
Abstract
The X chromosome–linked inhibitor-of-apoptosis protein (XIAP) contributes to apoptosis regulation after a variety of cell death stimuli. XIAP inhibits the caspase reaction via binding to caspases, and is inhibited via binding to the second mitochondria-derived activator of caspase (Smac)/DIABLO to tightly control apoptotic cell death. However, the interaction among XIAP, Smac/DIABLO, and caspases after in vivo cerebral ischemia is not well known. To clarify this issue, the authors examined time-dependent expression and interaction among XIAP, Smac/DIABLO, and activated caspase-9 by immunohistochemistry, Western blot analysis, and immunoprecipitation using an in vivo transient focal cerebral ischemia model. To examine the relationship of the XIAP pathway to the caspase cascade, a pan-caspase inhibitor was administered. XIAP increased concurrently with the release of Smac/DIABLO and the appearance of activated caspase-9 during the early period after reperfusion injury. The bindings of XIAP to Smac/DIABLO and to caspase-9 and the binding of Smac/DIABLO to caspase-9 reached a peak simultaneously after transient focal cerebral ischemia. Neither XIAP nor Smac/DIABLO expression was affected by caspase inhibition. These results suggest that the XIAP pathway was activated upstream of the caspase cascade and that interaction among XIAP, Smac/DIABLO, and caspase-9 plays an important role in the regulation of apoptotic neuronal cell death after transient focal cerebral ischemia.
The central players in the apoptotic pathway are the caspases, a family of cysteine aspartate-specific proteases (Slee et al., 1999). Caspases are activated in response to diverse stimuli and cleave multiple cellular substrates resulting in dismantling of the cell (Earnshaw et al., 1999). One important route to caspase activation involves the translocation of cytochrome c from the mitochondrial intermembrane space to the cytosol (Liu et al., 1996). Released cytochrome c interacts with Apaf-1 and caspase-9, both of which play essential roles in the cytochrome c–dependent mitochondrial pathway of apoptosis (Hakem et al., 1998; Kuida et al., 1998; Zou et al., 1997, 1999), by activating caspases such as caspases-2, 3, 6, 7, 8, and 10 (Slee et al., 1999), which results in apoptosis. Caspase-9 cleaves proteolytically to the large subunit p35 and the small subunit p12 after the assembly of apoptosomes (Zou et al., 2003). A proapoptotic molecule, second mitochondria-derived activator of caspase (Smac) /DIABLO, was recently identified (Du et al., 2000; Verhagen et al., 2000). In the early stages of apoptosis, Smac/DIABLO is released from mitochondria into the cytosol (concurrently with cytochrome c), eliminates the inhibitory effects of many inhibitor-of-apoptosis proteins (IAPs), and promotes caspase activation (Adrain et al., 2001; Du et al., 2000; Martins et al., 2002; Verhagen et al., 2000).
To avoid disease and inappropriate cell death, apoptotic mechanisms must be tightly controlled. Recent experimental evidence has revealed that apoptosis can be inhibited and controlled at several distinct points in the apoptotic pathway (Goyal, 2001; Wehrli et al., 2000). One group of naturally occurring inhibitors is the family of IAPs (Keane et al., 2001). The IAP family negatively regulates caspase activation (Deveraux et al., 1999; Miller, 1999). In mammals, this family includes cIAP1, cIAP2, X chromosome–linked IAP (XIAP), NAIP, Livin/KIAP, BRUCE/Apollon, and Survivin (Keane et al., 2001; Silke and Vaux, 2001). These proteins bind to and inhibit both initiator caspases such as caspase-9 and effector caspases such as caspases-3 and 7 (Martins et al., 2002). All IAPs contain one to three baculovirus IAP repeat (BIR) domains, which are characterized by a highly conserved (up to 70) amino acid domain (Duckett et al., 1996). Among the above IAPs, XIAP is the most potent inhibitor of caspases and apoptosis (Deveraux and Reed, 1999). The structure of XIAP is characterized by three tandem repeats of the BIR domain at its N-terminus and a RING finger domain near its C terminus (Suzuki et al., 2001). The second BIR domain (BIR2) of XIAP is sufficient to inhibit caspases-3 and 7, whereas the third BIR domain (BIR3) especially inhibits caspase-9 (Deveraux et al., 1999; Sun et al., 2000; Takahashi et al., 1998). Recently, in vitro experiments revealed that Smac/DIABLO interacts with the BIR2 and BIR3 domains of XIAP via its N-terminal amino acids. Moreover, mutations at the N-terminus of Smac/DIABLO abrogate its ability to inhibit XIAP (Srinivasula et al., 2001).
Many studies of in vivo cerebral ischemia have provided evidence that the mitochondrial pathway leads to caspase activation and plays an important role in the machinery of apoptotic neuronal cell death after ischemic injury (Chan, 1996; Colbourne et al., 1999; Du et al., 1996; Fujimura et al., 1998; Sugawara et al., 2002). However, to our knowledge, the mechanisms of XIAP and Smac/DIABLO activation remain unknown in in vivo cerebral ischemia studies. In our study, we hypothesized that both XIAP and Smac/DIABLO were activated reciprocally and that they had important roles in the caspase chain reaction in apoptotic neuronal cell death after focal cerebral ischemia. To determine how Smac/DIABLO and XIAP interact to regulate apoptotic neuronal cell death, we analyzed the spatial and temporal expressions and the direct bindings among Smac/DIABLO, XIAP and an activated initiator caspase after focal cerebral ischemia.
MATERIALS AND METHODS
Focal cerebral ischemia
Adult male mice (3 months old, 35–40 g) were subjected to transient focal cerebral ischemia (tFCI) by intraluminal middle cerebral artery (MCA) blockade with a nylon suture as described previously (Kondo et al., 1997). The mice were anesthetized with 1.5% isoflurane in 30% oxygen and 70% nitrous oxide using a facemask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Cannulation of a femoral artery allowed monitoring of blood pressure and arterial blood gases; samples for analysis were taken immediately after cannulation, 10 minutes after occlusion, and 10 minutes after reperfusion. Blood gas was analyzed with a pH/blood gas analyzer (Chiron Diagnostics Ltd., Essex, U.K.). After a midline skin incision, the left external carotid artery was exposed and its branches were electrocoagulated. A 11.0-mm 5–0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump. After 60 minutes of MCA occlusion, blood flow was restored by the withdrawal of the nylon suture. To examine the role of caspases in the interaction between XIAP and Smac/DIABLO, we intraventricularly injected a pan-caspase inhibitor, N-benzyloxycarbonyl-valinyl-alaninyl-aspartyl-(O-methyl)-fluoromethyl ketone (Z-VAD–FMK) using a 10-μL Hamilton syringe (Hamilton, Reno, NV, U.S.A.) after tFCI. Z-VAD–FMK was purchased from Sigma (St. Louis, MO, U.S.A.) and dissolved in 0.3% dimethylsulfoxide in phosphate-buffered saline. The dose of the drug was decided by referring to a previous intraventricular-injection study (Endres et al., 1998). The scalp was incised on the midline and the skull was exposed. Z-VAD–FMK (125 ng/μL in 0.3% dimethylsulfoxide in phosphate-buffered saline) and the vehicle (0.3% dimethylsulfoxide in phosphate-buffered saline alone) were injected intraventricularly (2 μL, bregma; 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep) 30 minutes after MCA occlusion.
Immunohistochemistry
Anesthetized animals and normal controls (n = 4 each) were perfused with 10-U/mL heparin and subsequently with 4% paraformaldehyde in 0.1-mol/L phosphate-buffered saline (pH 7.4) at 1, 2, 4, 8, and 24 hours of reperfusion. The brains were removed, postfixed for 12 hours, sectioned at 50 μm on a vibratome, and processed for immunohistochemistry. The sections were incubated with blocking solution and reacted with mouse monoclonal anti-XIAP antibody (StressGen Biotechnologies Corp., Victoria BC, Canada) at a dilution of 1:1,000. Immunohistochemistry was performed using the avidin-biotin technique and then the nuclei were counterstained with methyl green solution for 2 minutes. For histologic assessment, alternate slices from each brain section were stained with cresyl violet.
Immunofluorescent double-labeling staining
To evaluate colocalization of XIAP or Smac/DIABLO and neuron-specific nuclear protein (NeuN), we performed double immunofluorescent staining for these proteins. The sections fixed with 4% paraformaldehyde were immunostained with an anti-XIAP antibody or anti-Smac/DIABLO (Chemicon International, Temecula, CA, U.S.A.), as described above, with biotinylated goat anti–rabbit immunoglobulin G (IgG) (Vector Laboratories, Burlingame, CA, U.S.A.) followed by Fluorescein Avidin DCS (Vector Laboratories). The sections were then incubated with a blocking solution and reacted with an anti-NeuN antibody (Chemicon International), as described above, followed by Texas Red–conjugated donkey anti–mouse IgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, U.S.A.) at a dilution of 1:400. The sections were then placed on slides and subsequently, the slides were covered with VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Fluorescence of fluorescein was observed at excitation of 495 nm and emission of >515 nm, and fluorescence of Texas Red was observed at excitation of 510 nm and emission of >580 nm. Fluorescence of DAPI was also observed at excitation of 360 nm and emission of >460 nm.
Western blot analysis
Protein extraction of the cytosolic fraction was performed as described previously with some modification (Fujimura et al., 2000). Samples were obtained from the MCA territory brain tissue on the ischemic side and from nonischemic controls (n = 4 each). Fresh brain tissue was removed after 1, 2, 4, 8, and 24 hours (n = 4 each) of reperfusion and homogenized by gently douncing 35 times in a glass tissue grinder (Wheaton, Millville, NJ, U.S.A.) in 7 volumes of cold suspension buffer (20-mmol/L HEPES-KOH [pH 7.5], 250-mmol/L sucrose, 10-mmol/L KCl, 1.5-mmol/L MgCl2, 1-mmol/L EDTA, 1-mmol/L EGTA, 0.7% protease and phosphatase inhibitor cocktails [Sigma]). The homogenate was centrifuged at 750g for 10 minutes at 4°C and then at 8,000g for 20 minutes at 4°C. The 8,000g pellets were used to obtain the mitochondrial fraction. The supernatant was further centrifuged at 100,000g for 60 minutes at 4°C. This supernatant was used for cytosolic analysis. After adding the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA, U.S.A.) to the supernatant, equal amounts of the samples were loaded per lane. The primary antibodies were 1:1,000 dilution of rabbit polyclonal antibody against Smac/DIABLO (Chemicon), 1:1000 dilution of mouse polyclonal antibody against XIAP (BD Transduction Laboratories, San Diego, CA, U.S.A.), 1:400 dilution of rabbit polyclonal antibody against cleaved caspase-9 p12 subunit (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), 1:500 dilution of rabbit polyclonal antibody against cytochrome c (Santa Cruz Biotechnology) or 1:10,000 dilution of anti–β-actin monoclonal antibody (Sigma) and 1:2,000 dilution of anti–cytochrome oxidase (COX) IV monoclonal antibody (Molecular Probes, Eugene, OR, U.S.A.). Western blots were performed with horseradish peroxidase-conjugated anti–rabbit IgG (Cell Signaling Technology, Beverly, MA, U.S.A.) or anti–mouse IgG (Chemicon) using enhanced chemiluminescence Western blotting detection reagents (Amersham International, Buckinghamshire, U.K.). The film was scanned with a GS-700 imaging densitometer (Bio-Rad Laboratories, Hercules, CA, U.S.A.) and the results were quantified using Multi-Analyst software (Bio-Rad).
Coimmunoprecipitation
The protein extraction of the cytosolic fraction was performed essentially as described previously (Fujimura et al., 2000). The procedure for precipitation was performed as described previously with some modification (Springer et al., 2000). Samples were obtained from the MCA territory brain tissue on the ischemic side and from nonischemic controls (n = 4 each). Fresh brain tissue was removed after 1, 2, 4, 8, and 24 hours (n = 4 each) of reperfusion and was prepared for collection of the cytosolic fraction and the mitochondrial fraction in the same manner as for Western blotting. Protein concentrations were determined by the Bradford method (Bio-Rad). Three hundred micrograms of protein from the cytosolic fraction and 15 μg from the mitochondrial fraction were used for coimmunoprecipitation. Whole brain extract was included as a positive control. The protein sample was incubated with 50% slurry of protein G-Sepharose (Amersham Pharmacia Biotech, Uppsala, Sweden) for 1 hour at 4°C, and this mixed sample was centrifuged at 12,000g for 1 minute. The supernatant was incubated with 2 μg of polyclonal rabbit anti-Smac/DIABLO antibody (Chemicon) or polyclonal rabbit anti–cleaved caspase-9 antibody (Santa Cruz Biotechnology) and 15 μL of protein G-Sepharose (50% slurry) for 1 hour at 4°C. The negative control was prepared with protein G-Sepharose without antibody. The 14,000g pellets were washed three times and used as the samples bound to each antibody. After adding the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen) to the samples, they were boiled to remove Sepharose beads. After centrifugation at 14,000g for 1 minute, the supernatant was immunoblotted using 1:1,000 dilution of anti-XIAP antibody (BD Transduction Laboratories) as described for the Western blotting method.
Caspase-3 activity assay
For quantification of caspase-3 activation, we used a commercial enzyme immunoassay (Chemicon) to determine the cleaved bioluminescent substrate by active caspase-3 specifically (Bossy-Wetzel et al., 1998; Gill et al., 2002). Samples were obtained from the entire MCA territory on the ischemic side and from nonischemic controls (n = 4 each). Fresh brain tissue was cut into pieces after 1, 2, 4, 8, and 24 hours of reperfusion and homogenized with a Teflon homogenizer in 5 volumes of ice-cold buffer (50-mmol/L KH2PO4, 0.1-mmol/L EDTA, pH 7.8) and spun for 10 minutes at 750g. The supernatant was collected and spun for 20 minutes at 10,000g and was further centrifuged at 100,000g for 60 minutes at 4°C. The resulting supernatant was collected and the protein concentration was determined. A cytosolic volume containing 20 μg of protein was used for the enzyme-linked immunosorbent assay following the manufacturer's protocol.
Quantification and statistical analysis
The data are expressed as mean ± SD. Comparisons among multiple groups were performed using one-way analysis of variance with appropriate post hoc tests (SigmaStat software, Jandel Corporation, San Rafael, CA, U.S.A.). Comparisons between two groups were achieved using Student's t test. Significance was accepted with P < 0.05.
RESULTS
Physiologic data and cerebral infarction
Physiologic data showed no significant differences in body temperature, MABP, and arterial blood gas analysis between the groups. The preischemic physiologic values were as follows: body temperature, 36.4° ± 0.3°C; MABP, 83 ± 3.1 mm Hg; pH, 7.3 ± 0.1; Pao2, 164.9 ± 19 mm Hg; PaCO2, 30 ± 10 mm Hg (n = 4). There was no deviation from these values over the period of assessment. An ischemic lesion of the core of the caudate putamen was visible as a pale, slightly stained area in the ischemic hemisphere as early as 1 hour after reperfusion, and extended to the entire MCA territory at 4 hours (as evidenced by cresyl violet staining; data not shown). The time-dependent increase in infarction in the mouse brains using intraluminal suture blockade is consistent with previous reports that used the same focal cerebral ischemia model in mice (Kondo et al., 1997; Yang et al., 1994).
Expression of XIAP in the ischemic brain after transient focal cerebral ischemia
Double immunofluorescent staining for XIAP and NeuN showed that XIAP expression colocalized with neurons in the ischemic cortex 8 hours after reperfusion (Fig. 1A). The cytosolic immunostaining was clear and homogeneous. XIAP-immunopositive cells were seen in the cortex and the caudate putamen of both the nonischemic and ischemic hemispheres, and the expression was remarkably strong in the ischemic penumbra 8 hours after reperfusion compared with the nonischemic hemisphere (Fig. 1B). Western blot analysis showed that XIAP immunoreactivity was evident as a single band with a molecular mass of 57 kd in the cytosolic fraction from the MCA territory (Fig. 1C). XIAP expression significantly increased 4 and 8 hours after reperfusion (Fig. 1C). A consistent amount of β-actin was observed in control and ischemic samples, suggesting that the amount of the loaded protein was consistent (Fig. 1C). These results suggest that XIAP expression was observed in neurons and increased during the early period of reperfusion injury after tFCI.
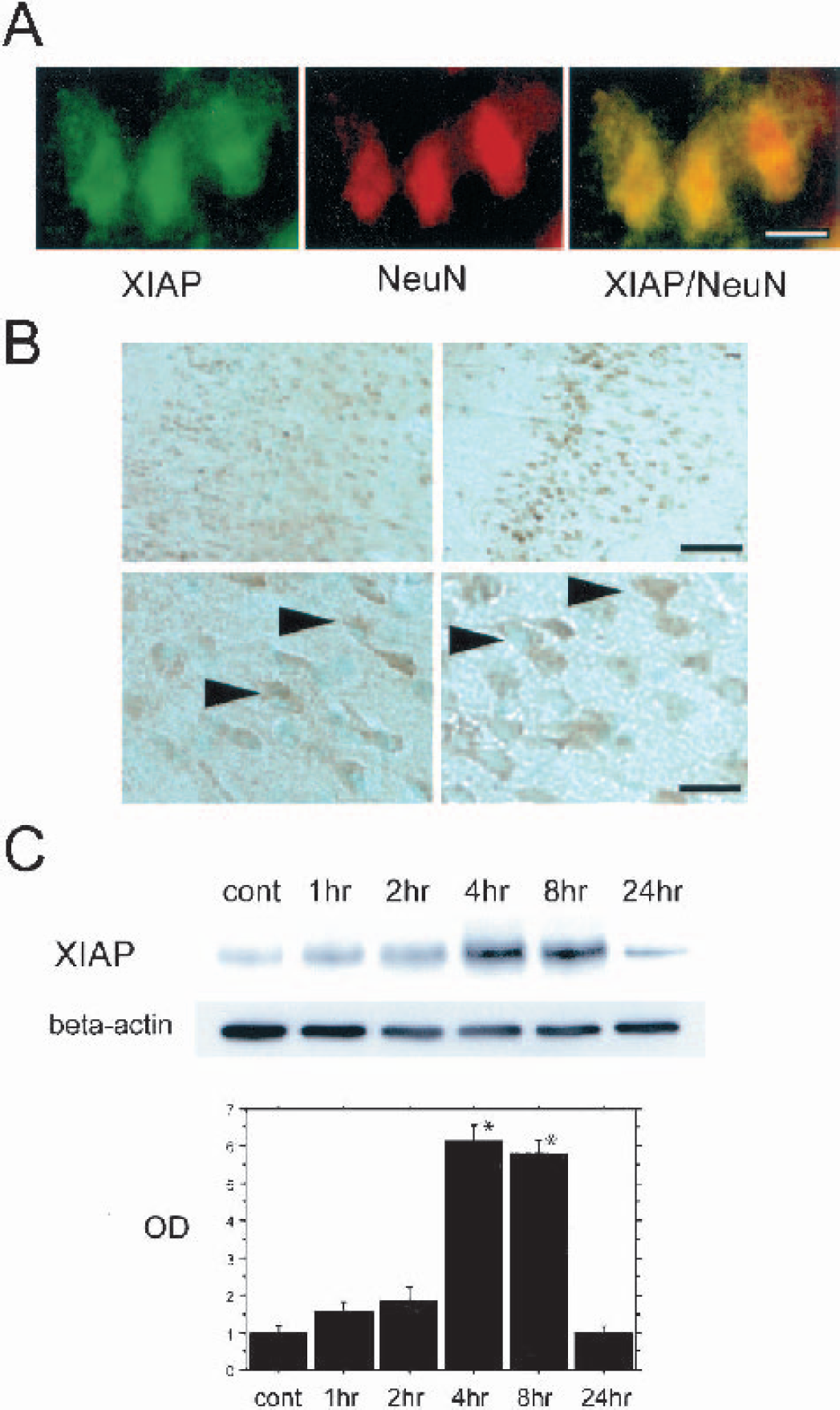
(
Smac/DIABLO expression was observed in neurons and translocated to the cytosol, corresponding to cytochrome c release before caspase-9 cleavage
Smac/DIABLO-immunopositive cells were colocalized with NeuN-positive cells in the ischemic hemisphere 24 hours after reperfusion (Fig. 2A). Smac/DIABLO-immunopositive cells were seen in the cortex and the caudate putamen of both the nonischemic and ischemic hemispheres. In the ischemic hemisphere, the Smac/DIABLO stain was slightly greater than in the nonischemic hemisphere at all time courses (data not shown). Western blot analysis revealed that Smac/DIABLO immunoreactivity was evident as a single band of molecular mass of 25 kd (Fig. 2B). Mitochondrial Smac/DIABLO expression transiently increased 1 hour after reperfusion and thereafter gradually decreased (Fig. 2B). In contrast, cytosolic Smac/DIABLO was barely detected in the control sample, but increased at 1 hour and gradually increased from 2 hours after reperfusion (Fig. 2B). We examined the activated form of caspase-9, the cleaved p12 subunit expression as an initiator of the caspase chain reaction, to detect the beginning of this chain reaction and the XIAP reaction. The cleaved caspase-9 p12 subunit was observed as a single band of 12 kd (Fig. 2C). The expression of cleaved caspase-9 was barely observed by 8 hours after reperfusion, but was prominently increased 24 hours after reperfusion (Fig. 2C). Cytochrome c expression was evident as a single band of molecular mass of 15 kd (Fig. 2D). Mitochondrial cytochrome c slowly decreased in a time-dependent manner and cytosolic cytochrome c gradually increased from 1 hour after reperfusion (Fig. 2D). Cytochrome c expression was consistent with previous results that used the same mouse tFCI model (Fujimura et al., 2000). A consistent amount of β-actin was observed in the control and ischemic samples. COX IV was used as a mitochondrial marker and no difference was observed between the samples, suggesting that the amount of the loaded protein was consistent (Figs. 2B–2D). These results show that neuronal Smac/DIABLO translocated from mitochondria to the cytosol during the early period after tFCI and that both Smac/DIABLO and cytochrome c translocation might progress simultaneously and begin before the appearance of the increase in the cleaved caspase-9.
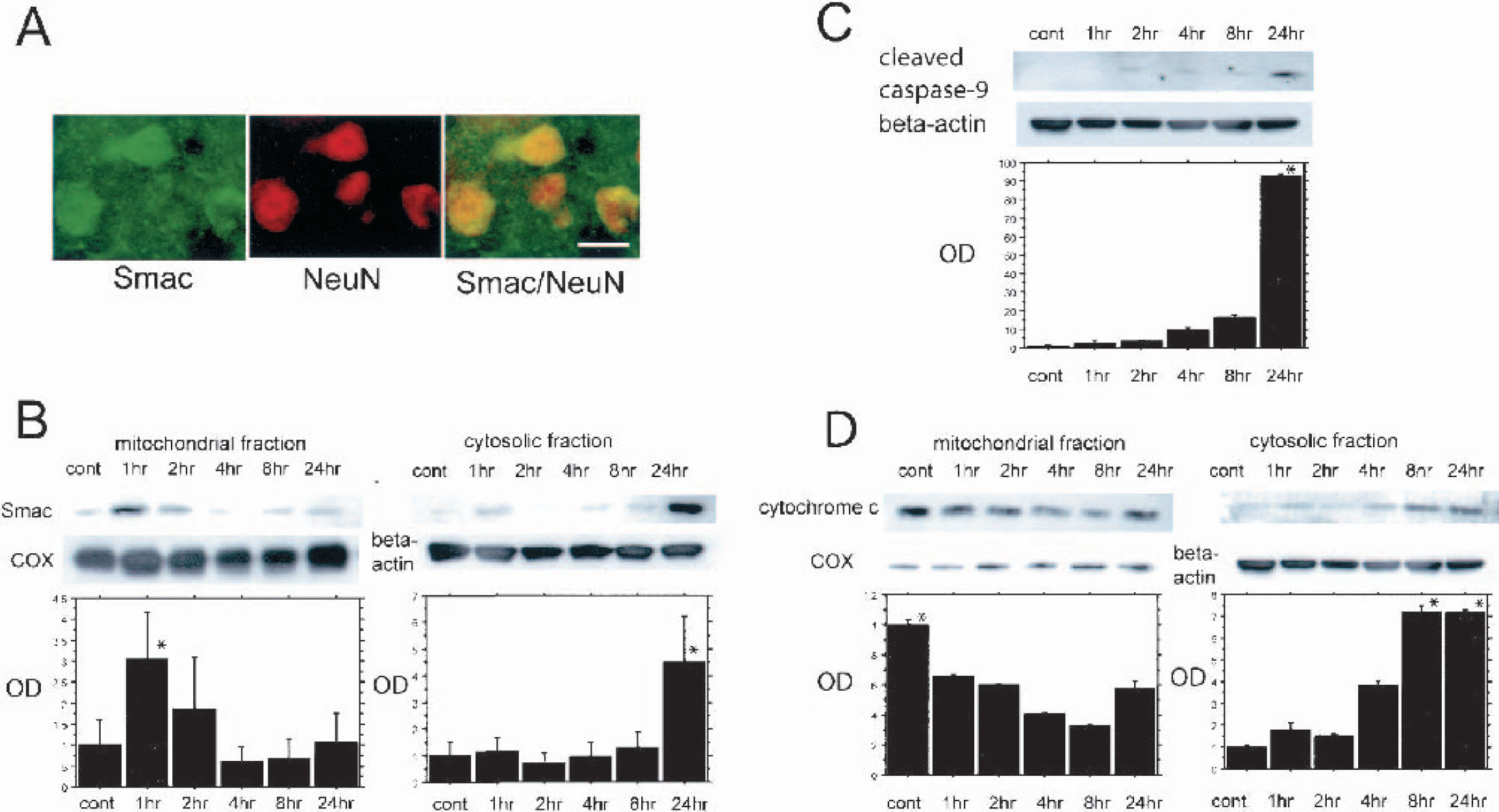
(
XIAP and released Smac/DIABLO expressions were not affected by caspase reaction
To investigate the effect of caspase reaction on XIAP and Smac/DIABLO expressions, we intraventricularly administered a pan-caspase inhibitor, Z-VAD–FMK. We examined caspase-3 activity, which is known as a critical effector of the caspase chain reaction, to investigate the effective administration of Z-VAD–FMK. Caspase-3 activity was diminished in the Z-VAD–FMK–treated mice compared with the vehicle-treated mice 24 hours after tFCI (Fig. 3A). Western blot analysis revealed that there was no difference in either XIAP or cytosolic Smac/DIABLO expression between the Z-VAD–FMK–treated mice and the vehicle-treated mice, respectively, after 8 and 24 hours of tFCI (Figs. 3B and 3C). These results show that the pan-caspase inhibitor did not affect XIAP and released Smac/DIABLO expression, and that both XIAP and Smac/DIABLO might react upstream of the caspase chain reaction.
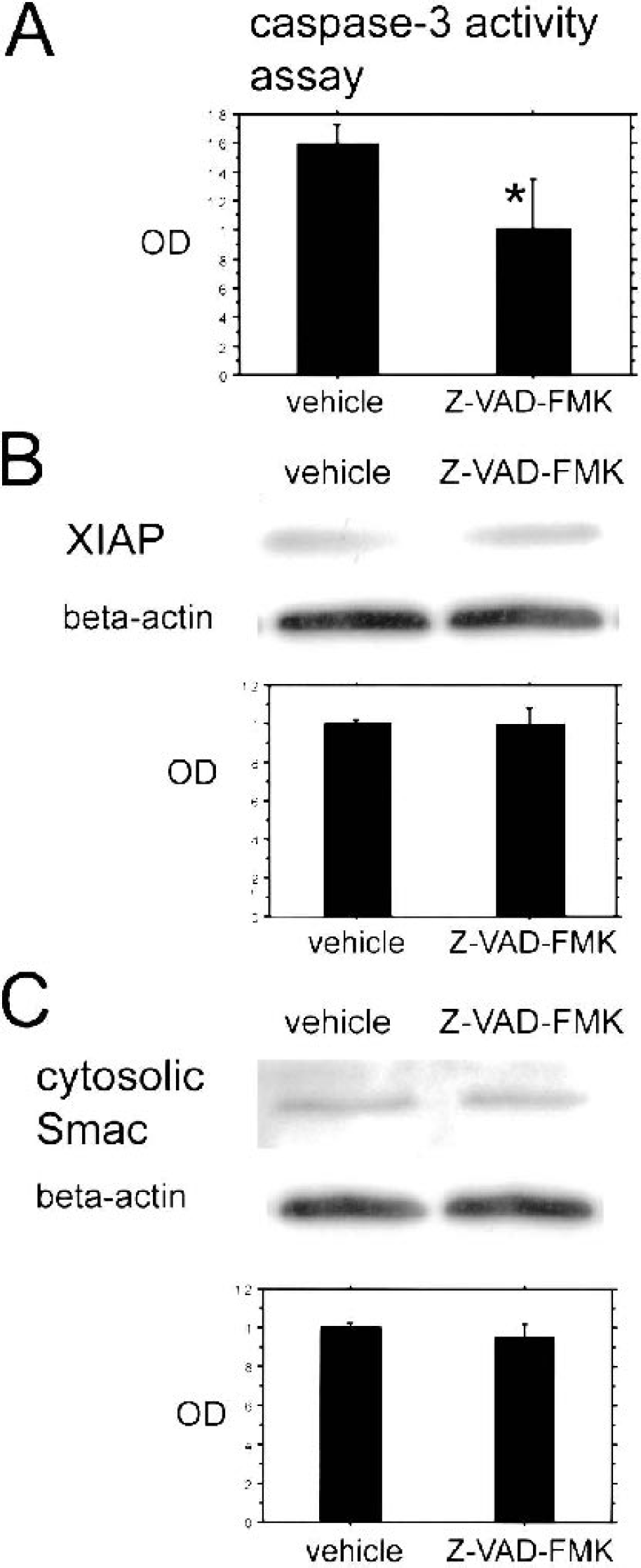
(
Direct interaction among XIAP, Smac/DIABLO, and caspase-9 was observed simultaneously
To investigate the existence of direct interaction among XIAP, Smac/DIABLO, and caspase-9, we performed coimmunoprecipitation and observed the time-dependent expressions. Positive and negative controls were examined to be certain of the specificity of each complex expression. Coimmunoprecipitation showed that the binding of XIAP to Smac/DIABLO (XIAP/Smac) slightly increased from 1 hour after reperfusion and that the peak period of interaction was 8 hours after reperfusion (Fig. 4A). The direct binding of XIAP to caspase-9 (XIAP/caspase-9) was also detected (Fig. 4B). The binding gradually increased from 1 hour and was greatly increased 8 and 24 hours after reperfusion (Fig. 4B). The peak expression was 8 hours after reperfusion and it began decreasing 24 hours after reperfusion (Fig. 4B). The direct binding of released Smac/DIABLO with caspase-9 (Smac/caspase-9) was also observed (Fig. 4C). These proteins bound gradually, increased from 2 hours, and reached a peak 8 hours after reperfusion (Fig. 4C). There was no clearly visible band in the nonischemic control sample among all these protein bindings (data not shown). These results show that XIAP, Smac/DIABLO, and caspase-9 directly interacted during the same time period after tFCI, and that 8 hours after reperfusion is a critical time point for regulation of caspase initiation mediated by XIAP and Smac/DIABLO.
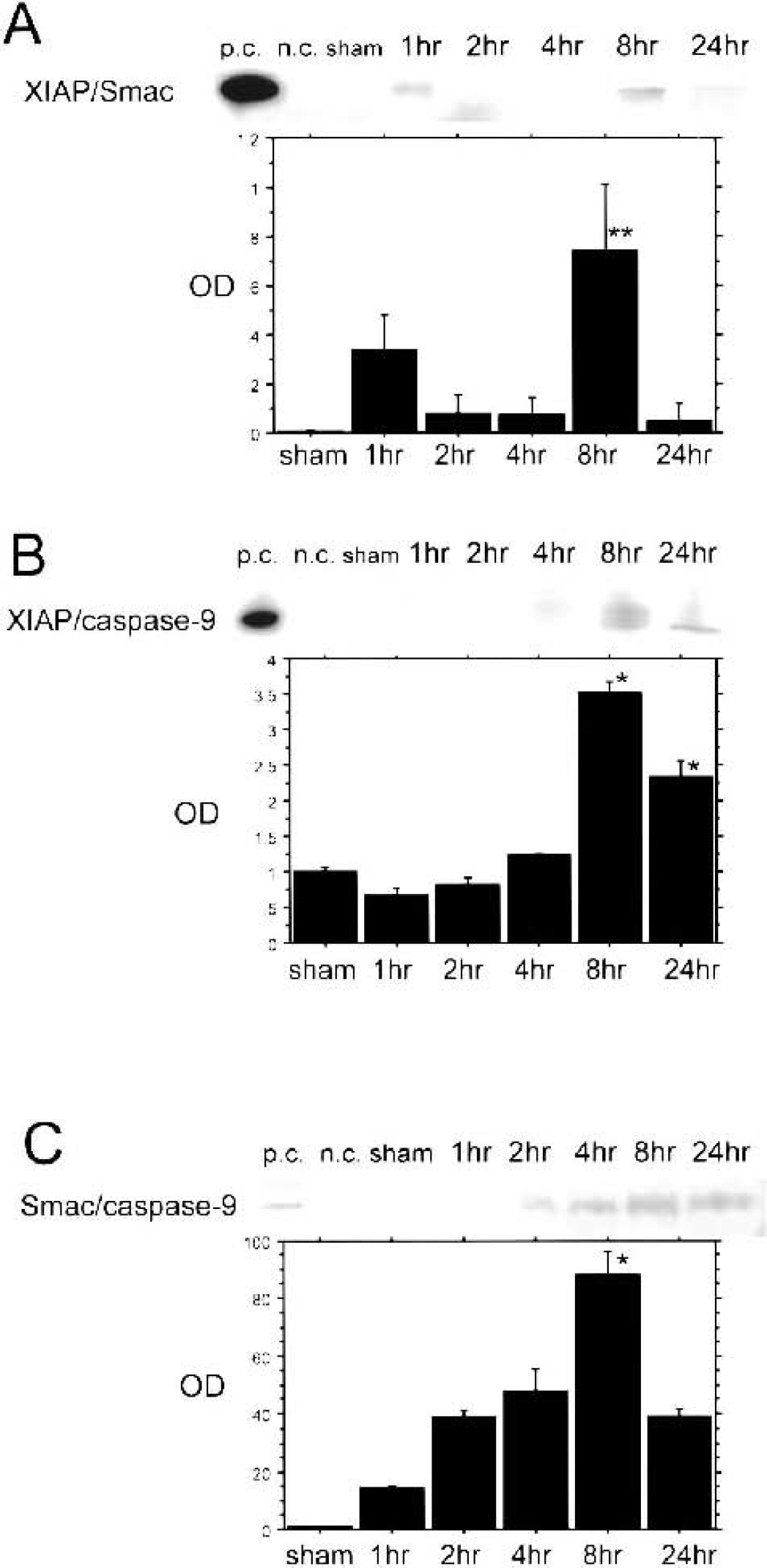
(
To compare the time-dependent expressions among XIAP, released Smac/DIABLO, released cytochrome c, activated caspase-9, and their bindings (XIAP/Smac, XIAP/caspase-9, and Smac/caspase-9) after reperfusion, we present summarized graphs in Fig. 5. Released cytochrome c increased from 8 hours after reperfusion and reached a peak 24 hours after reperfusion (Fig. 5A). Released Smac/DIABLO prominently increased 24 hours after reperfusion following the release of cytochrome c (Fig. 5A). Activated caspase-9 also prominently increased 24 hours after reperfusion (Fig. 5A). In contrast, XIAP increased temporally 4 and 8 hours after reperfusion before the peaks of released Smac/DIABLO, released cytochrome c, and activated caspase-9 (Fig. 5A). These proteins' direct bindings, XIAP/Smac, XIAP/caspase-9, and Smac/caspase-9, gradually increased and all reached their peaks 8 hours after reperfusion (Fig. 5B). The peak expression of XIAP was observed before the peak expressions of released cytochrome c, released Smac/DIABLO, and activated caspase-9, but the peak expression of XIAP/Smac, XIAP/caspase-9, and Smac/caspase-9 were in accord with the peak of XIAP (Figs. 5A and 5B). These bindings increased before the peak expressions of the individual proteins (Figs. 5A and 5B). These results show that the inhibitory reaction of XIAP via the direct bindings began before the proapoptotic progression during the early period of reperfusion injury.
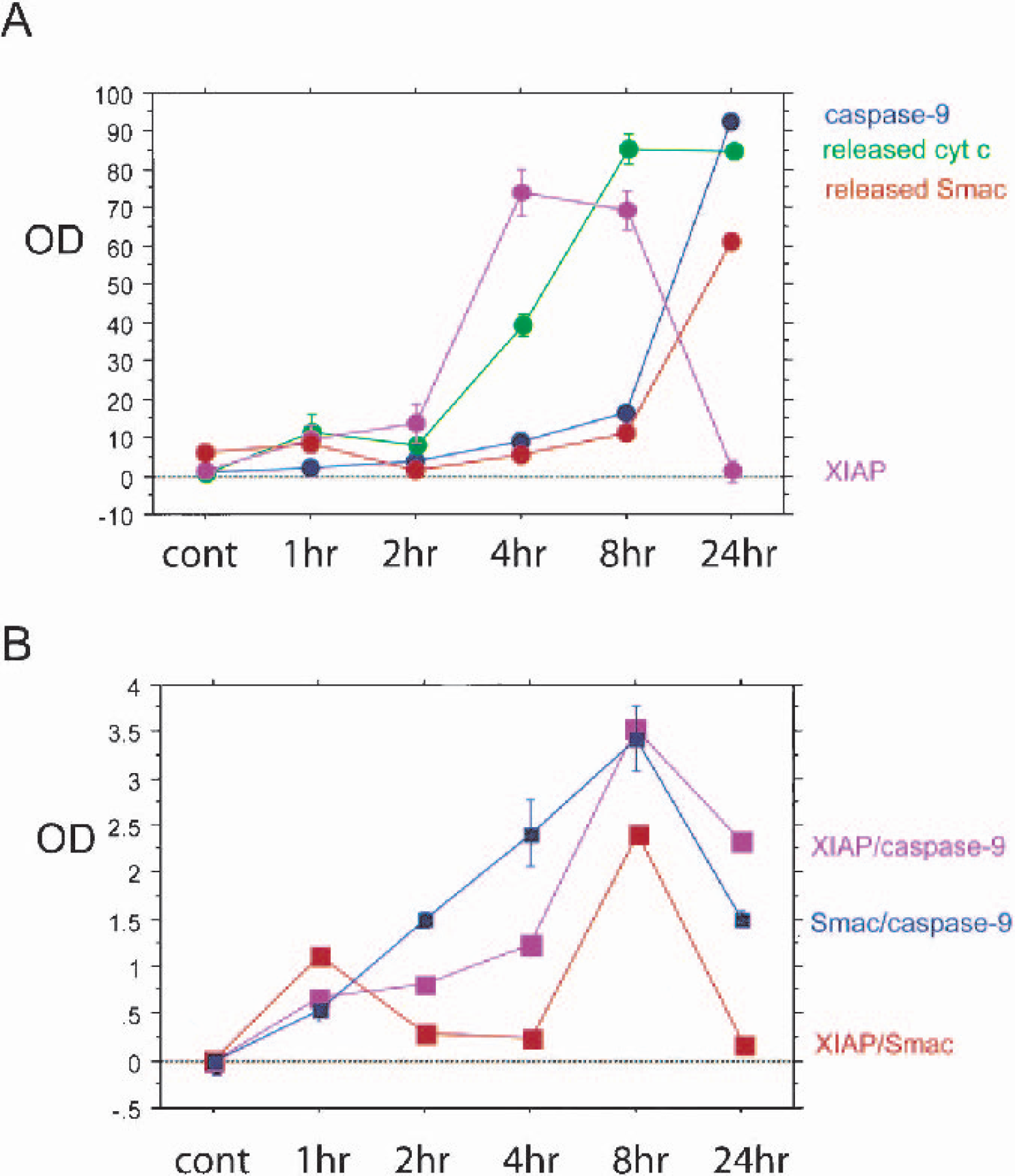
(
DISCUSSION
XIAP was expressed after transient focal cerebral ischemia
In the IAP family, XIAP is known as the most potent apoptotic inhibitor (Deveraux et al., 1999); however, little is known about its regulation in the CNS following cerebral ischemia. To our knowledge, there is only a limited number of reports about the XIAP machinery of in vivo CNS disorders: hypoxic-ischemia (Katz et al., 2001), traumatic brain injury (Keane et al., 2001), and transient cerebral ischemia (Shibata et al., 2002; Xu et al., 1999). In traumatic brain injury, XIAP was expressed in the nuclei and the cytosol of neural cells in the apoptotic lesion, and cytosolic immunoreactivity was increased time dependently before caspase activation (Keane et al., 2001). In the transient global cerebral ischemia model, adenovirally mediated XIAP overexpression reduced caspase-3 activation and apoptosis in hippocampal CA1 neurons (Xu et al., 1999). Katz et al. (2001), who evaluated the expression of XIAP in the hypoxic brain after asphyxial cardiac arrest by using immunohistochemistry, reported that immunoreactivity was observed 72 hours after reperfusion in CA1 and Purkinje cells of the cerebellum, where caspase-1 and 3 expression was also shown at the same time (Katz et al., 2001). XIAP expression in these vulnerable brain regions was reduced in animals treated with Z-VAD–FMK, a pan-caspase inhibitor (Katz et al., 2001). In tFCI, XIAP was induced in the ischemic cortical region and the time-dependent expression did not differ 2, 5, 11, or 23 hours after reperfusion (Shibata et al., 2002). In this study, we observed the peak expression of XIAP after reperfusion by examining the detailed time-dependent expressions. XIAP expression was observed in the ischemic lesion and was especially strong in the cortical penumbra 8 hours after reperfusion, according to the results of immunohistochemistry and Western blot analysis (Fig. 1). These results show that peak XIAP expression was observed during the early period of delayed neuronal cell death after ischemic injury. Moreover, XIAP increased concurrently with Smac/DIABLO release and the peak expression of activated caspase-9. In our model, XIAP activation was not affected by a caspase chain reaction. The transient upregulation of XIAP might play an important role in the inhibitory regulation of excessively rapid apoptotic neuronal cell death progression.
Smac/DIABLO was expressed and translocated after transient focal cerebral ischemia
Mitochondria are the major site of superoxide production under normal or pathologic conditions, such as tFCI (Piantadosi and Zhang, 1996). Excessive superoxide production may cause mitochondrial injury that leads to the release of proteins, such as cytochrome c and Smac/DIABLO, from their intermembrane space (Adrain et al., 2001; Madesh et al., 2002; Sugawara et al., 2002). Based largely on in vitro studies, the supposition is that Smac/DIABLO accumulation within the cytosol is readily detected in cells stimulated to die in response to diverse proapoptotic agents, including death receptor ligation, cytotoxic drugs, and DNA-damaging agents (Adrain et al., 2001). In experimental in vivo animal models, the machinery of Smac/DIABLO activation is not well known. We previously detected Smac/DIABLO expression in the hippocampal CA1 subregion of rat brains in a transient global cerebral ischemia model (Sugawara et al., 2002). In this previous study, immunoreactivity of Smac/DIABLO appeared from the early period after reperfusion and increased concomitantly with cytochrome c expression. The release of Smac/DIABLO to the cytosolic space was shown at upstream positions in effector caspase activation because it was not prevented by caspase-3–inhibitor administration. The subcellular localization of Smac/DIABLO remains unclear. We observed a slight nuclear immunoreactivity of Smac/DIABLO in the mouse brain after cerebral ischemia in this experiment (Fig. 2). We suggest the possibility that Smac/DIABLO may exist and function in nuclei after cerebral ischemia. In the current study, Smac/DIABLO was released from mitochondria to the cytosolic space during the early period of reperfusion injury, and this translocation was concurrent with cytochrome c release and the appearance of activated caspase-9 (Fig. 2). Activated caspase-9 regulates the initiation of the mitochondrial caspase chain reaction, and expression of released Smac/DIABLO was not prevented by the pan-caspase inhibition. This finding suggests that similar to cytochrome c activation, Smac/DIABLO might also regulate caspase initiation upstream of the caspase chain reaction. Smac/DIABLO might play a critical role in the early processes of the mitochondrial cell death pathway, which is followed by the caspase chain reaction.
Direct binding among XIAP, Smac/DIABLO, and caspase-9 was detected after transient focal cerebral ischemia
Concurrent with cytochrome c release, Smac/DIABLO is also released from the mitochondria into the cytosol, where it eliminates the inhibitory effect of many IAPs after direct binding to IAPs, including XIAP, and this phenomenon has been observed in a variety of in vitro experiments (Chai et al., 2000; Du et al., 2000). During mitochondrial import, the amino-terminal 55 residues derived from the N-terminus of Smac/DIABLO are removed to generate a mature form of the molecule (Chai et al., 2000; Du et al., 2000). Mature Smac/DIABLO exists as a dimer, mediated by a hydrophobic interface within the N-termini of individual Smac/DIABLO molecules (Chai et al., 2000). In the cytosol, a functionally active nine-residue peptide derived from the N-terminus of Smac/DIABLO binds BIR domains of XIAP to eliminate the inhibitory effect (Liu et al., 2000). The complex is stabilized by four intermolecular hydrogen bonds, an electrostatic interaction involving the N-terminus of the peptide, and several hydrophobic interactions (Liu et al., 2000). Interaction between caspase-9 and Apaf-1 to form the apoptosome occurs in response to cytochrome c release from the mitochondria (Zou et al., 1997). The apoptosome is assembled when seven Apaf-1:cytochrome c heterodimers oligomerize to form a symmetrical wheellike structure and procaspase-9 molecules become associated noncovalently to Apaf-1 via caspase-9 CARD/Apaf-1 CARD heterophilic interaction (Zou et al., 2003). The function of the apoptosome is to cleave and activate the effector caspases such as caspases-3, 6, and 7 (Srinivasula et al., 1998). Binding of procaspase-9 to Apaf-1 increases the intrinsic catalytic activity of the caspase-9 protease leading to the autolytic cleavage of the proform of caspase-9, yielding active subunits (Zou et al., 1999, 2003). The cleavage of procaspase-9 exposes an epitope comprising the NH2-terminal four amino acids of the p12 subunit, which has a crucial role in binding to the BIR3 domain of XIAP (Srinivasula et al., 2001; Zou et al, 2003). In in vivo experiments, the direct interaction of XIAP, Smac/DIABLO, and caspase-9 after apoptotic insults is not well known. In this study, we used the XIAP antibody, which is specific to the BIR3 domain and the RING motif. This antibody is appropriate for detecting the specific binding of XIAP to Smac/DIABLO and caspase-9.
We specifically detected, for the first time, the direct bindings of XIAP, Smac/DIABLO, and caspase-9 and the time-dependent expressions of these complexes after cerebral ischemia. Coimmunoprecipitation data revealed that these bindings exist in in vivo cerebral ischemia and that they occur simultaneously during the early period of reperfusion injury after cerebral ischemia. We suggest that XIAP might play an important inhibitory role in the initiation of the mitochondrial caspase pathway via the binding to caspase-9, and that XIAP might be inhibited via the binding to Smac/DIABLO simultaneously after cerebral ischemia.
The rapid increase of released Smac/DIABLO might follow cytochrome c release and it might promote the proapoptotic cascade by overcoming the inhibitory reaction of XIAP via the binding to XIAP. There is a discrepancy between the individual expression of released Smac/DIABLO and activated caspase-9 expression and that of XIAP/Smac after its peak. Expression of XIAP/Smac might be strongly affected by the temporal XIAP increase to inhibit the proapoptotic reaction. In contrast, the proapoptotic regulators such as released cytochrome c, released Smac/DIABLO, and activated caspase-9 might continuously accumulate in the cytosolic space after mitochondrial oxidative stress caused by reperfusion injury. Moreover, there is evidence that Smac/DIABLO functions not only at the level of XIAP binding, but also directly with both the Apaf-1 apoptosome and the effector caspases after caspase initiation (Srinivasula et al., 2000). There is a possibility that Smac/DIABLO also might have a variety of important roles in the progress of the caspase reaction after binding to IAPs in reperfusion injury after tFCI.
The peak expression of XIAP/caspase-9 was also in accord with the peak expression of XIAP, and these expressions were observed before the peak expression of activated caspase-9. We suggest that XIAP might inhibit the progression of the proapoptotic cascade before the initiation of the cascade reaction via direct binding to caspase-9. Both released Smac/DIABLO and activated caspase-9, which are key factors for the mitochondrial proapoptotic pathway after tFCI, might be controlled by XIAP before they accumulate in the cytosol to the maximum level, and their direct binding to XIAP might be strongly affected by XIAP expression. The XIAP pathway might be a key factor in regulating the balance between progression and inhibition of the mitochondrial caspase pathway.
CONCLUSION
Our results show that the XIAP pathway was mediated by released Smac/DIABLO and activated caspase-9 in mouse brains after tFCI. The interaction among XIAP, Smac/DIABLO, and caspase-9 may play a critical role in the regulation of apoptotic neuronal cell death after cerebral ischemia. We suggest that this endogenous signaling pathway in neuronal cell death warrants further elucidation and possible therapeutic consideration in cerebral ischemia.
Footnotes
Acknowledgments:
The authors thank Dr. Charles J. Epstein (Department of Pediatrics, University of California at San Francisco School of Medicine), for the breeding pairs of SOD1 transgenic mice. They also thank Cheryl Christensen for editorial assistance, Liza Reola and Bernard Calagui for technical assistance, and Elizabeth Hoyte for figure preparation.