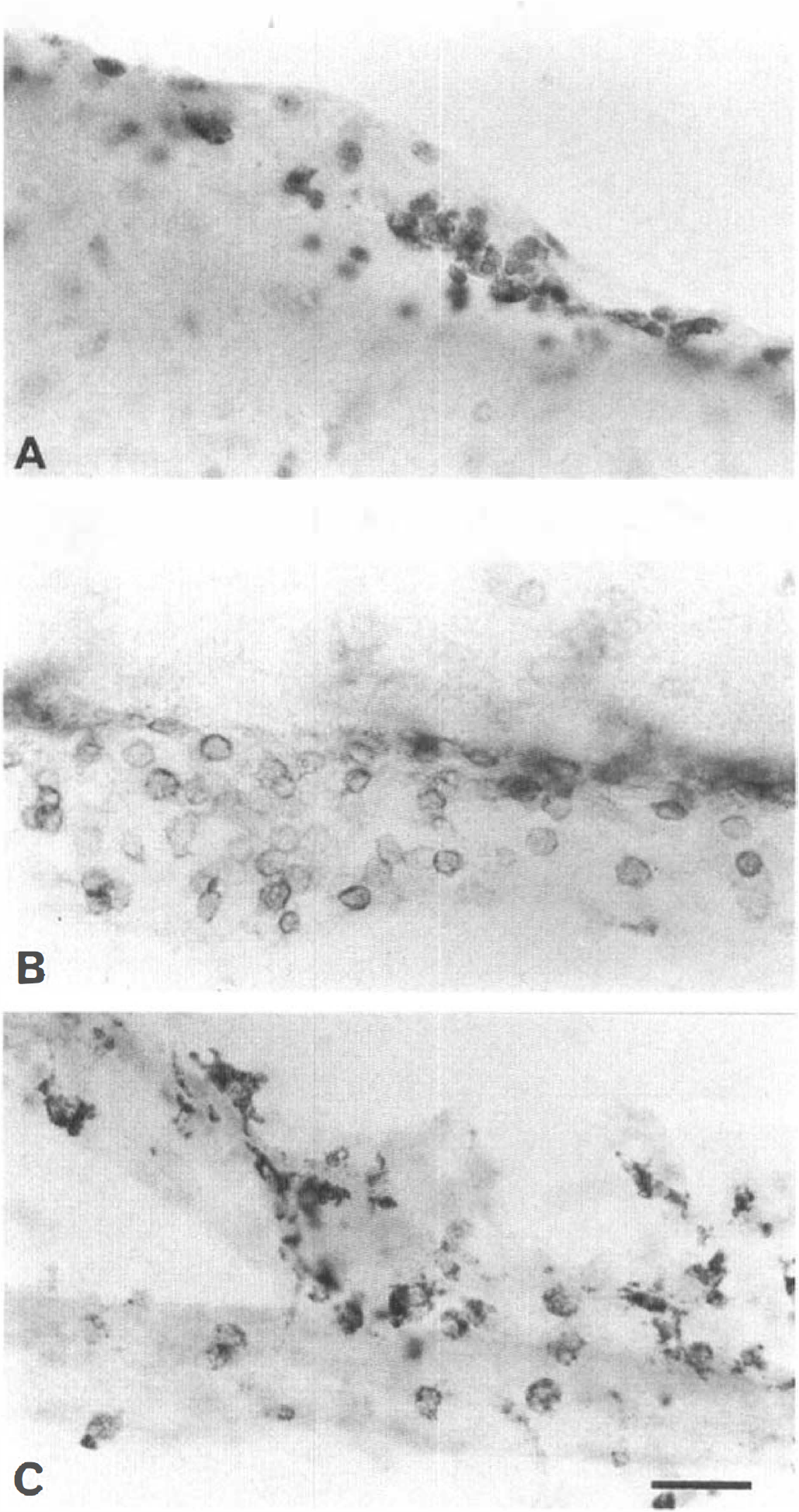
Comparison of cortical meningeal cells labeled with pro/mature interleukin-1β (IL-1β) antibody and specific markers for macrophages (OX-42, ED-1).
Ischemic damage 0.5, 1, 2, and 6 hours after middle cerebral artery occlusion
Sham-operated rats showed some dark, slightly shrunken neurons, scattered sparsely in the cerebral cortex, but no focal cell damage was present in the territory of the MCA. The topographic distribution of ischemic cell damage was similar at 0.5 and 1 hour after MCAO and was restricted to the ipsilateral hemisphere. The damage was located rostrally (Bregma 1.7 mm and Bregma 0.2 mm; Paxinos and Watson, 1986) in the insular and lateral piriform cortex. By 30 minutes after ischemia, slightly shrunken, angular neurons surrounded by a space or vacuoles were observed in the outer insular and piriform cortex, but no damaged neurons were apparent caudally (Bregma −1.8 mm and Bregma −3.3 mm; Paxinos and Watson, 1986). By 1 hour, more shrunken neurons were apparent, some with vacuolated cytoplasm, throughout the insular and lateral piriform cortex and in the adjacent parietal cortex. The ischemic caudoputamen was enlarged and contained slightly condensed neurons dorsolaterally. The total area of damage had spread caudally as far as Bregma −1.8 mm.
Two hours after MCAO, the total area of ischemic damage had increased greatly to encompass most of the sampling area rostrocaudally (Bregma 1.7 to −3.3 mm), although fewer neurons were affected toward the caudal part of the infarct. Shrunken neurons surrounded by vacuolated neuropil were seen in the frontal, parietal, insular, perirhinal, and lateral piriform cortex, and also in the dorsolateral caudoputamen. The worst affected neurons were those in the upper layers of the parietal cortex and the insular, perirhinal, and lateral piriform cortex.
By 6 hours after MCAO, the infarcted area had become more obvious by the presence of many triangular, shrunken neurons with large perineuronal spaces or cytoplasmic vacuoles throughout the cortex. The ischemic damage in the caudoputamen extended more medially, most of the neurons appearing condensed. Neutrophils were observed in the blood vessels in the ipsilateral outer insular/piriform cortex.
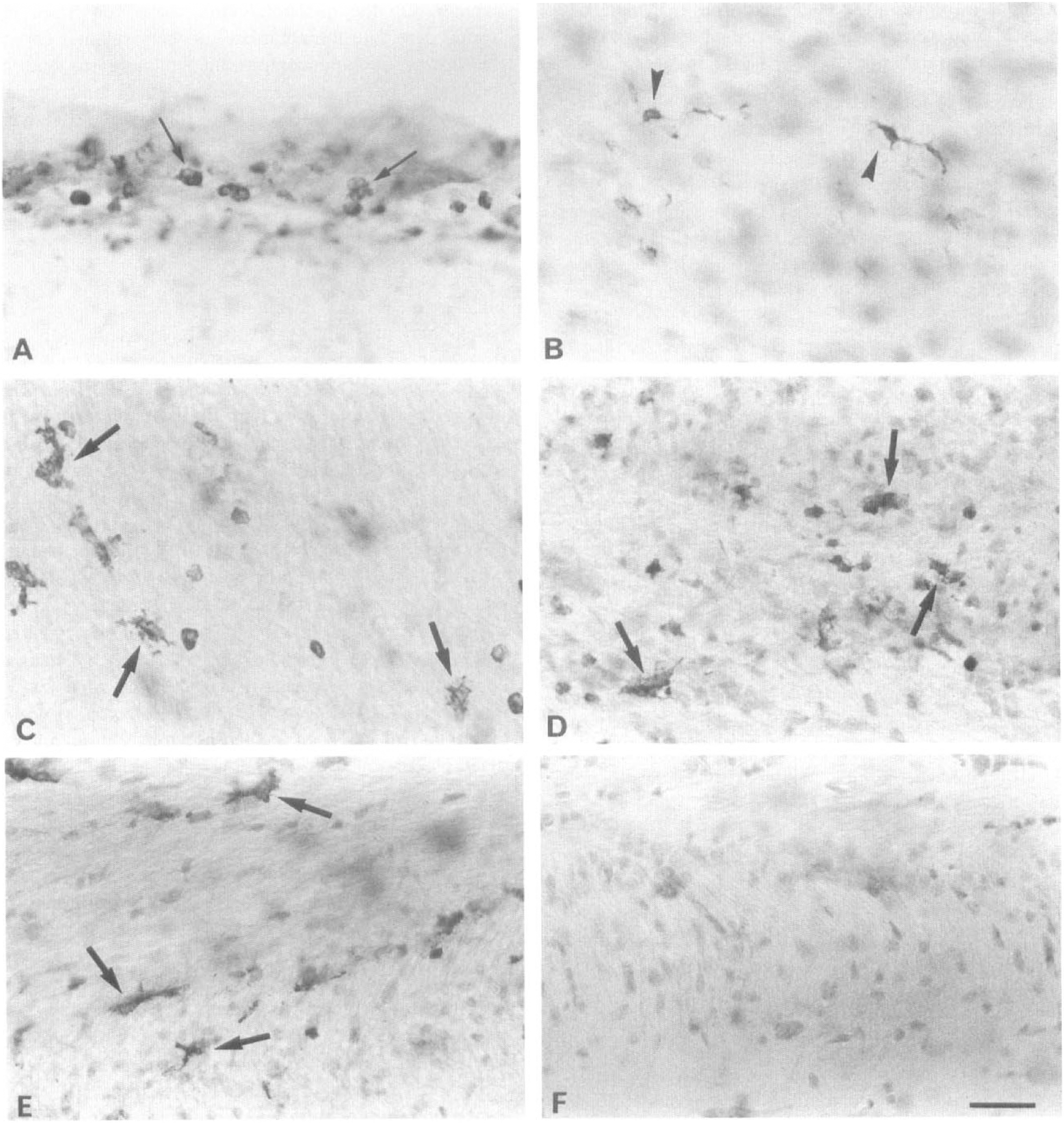
Interleukin-1β-immunoreactive cells 0.5, 1, 2, and 6 hours after middle cerebral artery occlusion
No IL-1βir cells were detected in any of the sham-operated rats, or in those killed 0.5, 1, or 2 hours after MCAO. However, immunopositive macrophage-like cells were seen in the ipsilateral meninges above the frontal and parietal cortex in rats killed 1 or 2 hours after MCAO (Fig. 5A), but not at 0.5 hours after MCAO.
Pro/mature interleukin-1β (IL-β)-immunopositive cells in vibratome sections (80 μm) at various times after middle cerebral artery occlusion (MCAO). Sections are counterstained with toluidine blue.
Scattered IL-1βir cells resembling ramified microglia were first observed 6 hours after MCAO in the ipsilateral frontal and parietal cortex and in the corpus callosum in all four sampling areas (Fig. 1A through D). These cortical immunopositive cells often were perivascular and were located in layers I through VI (Fig. 5B). The topography of IL-1βir cells was consistent among animals and was restricted to the ischemic hemisphere of the brain. In the rostral half of the lesion (Fig. 1A and B), a few immunopositive microglia-like cells also were present at the junction of the dorsal caudoputamen and the white matter, and ventrally in the insular cortex and the fundus striati. The IL-1βir cells resembling macrophages were observed in the ipsilateral meninges above the frontal and parietal cortex, as at 2 hours.
Ischemic damage 24 hours after middle cerebral artery occlusion
The overall topographic distribution of the neuronal damage 24 hours after MCAO was similar to that observed in animals killed 6 hours after surgery (Fig. 1E through H). However, the number and degree of damaged neurons had markedly increased by 24 hours, with many neurons showing necrotic changes, especially in the upper layers of the parietal (Pa2) (Fig. 1), insular, perirhinal, and piriform cortex and also in the dorsal and lateral caudoputamen (i.e., the lesion core). The margins of the infarct had become well defined, with severely condensed disintegrating neurons adjacent to slightly shrunken ones in the peri-infarct zone. Toward the most caudal part of the lesion (Fig. 1H), the neuronal damage generally was less severe. A major difference between the earlier time points and 24 hours after MCAO was the commencement of infiltration of neutrophils and macrophages into the core of the lesion. The neuropil in the ischemic zone was markedly vacuolated because of the enlarged processes of astrocytes and neurons. In the ipsilateral alveus at the junction of the hippocampus and corpus callosum, the axons from the CA1 hippocampal pyramidal neurons were swollen, but the cell bodies were normal.
Interleukin-1β-immunoreactive cells 24 hours after middle cerebral artery occlusion
The IL-1βir cells were far more numerous and more intensely stained 24 hours after MCAO than at 6 hours. Again, immunolabeled cells, presumed to be microglia and macrophages, were confined to the ischemic hemisphere. The pattern of distribution was consistent among animals along the margins of the infarct and in the adjacent penumbra (Fig. 1E through H). Within the lesion, the interanimal topography and number of IL-1βir cells seen was more variable, partly because of the differing amounts of tissue loss from the ischemic core. In the rostral half of the lesion, immunopositive microglia-like cells were seen in all layers of the frontal cortex, the corpus callosum (Fig. 5C), the caudoputamen (Fig. 5D), the fundus striati, and the piriform cortex (Fig. 1E and F). Interleukin-1βir cells resembling activated microglia and macrophages also were present in the core of the lesion.
In the caudal part of the lesion, IL-1βir microglia-like cells were present in all layers of the frontal cortex, the parietal cortex, and the underlying white matter (Fig. 1G and H). Cells resembling microglia also were immunostained in the stratum oriens in the CA1 region of the hippocampus (compare Fig. 5E and F), the lateral caudate, the amygdaloid nuclei, the optic tract, and the perirhinal and piriform cortex. A few of these putative microglia were ramified in appearance, whereas others were more activated with enlarged, irregularly shaped cell bodies and short, thick processes, particularly in the most severely affected ischemic areas (Fig. 5C and D). The IL-1βir macrophage-like cells were present in these same areas, but since the infiltration of blood-borne macrophages had commenced, it was not possible to differentiate between IL-1β-immunopositive resident phagocytic microglia and invading macrophages.
Many IL-1βir cells, presumed to be macrophages, were observed in the cortical meninges (Fig. 4A) above the ipsilateral insular, parietal, and frontal cortex, but usually only a few contralaterally in the meninges of the frontal cortex.
Ischemic damage 48 hours after middle cerebral artery occlusion
The progression of the ischemic damage between 24 and 48 hours was marked by the massive invasion of neutrophils and macrophages into the core of the lesion. Mass necrosis was by now widespread in the frontal cortex, and shrunken neurons were present more medially in the previously undamaged frontal 2 region, rostrally (F2, Fig. 1I and J). The axons of the CA1 pyramidal neurons in the alveus were distended in the contralateral hemisphere (unlike at 24 hours), as well as ipsilaterally (as at 24 hours). No damage to neuronal cell bodies was observed contralaterally.
Interleukin-1β-immunoreactive cells 48 hours after middle cerebral artery occlusion
The topographic distribution of IL-1βir was similar but more extensive ipsilaterally than at 24 hours, and was apparent contralaterally throughout the sampling areas (Fig. 1I–L). The interanimal pattern of labeling was consistent, but numbers of IL-1βir cells varied between rats in the contralateral hemisphere, where stained cells were more sparse.
Ipsilateral immunostaining. The IL-1βir cells resembling microglia were present more medially than at 24 hours in the frontal 2 region of the cortex (F2) (Figs. 1I and J, and 6A) and the cingulate cortex rostrally (Fig. 1I and J), and in the retrosplenial cortex caudally (Fig. 1K and L). Additionally, similar IL-1βir cells were seen in the septum and accumbens rostrally, and throughout the hippocampus (Fig. 6C), dentate gyrus, and the thalamus, caudally. Faintly IL-1βir cells resembling astrocytes were seen only in the undamaged medial caudoputamen in two of four brains.
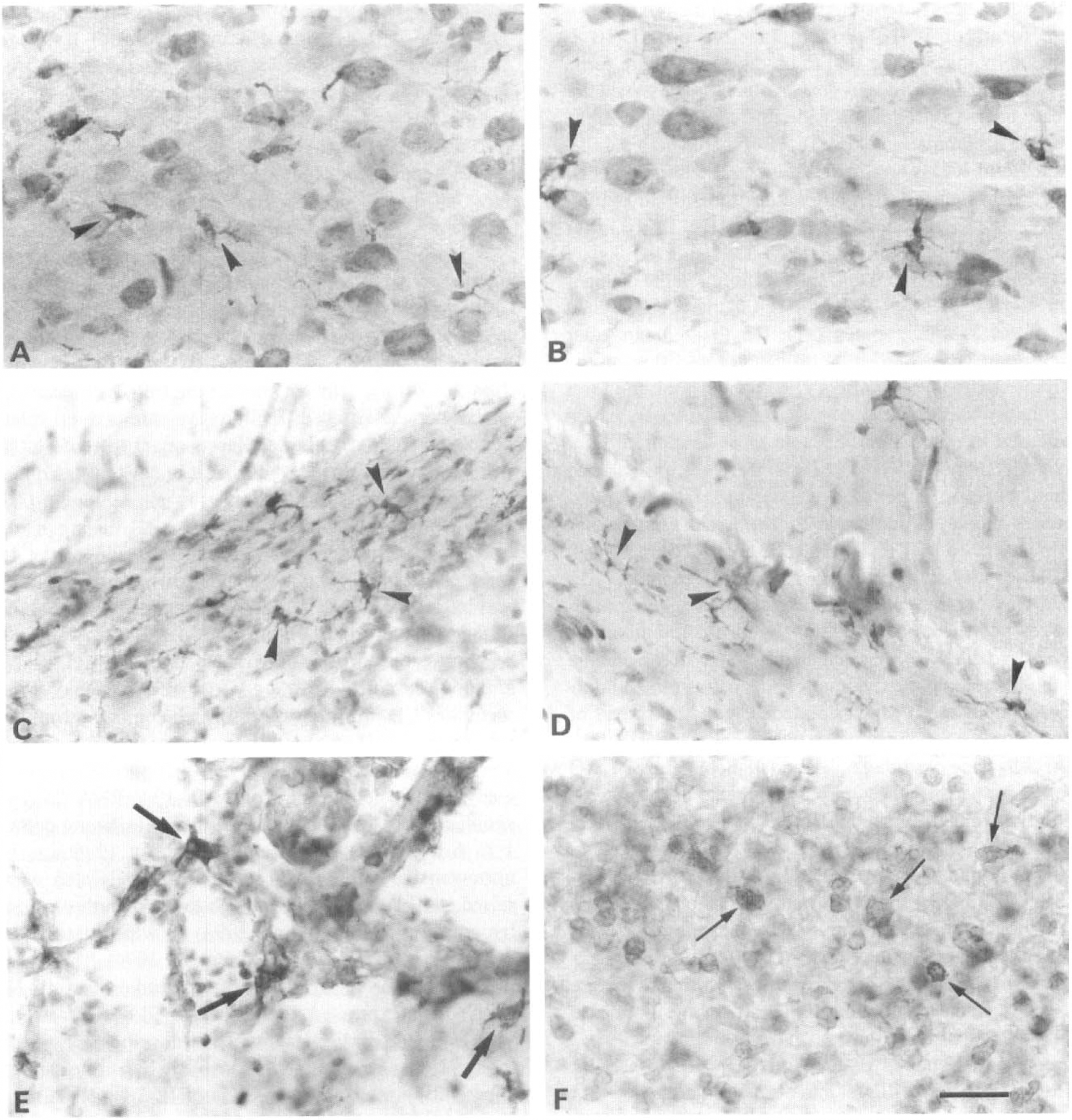
Pro/mature interleukin-1β (IL-1β)-immunopositive cells in vibratome sections (80 μm) 48 hours after middle cerebral artery occlusion (MCAO). Sections are counterstained with toluidine blue. The IL-1βir partly activated cells resembling microglia (arrowheads) in the ipsilateral frontal cortex adjacent to the infarct
Contralateral immunostaining. The IL-1βir microglia-like cells were observed in the cingulate, retrosplenial, frontal (Fig. 6B), and parietal cortex, particularly caudally (Fig. 1I through L). There also was immunostaining in the dorsal caudoputamen, septum, corpus callosum and all regions of the hippocampus (Fig. 6D), although there always were more IL-1βir cells ipsilaterally.
Most of the immunopositive putative microglia were activated (Fig. 6E), with typical hypertrophic cell bodies and thick, shortened processes, a notable exception being some in the hippocampus that were ramified, especially on the contralateral side (Fig. 6D). In addition to these activated cells, there were many immunopositive cells, presumed to be macrophages, particularly in the infarcted parietal, insular (Fig. 6F), perirhinal, and piriform cortex; lateral caudoputamen; and in the corpus callosum (Fig. 3E). In the meninges, many more IL-1βir macrophages were present ipsilaterally than contralaterally, as observed 24 hours after MCAO.
Summary of relation between interleukin-1β-immunoreactive cells and ischemic damage
In the brains of animals killed 6 and 24 hours after MCAO, the IL-1βir putative microglia were located in regions in which neuronal shrinkage was marked in the ischemic core, but mostly in adjacent regions in which neurons apparently were only slightly damaged. By 48 hours, mass necrosis in the core coincided with the influx of neutrophils and macrophage-like cells, most of the latter being IL-1βir. Immunopositive microglia and macrophages also were found ipsilaterally and contralaterally in regions where there was no obvious ischemic cell damage. On the ipsilateral side, these comprised the frontal 2 region, the cingulate and retrosplenial cortex, the septum, the hippocampus, the dentate gyrus, and the thalamus. The same undamaged regions in the contralateral hemisphere contained IL-1βir microglia, which were more sparse and less activated than on the ipsilateral side.
DISCUSSION
One of the aims of this study was to identify the cellular source of IL-1β protein in the brain induced by permanent focal MCAO in the rat. Interleukin-1β was first detected in a few meningeal macrophages by 1 hour after occlusion. By 6 hours, scattered IL-1βir ramified microglia were present within the ischemic area in the cerebral cortex, caudoputamen, and corpus callosum, some in close association with blood vessels. Twenty-four and 48 hours after the ischemic insult, there was a massive increase in the distribution and number of immunopositive cells, as the core of the infarct became infiltrated with blood-borne macrophages in addition to many activated IL-1βir microglia, particularly in the peri-infarct zone. The identity of the immunopositive cells was confirmed by both double labeling and staining consecutive sections with IL-1β antibody or a known marker (GSA I-B4) for microglia and macrophages. Interleukin-1βir has been located previously in meningeal macrophages, infiltrated macrophages, and both ramified and activated microglia in endotoxin-treated rats (Van Dam et al., 1992) and in rats with experimental allergic encephalomyelitis (Bauer et al., 1993). When the latter condition had reached its full clinical appearance, Bauer and colleagues (1993) reported a massive infiltration of ED-1ir macrophages into the lesions, similar to our own observations in response to MCAO. In the current study, no IL-1β-immunopositive neurons were apparent after MCAO, which concurs with the effects of endotoxin administration (Van Dam et al., 1992) and observations in rats with experimental allergic encephalomyelitis (Bauer et al., 1993). Neurons, especially their processes, have been reported to express IL-1β protein in normal human and rat hypothalamus and hippocampus (Breder et al., 1988; Lechan et al., 1990; Van Dam et al., 1992), as have hippocampal pyramidal cells after excitotoxic damage in perinatal rats (Hagan et al, 1996). However, neither immunopositive neuronal somata nor processes were detected in our sham-operated or MCAO rats, which probably results from the use of a notably different protocol from these authors. Another explanation could be related to the specificity of the respective antibodies used. For instance, in our preliminary studies using a polyclonal rat IL-1β antibody (Cytokine Sciences, Inc., Boston, MA, U.S.A.), we observed IL-1βir astrocytes in MCAO, sham-operated, and normal rats, but the staining was not abolished by preabsorption of this antibody with recombinant rat IL-1β (data not shown). In the current study, not only was immunolabeling abolished by preabsorption, but also the IL-1β antibody did not recognize IL-1α or IL-1ra, with which IL-1β has some homology (≈20% to 25%). Furthermore, in damaged tissue, the identification of immunopositive cells can be difficult because necrotic or crushed cells always stain nonspecifically with immunoperoxidase methods (Bourne, 1983, and our unpublished observations).
The polyclonal sheep anti-rat antibody used in the current study, in common with all other available IL-1β antibodies, does not distinguish between the pro and mature forms of IL-1β protein. Inactive pro IL-1β is cleaved by the enzyme ICE to produce the mature form. Therefore, the possibility exists that the observed increase in IL-1β production with time could have resulted from an accumulation of pro IL-1β if there was a parallel decrease of ICE. There are no data on the immunolocalization of mature IL-1β, but we have identified by bioassay significant increases in bioactive IL-1β after MCAO in the rat.
In a comparison of the timing of morphologic changes and IL-1β expression, ischemic cell damage was seen at all of the sampling times from 0.5 to 48 hours after occlusion. As assessed from 80-μm thick sections in the current study, these morphologic changes took the form of slightly shrunken neurons at 0.5 and 1 hour, which increased in number and became more condensed by 2 hours. The first affected regions were the insular and lateral piriform cortex and the dorsolateral caudoputamen. A similar chronology of ischemic cell changes has been described by Garcia and colleagues (1993, 1995a). At these early stages in the evolution of the infarct, IL-1βir was present in the ipsilateral, meningeal macrophages only, from 1 hour after the insult. Immunopositive microglia were first detected 6 hours after ischemia in the infarcted cortex, dorsal corpus callosum, and dorsal caudoputamen, which coincided with the spread of damaged neurons demarcating the prospective lesion. IL-1βir cells were observed 6, 24, and 48 hours after MCAO within the peri-infarct zone, in which the neurons were slightly shrunken, as well as in the core of the lesion. In addition by 48 hours, IL-1βir cells were localized in apparently undamaged tissue both ipsilaterally and contralaterally in the cortex, corpus callosum, caudoputamen, hippocampus, and septum. The causes of this IL-1β immunolabeling beyond the territory of the MCA is unclear, although again, this may represent pro and mature IL-1β. It is conceivable that IL-1β alone does not cause neuronal death unless the cells have been previously exposed to an ischemic or excitotoxic insult. Consistent with this, infusion of IL-1β into normal rat brain does not cause neurodegeneration, but markedly exacerbates ischemic damage if administered at the time of MCAO (our unpublished data). Alternatively, the neurons may have undergone slight deterioration (feasibly, undetectable in thick vibratome sections), caused perhaps by increased levels of excitatory amino acids, which, in turn, could have resulted in the induction of IL-1β. The inflammatory response to ischemia includes the influx of neutrophils into the damaged tissue, and one of the proinflammatory effects of IL-1 is leukocytosis (Dinarello, 1994). Therefore, it follows that the spread of the inflammatory response to the contralateral dorsomedial corpus callosum, evident at 48 hours in the current study, could have been caused by the induction of IL-1β in the contralateral hemisphere. In accordance with this suggestion, the inhibition of IL-1β by the administration of IL-1ra or IL-1β neutralizing antibody to rats after focal MCAO resulted in fewer neutrophils and necrotic neurons (Garcia et al., 1995b; Yamasaki et al., 1995). Other studies also document the induction of cytokines in the contralateral hemisphere of injured brains, which the authors have attributed to the diffusion of cytokines through edematous pathways, or in response to pyramidal cell loss in the hippocampus after fluid percussion injury (Cortez et al., 1989; Taupin et al., 1993).
It was considered worthwhile to compare the expression of IL-1β protein to published data on IL-1β gene expression induced by permanent MCAO (Buttini et al., 1994; Liu et al., 1993), although the full significance of such a comparison is difficult to evaluate because it has been shown that although large quantities of IL-1β mRNA can be induced, much of it is not translated and undergoes degradation (Schindler et al., 1990a,b). One study reports the induction of IL-1β mRNA using in situ hybridization histochemistry in the ischemic cortex 15 minutes after MCAO in spontaneously hypertensive rats, and by 1 hour, IL-1β mRNA was detected in the whole of the MCA territory (Buttini et al., 1994); the peak level of expression occurred at 3 hours after MCAO. These authors also noted a marked IL-1β mRNA signal in the meninges 1 hour after MCAO, which coincided with the localization of IL-1β protein in our study. By comparison, Liu and associates (1993), using Northern blot analysis, detected an increase in IL-1β mRNA (also in spontaneously hypertensive rats) in the ipsilateral cortex 1 to 6 hours after occlusion, peaking at 12 hours. In normotensive rats, the IL-1β mRNA levels also were raised at 12 hours but were much lower than those from hypertensive rats (Liu et al., 1993), in which the size of the lesion is 60% larger (Buttini et al., 1994). Therefore, it can be inferred from these two studies that IL-1β mRNA was expressed during the period of necrosis, as well as in the earlier stages in the development of the infarct. Considering that spontaneously hypertensive rats apparently produce higher levels of IL-1β mRNA than normotensive ones (Liu et al., 1993) and that much of the message may not be translated (Schindler et al., 1990a,b), the timing of IL-1β gene expression after MCAO is compatible with that of the protein found in our study. The regional expression of IL-1β mRNA (Buttini et al., 1994) correlated closely with that of IL-1β protein ipsilateral to the occlusion and was confined to the territory of the MCA. Moreover, consistent with the localization of IL-1β protein in microglia, Buttini and coworkers (1994) concluded that IL-1β mRNA was probably expressed in microglia.
Measurements of IL-1β protein content by immunoassay in rat brain (Iannotti et al., 1993) showed increasing levels between 0.5 and 48 hours after permanent focal MCAO in samples taken from the ischemic cortex, agreeing well with the timing of the increase in number of IL-1βir cells found in the current study. Iannotti and associates (1993) also observed a rise in IL-1β content in the contralateral cortex 48 hours after the occlusion. Again, the findings of this study and those of Iannotti and others (1993) indicate that IL-1β protein is produced early after the occlusion, as well as throughout the later, necrotic stages in the evolution of the infarct.
The current data are consistent with previous studies implicating IL-1 in neurodegeneration. These include the exacerbation of ischemic brain damage by injection of recombinant IL-1β (Loddick and Rothwell, 1996; Yamasaki et al., 1995) and the inhibition of IL-1β action by the administration of IL-1ra (Garcia et al., 1995b; Loddick and Rothwell, 1996; Relton and Rothwell, 1992) or IL-1β neutralizing antibody (Yamasaki et al., 1995), and also, inhibition of IL-1β synthesis by the injection of ICE inhibitor (Loddick et al., 1996), all of which attenuate ischemic brain damage. Localization of IL-1β in microglia and macrophages is in accordance with a recent report on the induction of ICE gene and protein expression in the hippocampus and ICE-like immunopositive microglia after global cerebral ischemia in the gerbil (Bhat et al., 1996). Overexpression of ICE has been shown to induce programmed cell death in cultured cells (Gagliardini et al., 1994; Miura et al., 1993). However, Bhat and coauthors noted that ICE-like immunoreactivity was not altered in the hippocampal CA1 neurons, which were apoptotic after ischemia. They concluded, therefore, that ICE has an indirect role in ischemic damage to cause the release of IL-1β. Although other studies implicate ICE (e.g., Miura et al., 1993) or ICE-homologues (e.g., Kumar, 1995) in programmed cell death and apoptosis in neuronal death after cerebral ischemia (Johnson et al., 1995; Linnik et al., 1993), it appears that after focal cerebral ischemia most neuronal death probably occurs through the process of necrosis (DeGirolami et al., 1984; Garcia et al., 1993; 1995a; Van Lookeren Campagne and Gill, 1996) rather than apoptosis. Nevertheless, the possibility remains that apoptotic neurons might be difficult to detect if they were phagocytosed rapidly.
In summary, the results of this study defining the chronologic and topographic distribution of pro/mature IL-1β protein after permanent focal cerebral ischemia add to the mounting evidence indicating that the inhibition of IL-1 in the brain might be of therapeutic benefit after various types of neurodegenerative insult, and indicate that the predominant cell sources of IL-1β comprise resident microglia and infiltrated macrophages.