
Abstract
Our previous studies indicate that apoptosis in endothelial cells of major cerebral arteries contributes to cerebral vasospasm after subarachnoid hemorrhage (SAH). This study examined the pathologic roles of tumor suppressor p-53 in cerebral vasospasm using an established dog double-hemorrhage model. Twenty mongrel dogs were divided into four groups: (1) control, (2) SAH, (3) SAH+DMSO (vehicle), and (4) SAH+pifithrin-α (PFT) (p53 inhibitor). The p53 inhibitor (200 nmol/L) was injected into the cisterna magna daily from Day 0 through Day 3. Angiogram was performed on Day 0 and Day 7. Western blot, cell proliferation assay, histology, and TUNEL staining were conducted on the basilar arteries collected on Day 7 after SAH. The arterial diameter on Day 7 was 42%±4%, 40%±5%, and 59%±4% for SAH, SAH+DMSO, and SAH+PFT, respectively. In addition, positive staining of TUNEL and increased protein expression of p53, Bax, and PCNA in the basilar artery were observed on Day 7. PFT suppressed apoptosis in endothelial cells and proliferation in smooth muscle cells, and attenuated angiographic vasospasm. In conclusion, p53 may be a key factor in endothelial apoptosis and smooth muscle proliferation after SAH. Inhibition of p53 may potentially reduce or even prevent cerebral vasospasm.
Introduction
Two major vascular wall structural changes have been reported in cerebral vasospasm: endothelial apoptosis and smooth muscle proliferation (Findlay et al, 1991; Mayberg, 1998; Zhang et al, 1998; Dumont et al, 2003). Apoptosis in cerebral endothelial cells has been established in vitro (Ogihara et al, 1999; Meguro et al, 2000, 2001), in vivo (Zubkov et al, 2000b, 2002), in patients (Zubkov et al, 2000a), and caspase inhibitors have reduced cerebral vasospasm (Zhou et al, 2004). In the mean time, vascular remodeling occurs in the spastic vessels (Borel et al, 2003), probably promoted by endothelial apoptosis and smooth muscle proliferation (Li et al, 1997; Malik et al, 1998; Durand et al, 2002). Smooth muscle proliferation contributes to arterial wall thickening and vascular stiffening and enhances cerebral vasospasm (Borel et al, 2003). Thus, both apoptosis and proliferation are involved in cerebral vasospasm.
The tumor suppressor p53 is a transcription factor that plays an essential role in the death of many cell types by stimulating synthesis and mitochondrial translocation of Bax, resulting in cytochrome c release and apoptosis. P53 was also reported to regulate proliferation of smooth muscle cells (Bennett et al, 1998; Hsieh et al, 2000), because transferring of wild-type p53 gene inhibited smooth muscle proliferation (Yonemitsu et al, 1998). A chemical inhibitor of p53, pifithrin-α (PFT), selectively inhibits p53 transcriptional activity in various cell lines and prevents DNA damage-induced apoptosis (Komarov et al, 1999). Pifithrin-α protects brain tissue against ischemic damage by decreasing p53 DNA binding and reducing expression of the p53 target gene Bax, preserves mitochondrial functions, and blocks activation of caspase-3 (Culmsee et al, 2001, 2003; Leker et al, 2004). Pifithrin-α inhibits both p53 transactivation activity and downstream events including the upregulation of the proapoptotic protein Bax and the activation of caspases in vascular endothelial cells (Lorenzo et al, 2002). In this study, we examined the hypothesis that p53 is involved in endothelial apoptosis and smooth muscle proliferation in the basilar artery after experimental subarachnoid hemorrhage (SAH) and that PFT attenuates vasospasm.
Materials and methods
All experiments were performed according to the Rules of Animal Experimentation and Guide for the Care and Use of Laboratory Animals of Louisiana State University.
Twenty adult mongrel dogs (either sex, weighing 17 to 20 kg) were used. Dogs were randomly assigned to four groups: (1) normal control (without SAH, n=2); (2) SAH (without treatment, n=6); (3) SAH treated with vehicle of dimethyl sulfoxide (DMSO) (n=6); and (4) SAH treated with PFT (n=6). Dimethyl sulfoxide and PFT were injected into the cisterna magna daily, beginning on Day 0 and ending on Day 3. This study was semimasked, meaning that the investigators knew the normal and SAH groups because since no treatment was applied, but they did not know the content of treatment in the two treated groups (DMSO or PFT, prepared and recorded by a technician).
Experimental Dog Double-Hemorrhage Model
Dogs were preanesthetized with acepromanize (0.1 to 0.5 mg/kg), atropine (0.05 mg/kg), and xylazine (1.1 mg/kg), and anesthesia was maintained by isoflurane 1.25%, O2 2L/min after being mechanically ventilated. The body temperature of the dogs was kept at 37°C with a heating blanket. The mean arterial blood pressure and blood gas levels were monitored through a catheter inserted into the femoral artery. The flow of isoflurane was adjusted during surgery according to the values of the mean arterial blood pressure and blood gas and to control the depth of anesthesia. Subarachnoid hemorrhage was induced according to the method described in our previous publications (Kusaka et al, 2003; Zhou et al, 2004). For cerebral angiogram, the left vertebral artery was catheterized with a No. 4 French catheter via the right femoral artery. A baseline vertebro-basilar artery angiogram was obtained. The cisterna magna was punctured transcutaneously, and 0.4 mL/kg of cerebrospinal fluid (CSF) was withdrawn. An equal amount of arterial blood was withdrawn from the femoral artery and immediately injected into the cisterna magna within 1 min to avoid possible blood clotting in the syringe. The dogs were then tilted at a 20° for 20 mins with their heads down, in a prone position, to permit pooling of blood around the basilar artery.
The first blood injection was considered to be Day 0. On Day 2, the same volume of blood was injected under identical anesthesia conditions (with similar mean arterial blood pressure and blood gas levels) without performing angiography. The angiogram was repeated on Day 7 before all dogs were killed.
P53 Inhibitors (Pifithrin-α)
Pifithrin-α, a p53 inhibitor, was purchased from BIOMOL Inc. It is a small, stable, and lipophilic molecule that has an octanol/water partition coefficient, which predicts a high permeability at the blood–brain barrier (Komarov et al, 1999). The dosage of the inhibitor was individually calculated for each dog to reach similar drug levels in the dogs' CSF, taking into account the relative size of the CSF space to obtain the final concentration in the CSF, assuming that canine CSF volume is 2 mL/kg (Zoghbi et al, 1985). Pifithrin-α was diluted in DMSO (Culmsee et al, 2003). One milliliter of CSF was withdrawn and mixed with the PFT (about 200 μL). The inhibitor containing CSF was intracisternally injected within 30 secs to obtain a final concentration of PFT in the CSF of 200 nmol/L (Culmsee et al, 2001). The first injection was administered at 30 mins after the first blood injection and was repeated daily for an additional 3 days.
Measurements of Arterial Diameter
Arterial diameters were measured in a double-blind fashion on magnified angiograms (Satoh et al, 2002; Kusaka et al, 2003; Zhou et al, 2004). To eliminate magnification differences on the angiograms, a radio detectable scale was placed on the dog's chin during the angiogram run. The same scale was used consistently and was put at the same point on each dog's chin. Relative to the size of this scale as a standard, all arterial diametric values were adjusted. Two researchers (Drs Zhou and Yamaguchi) independently measured the arterial diameters on the magnified angiograms at three points: the distal (just before the bifurcating superior cerebellar arteries), the proximal (just after basilar union), and the central (the midpoint between the previous two points) portions of the basilar artery. The mean of these three measurements was calculated to yield the arterial diameter. The mean of the values measured by the two researchers was taken as the final diameter of the basilar artery. The caliber of the basilar artery on Day 7 was calculated as a percentage of the mean basilar artery diameter on Day 0 before the blood injection in each dog.
Clinical Assessment
Clinical scores were recorded based on the independent observations of a veterinarian who was masked to the study, and one of the researchers (Dr Zhou). The scores were recorded daily. Three clinical observations were recorded and scores were given (Zhou et al, 2004):
Appetite: Finished meal=0; left meal unfinished=1; scarcely ate=2.
Activity: Active, barking, or standing=0; lying down, will stand and walk with some stimulation=1; almost always lying down=2.
Neurological deficit: No deficits=0; unstable walk because of ataxia or paresis=1; impossible to walk and stand because of ataxia or paresis=2.
Morphological Assessment
After being euthanized with an overdose of pentobarbital (150 mg/kg), the dogs were perfused with 200 mL of 0.1 mol/L PBS and then 500 mL of 4% paraformaldehyde in 0.1 mol/L PBS (pH 7.4) via both common carotid arteries. The brain was removed and postfixed with the same fixative solution (Zhou et al, 2004). The basilar artery was sectioned at 6 μm thick by a cryostat (Leica CM3050 S). A series of sections were obtained from dogs in the normal control, SAH, SAH+DMSO, and SAH+PFT groups, and were divided into several subgroups for H&E staining, immunohistochemistry, TUNEL staining, and treble fluorescence staining, respectively. They were observed under an OLYMPUS BX51 microscope.
H&E staining:
Sections were stained in hematoxylin for 2 mins and eosin for 1 min. They were then dehydrated and coverslipped with permount as described previously (Zhou et al, 2004).
TUNEL staining:
Sections were stained by TUNEL staining Kit (Roche Inc., Basel, Switzerland), the TUNEL-positive cells were expressed by fluorescein-dUTP with dNTP or POD with 3–3′ diaminobenzidine (DAB) (manufacturer's protocol for the in situ Apoptosis Detection Kit [Roche Inc.]) as described previously (Zhou et al, 2004).
Immunohistochemistry staining:
The methods for immunohistochemical staining have been described previously (Zhou et al, 2004; Sun et al, 2004). Four series of sections were incubated in 3% hydrogen peroxide (H2O2) diluted in PBS (10 mins) to prevent reaction with endogenous peroxidases. After 30 mins in 3% normal serum in PBS, the sections were incubated with primary antibodies overnight at 4°C. The following primary antibodies were purchased from Santa Cruz Inc., California USA: (1) rabbit polyclonal anti-p53 (FL-393), 1:200; (2) goat polyclonal anti-p53AIP1 (G-20, p53 regulated apoptosis-inducing protein), 1:200; (3) rabbit polyclonal anti-cytochrome c (C-20), 1:200; (4) goat polyclonal anti-PCNA (C-20, proliferating cell nuclear antigen), 1:200, respectively. After rinsing them with PBS, the sections were treated with a rabbit (or goat) ABC kit (Santa Cruz Inc., CA, USA). Sections were then incubated with goat anti-rabbit (or donkey anti-goat) IgG as secondary antibody (1:200) for 30 mins and were placed in avidin–peroxidase complex solution containing avidin–peroxidase conjugate for 30 mins. Peroxidase activity was revealed by dipping the sections for 5 mins in a mixture containing DAB and H2O2 (ABC kit, Santa Cruz Inc.) at room temperature. Sections were air-dried, dehydrated, and coverslipped with permount. Application of control serum, instead of the primary antibody, on another section of the same artery provided a negative control for each staining.
Treble fluorescence staining:
The methods for double-fluorescence labeling have been described previously (Zhou et al, 2004; Sun et al, 2004). For treble labeling, sections from SAH dogs were used. After TUNEL fluorescence staining (green, fluorescein dUTP and dNTP Kit, Roche Inc., protocol), the sections were performed immunoflouorescence staining with primary antibodies, rabbit anti-p53, 1:200 and goat anti-PCNA, 1:200 overnight at 4°C, after goat anti-rabbit IgG Texas Red 1:200 (Santa Cruz Inc.), and donkey anti-goat IgG-AMCA (blue aminomethylcoumarin acetate) 1:200 (Jackson Immuno Research Inc., Pennsylvania, USA) second antibodies, respectively. Sections were coverslipped with 30% glycerin and observed under an OLYMPUS BX51 fluoresce microscope. The TUNEL green fluorescein absorption is at 492 nm and was emitted at 520 nm. The absorption of Texas Red is 596 nm, and the emission peak is at 620 nm. The absorption of AMCA blue flourescence is 350 nm, and the emission peak is at 450 nm.
Western Blotting Analysis
The method for Western blot in canine basilar arteries has been described previously (Kusaka et al, 2003; Sun et al, 2004). The frozen arteries were homogenized for 20 min at 4°C with an ultrasonic wave (10 sec, 3 times) in 200 μL of an extraction buffer containing 50 mmol/L Tris-HCl (pH 7.6); 1% nonylphenol ethoxylate (Igepal); 0.25% sodium deoxycholate; 150 mmol/L NaCl; 1 mmol/L EGTA; 1 mmol/L phenylmethylsulfonyl fluoride; 1 μg/mL aprotinin, leupeptin, pepstatin; 1 mmol/L Na3VO4; and 1 mmol/L NaF. The insoluble material was removed by centrifugation at 16 000g at 4°C for 15 mins. The samples (20 μg protein) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis with 10% polyacrylamide gel. After electrophoretic transfer of the separated polypeptides to nitrocellulose membranes, the membranes were blocked with 5% nonfat milk in Tween-TBS (TBST). The membranes were then washed and incubated with the primary antibodies 1:400 with 1% nonfat milk in TBST at 4°C overnight.
The following primary antibodies were used: goat polyclonal anti-TNF-α, rabbit polyclonal anti-p53 antibody, mouse molyclonal anti-Bax, rabbit polyclonal anti-Bcl2, goat polyclonal anti-PCNA, and rabbit polyclonal anti-β actin antibody (Santa Cruz Inc.). After incubation with the primary antibodies, the nitrocellulose membranes were washed with TBST and incubated with the appropriate horseradish peroxidase-labeled secondary antibodies 1:800 (Santa Cruz Inc.) using 1% nonfat milk in TBST for 1 h at room temperature. An enhanced chemiluminescence system (Amersham) was used to visualize the protein bands. The results were quantified by Quantity One Software (Biorad).
Data Analysis
Data are expressed as mean±s.d. Statistical differences between the control and other groups were compared by one-way ANOVA and then the Tukey–Kramer multiple comparison procedure, if a significant difference had been determined by ANOVA. The clinical behavior scores were compared by Kruskal–Wallis one-way ANOVA on ranks, and then, if significant differences were found, the t-test procedure was used. A probability value of P<0.05 was considered statistically significant.
Results
Angiography
Severe angiographic vasospasm was observed in the basilar arteries in the SAH and SAH+DMSO groups, and moderate vasospasm was observed in the group of dogs treated with PFT (Figure 1A). The mean residual diameters of the basilar arteries on Day 7, as a percentage of that on Day 0 before the blood injection, were SAH, 42.02%±4.34%; SAH+DMSO, 40.54%±5.09%; and SAH+PFT, 59.81%±4.92% (Figure 1B). The diameters of the basilar arteries in the PFT-treated group were significantly larger than those of the SAH and SAH+DMSO groups (P<0.05, ANOVA). No statistical difference was noted between the SAH and SAH+DMSO groups (P>0.05, ANOVA, Figure 1B).
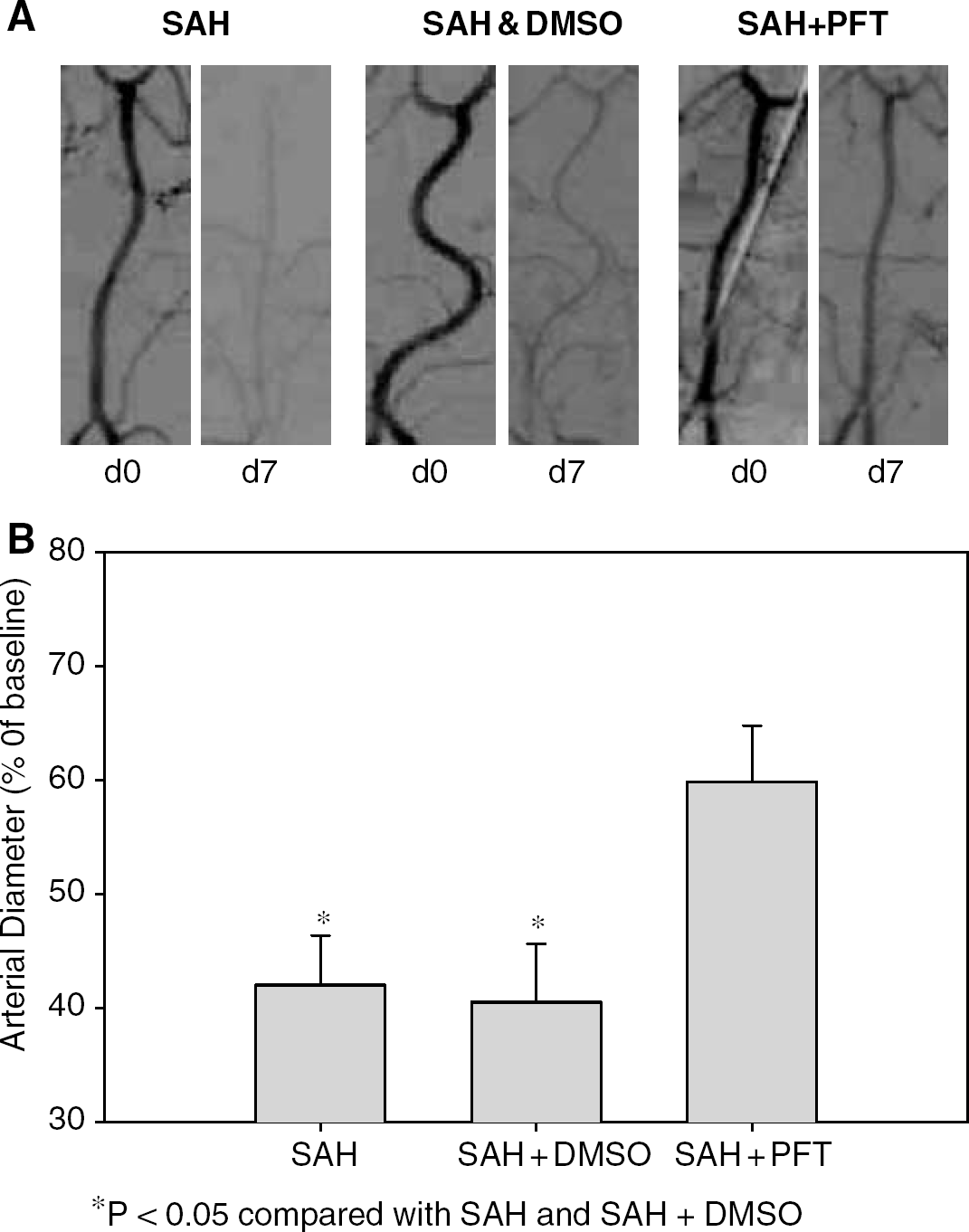
Representative angiograms and summary of diameters. (
Clinical Behavior Scales
The clinical behavior scales on appetite, activity, and neurologic deficit of the three groups of dogs are shown in Figure 2. There was no significant difference in the first 3 days in appetite and activity, and in all 7 days in neurologic deficit among the three groups. The appetite in PFT treatment group was significantly better than the SAH groups from Day 4 to Day 7 (P<0.05, Figure 2A) and better than the SAH+DMSO groups at Day 5. There is no significant difference when the SAH group was compared with the SAH+DMSO group (P>0.05, Figure 2A). The activity of the dogs in the PFT treatment group was significantly better than those in the SAH+DMSO groups at Day 4, Day 6, and Day 7 and SAH at Day 4 (P<0.05, Figure 2B). Most dogs did not have neurologic deficits, and no significant difference was observed among the groups (P>0.05, Figure 2C).
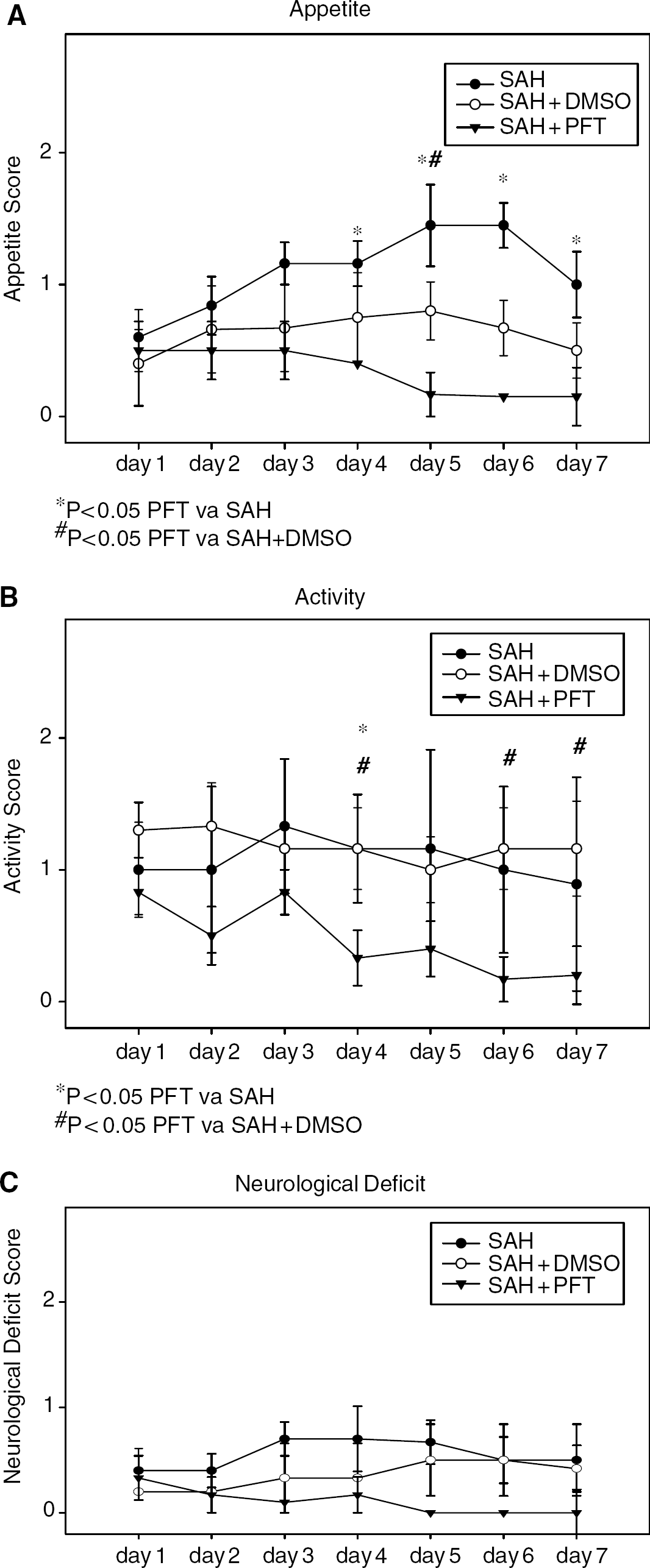
Clinical behavior scales on appetite, activity, and neurologic deficit. (
Morphologic Assessment
Morphologic assessment was peformed on samples from the basilar arteries at the pons with H&E, TUNEL, and immunohistochemistry staining.
H&E staining:
No blood clots were observed around the basilar artery in the normal control dogs (Figure 3A1). Subarachnoid blood clots (black ‘star’ signs) were found under the arachnoid membrane at pons and medulla in all three SAH groups (Figures 3B1, 3C1, 3D1). Severe arterial contraction was observed in the SAH and SAH+DMSO groups based on the histologic observation, which was named as ‘histological vasospasm’, and is characterized by a decreased vessel lumen, a thickened vessel wall, and a corrugated internal elastic lamina (IEL) (Figures 3B1, 3B2, 3C1, 3C2), contracted smooth muscle cells, and a shrunken endothelium, some of which were desquamated to reveal a denuded IEL (Figures 3B2, 3C2). However, in the PFT-treated group, histological vasospasm was not obviously observed, and arterial rings showed a bigger lumen, thinner vessel wall, smooth IEL, normal shaped endothelial cells, and elongated smooth muscle cells (Figures 3D1, 3D2), despite the same volume of blood in the subarachnoid space.
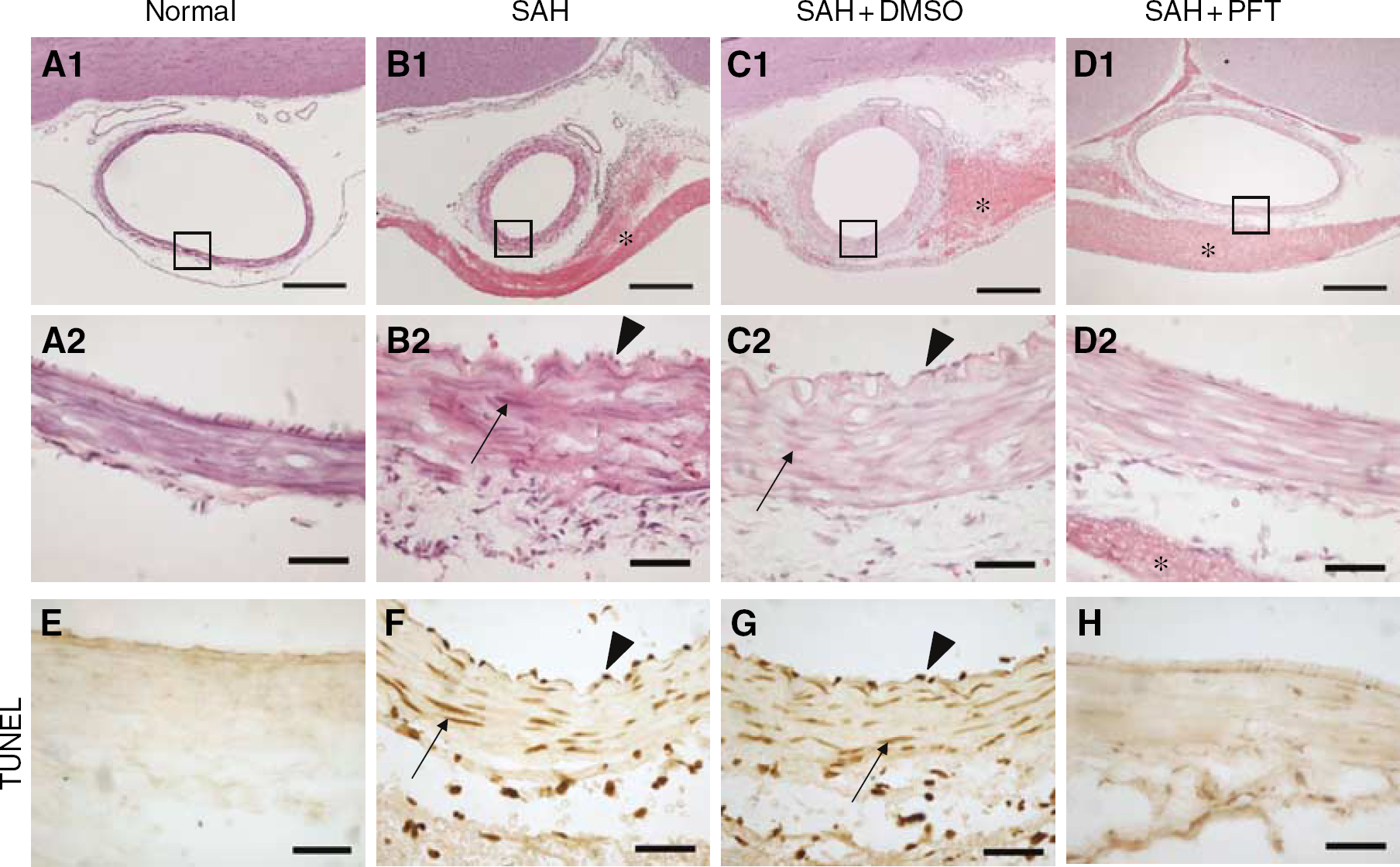
H&E staining and TUNEL staining of the basilar artery transect sections on the surface of pons. (
TUNEL staining:
The TUNEL-positive material was clearly observed in the nucleus of the endothelial cells and vascular smooth muscle cells in the SAH and SAH+DMSO groups (Figures 3F, 3G). The TUNEL-positive endothelium was localized on the inner surface of the corrugated IEL. TUNEL-positive nuclei of smooth muscle cells were localized in the tunica media (Figures 3F, 3G). In the PFT-treated group, no positive TUNEL staining was obtained in endothelial or smooth muscle cells (Figure 3H), comparable to what was observed in samples from the normal control dogs (Figure 3E).
Immunohistochemistry staining:
Tumor necrssis factor-α (TNF-α) immunohistochemistry in the basilar artery is shown in Figure 4. The fibroblasts in the adventitia and endothelial cells expressed strong TNF-α immunoreactivity, while smooth muscle cells showed negative staining in the SAH and SAH+DMSO groups (Figures 4B, 4C). In PFT-treated animals, only the adventitia show a positive staining; the endothelial cells and smooth muscle cells show negative staining (Figure 4D), comparable to normal artery (Figure 4A).
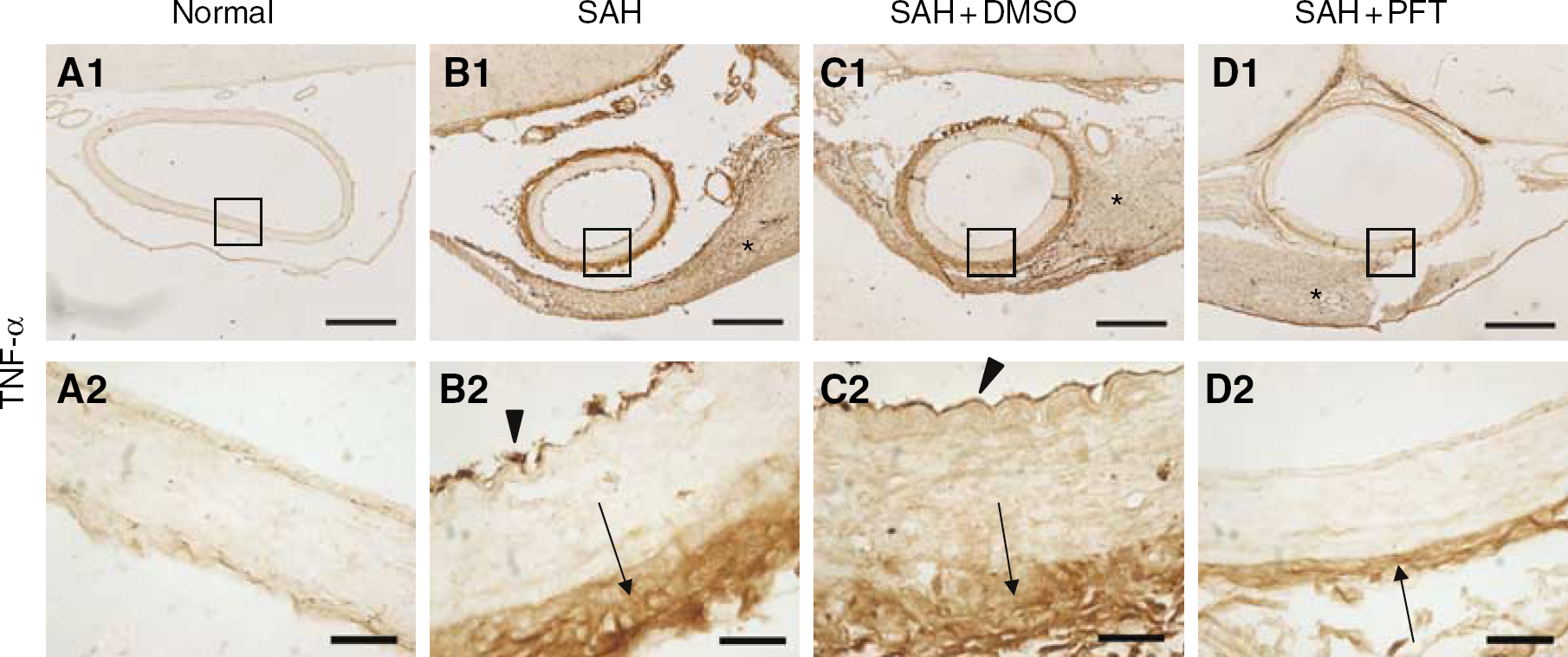
TNF-α immunohistochemistry in the basilar artery. (
Strong staining of p53, p53AIP-1 (p53 regulated apoptosis inducing protein 1), and cytochrome c was observed in endothelial and smooth muscle cells (Figures 5B, 5C, 5F, 5G, 5J, 5K). p53 staining was noted in the cytoplasm and nucleus.
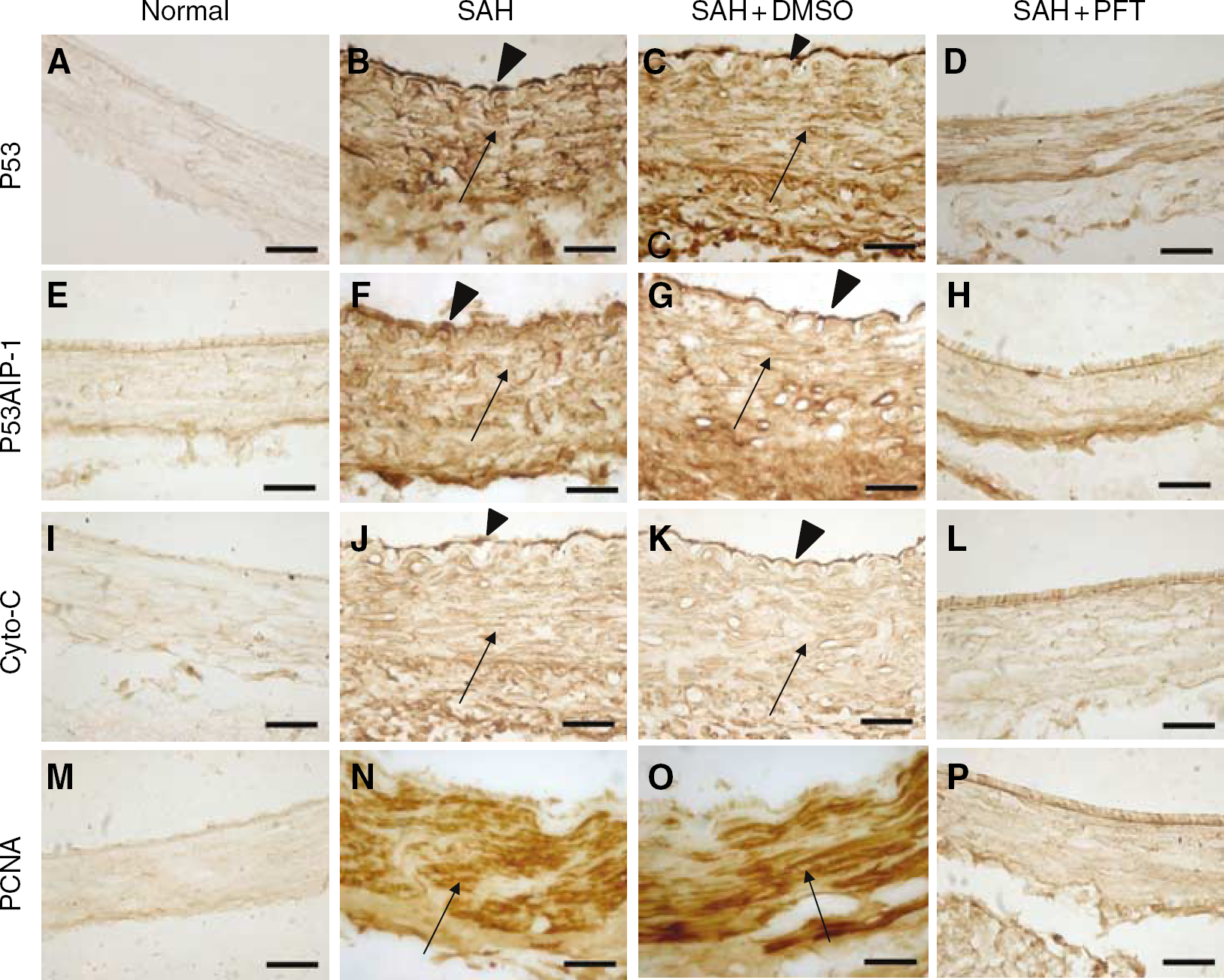
p53, p53AIP-1, cytochrome c, and PCNA immunohistochemistry in the basilar artery. P53
Proliferating cell nuclear antigen (PCNA), the marker of proliferation was clearly present in the nucleus of the smooth muscle cells in both the SAH and SAH+DMSO groups (Figures 5N, 5O), but expressed only mildly in the PFT-treated group (Figures 5D, 5H, 5L, 5P), comparable to the normal tissues (Figures 5A, 5E, 5I, 5M).
Treble fluorescence labeling is shown in Figure 6. The TUNEL-positive nuclei of endothelial and smooth muscle cells were expressed in green fluorescence color (Figure 6A), the p53 immunoreactive-positive endothelial and smooth muscle cells (cytoplasm and nucleus) were expressed by Texas Red (Figure 6B), and the PCNA immunoreactive positive nuclei of smooth muscle cells were expressed by AMCA blue (Figure 6C). Color merging shows that the nuclei of endothelial cells were colocalized by TUNEL and p53, and the nuclei of the smooth muscle cells were colocalized by the TUNEL, p53 and PCNA (Figures 6D–6F). In addition, p53 was also localized in the cytoplasm. It was noted the IEL was autofluorescent in each of the green, red and blue fluorescence (Figure 6).
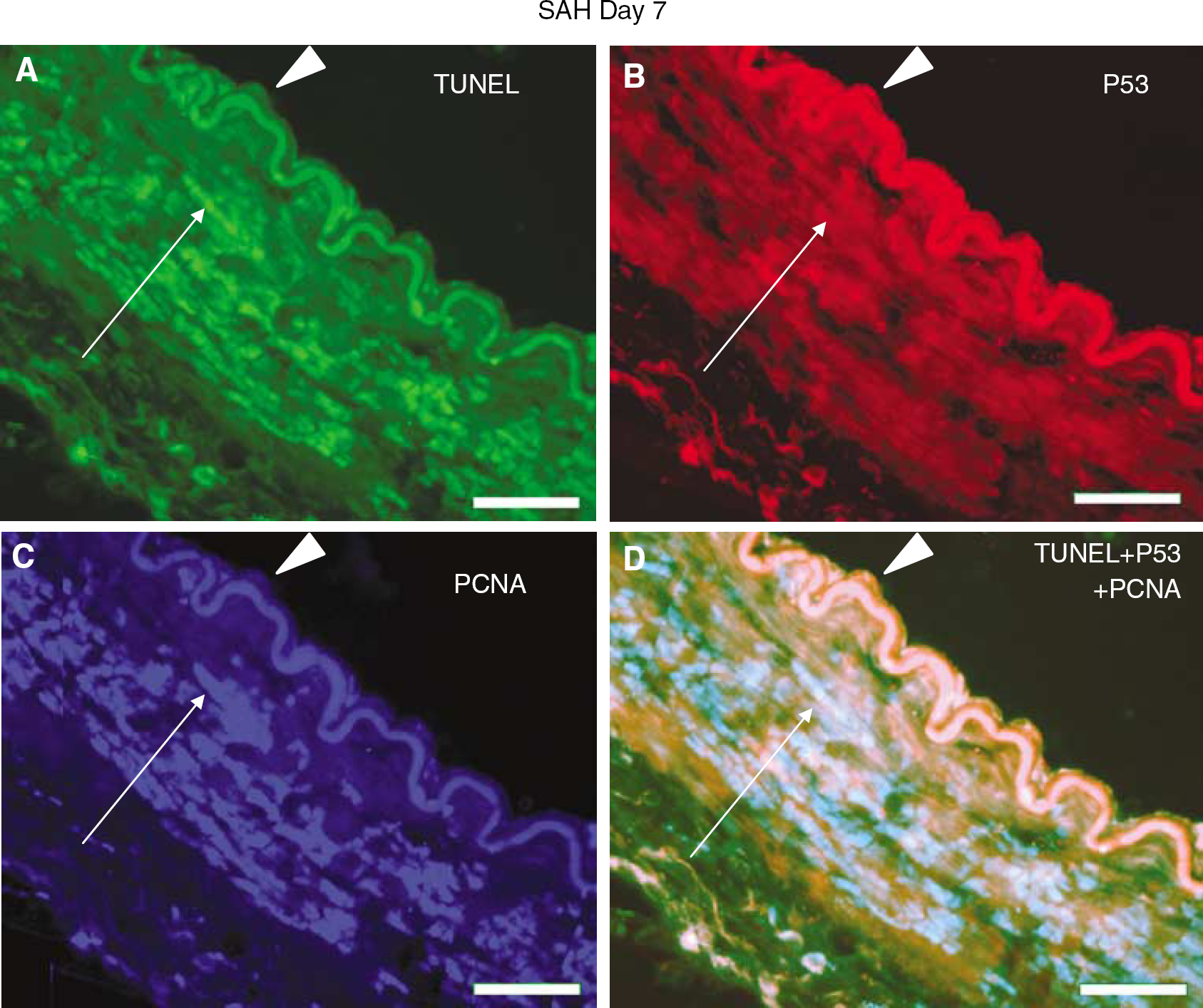
Treble fluorescence staining in a subarachnoid hemorrhage (SAH) basilar artery. (
Western Blot Analysis
The expressions of p53, Bax, Bcl-2, and PCNA in the basilar artery samples in control, SAH, SAH+DMSO, and SAH+PFT groups are shown in Figure 7. TNF-α expression in SAH, SAH+DMSO, and SAH+PFT were significantly higher than that of normal samples (P<0.05) and no significant differences in SAH, SAH+DMSO, and SAH+PFT were observed (Figure 7A). p53 expression in the SAH and SAH+DMSO groups was significantly higher than that of the normal group (P<0.05) and in the SAH+PFT treatment group was significantly lower than that of the SAH and SAH+DMSO groups (P<0.05, Figure 7B). The expression of Bax and Bcl2 in the basilar artery in different groups is shown in Figure 7C. Bax expression in the SAH and SAH+DMSO groups was significantly higher than that of the normal and SAH+PFT groups. Bcl2 expression in the SAH and SAH+DMSO groups was significantly lower than that of normal and SAH+PFT groups (P<0.05, Figure 7C). The expression of PCNA in the basilar artery in different groups is shown in Figure 7D. The quantitative analysis shows that PCNA expression in the SAH and SAH+DMSO groups was significantly higher than that of control, and the SHA+PFT treatment group were significantly lower than that of the SAH and SAH+DMSO groups (P<0.05).
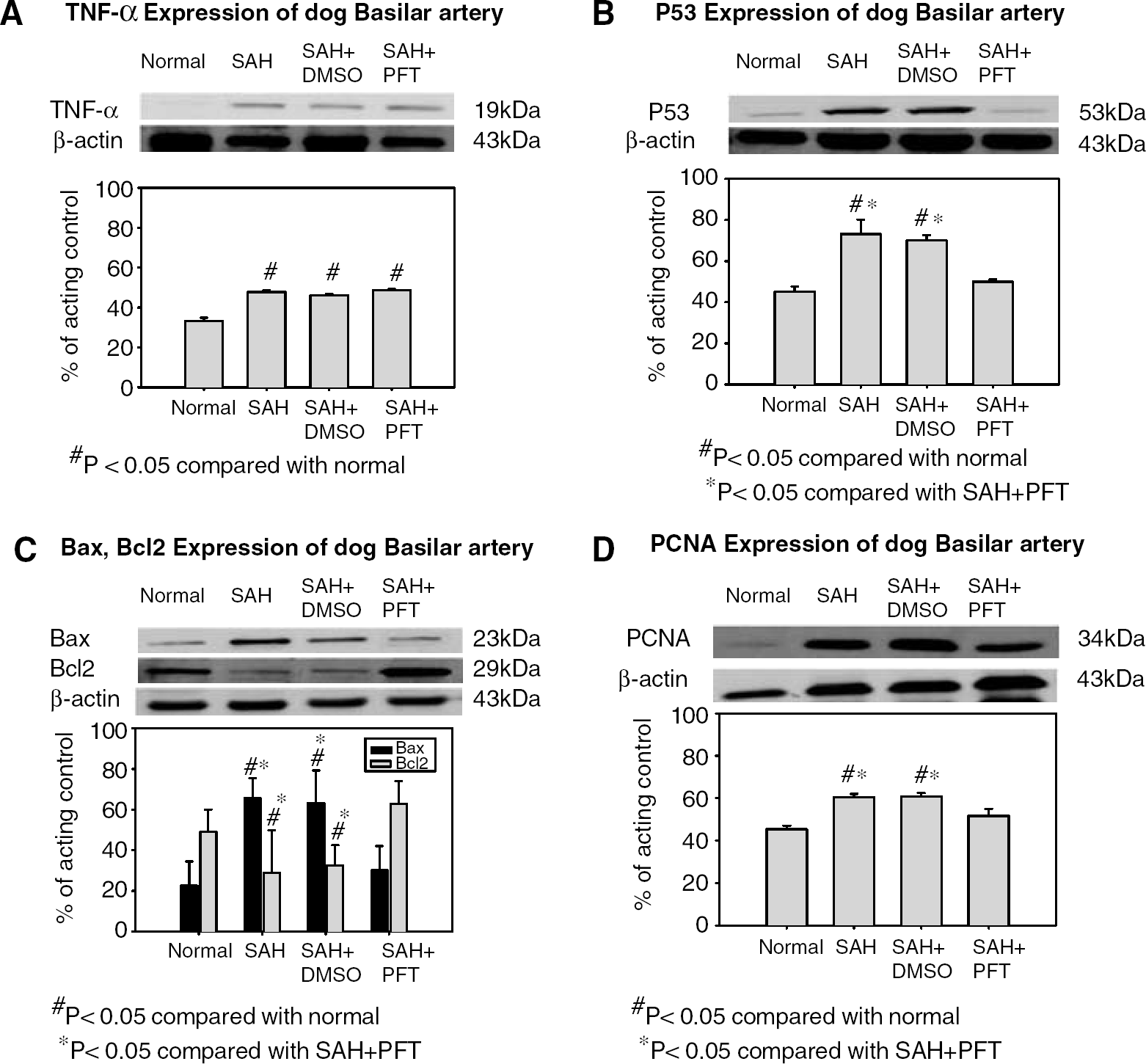
Western blot analysis and quantification. (
Discussion
There are two major observations in the present study. First, endothelial apoptosis and smooth muscle proliferation were noted at Day 7, the peak period of cerebral vasospasm after SAH in dogs. In addition, p53 appears involved as one of the central regulators in the apoptosis and proliferation, essential components for vascular remodeling. Second, a p53 inhibitor, PFT, significantly improved angiographic and histologic vasospasm by attenuating the apoptosis in endothelial cells and proliferation in smooth muscle cells.
Role of p53 in Endothelial Apoptosis
In one of our previous studies, we have shown that histologic vasospasm is accompanied by endothelial damage with features of apoptosis, and the signaling pathways for apoptosis after SAH in endothelial cells are mediated, at least partially, by TNFα-receptor-1, which recruits caspase-8, which in turn activates caspase-3 (Zhou et al, 2004). A broad caspase inhibitor Z-VAD-FMK and a selective caspase-3 inhibitor Ac-DEVD-CHO reduced apoptosis and cerebral vasospasm in this dog SAH model (Zhou et al, 2004).
Tumor suppressor protein p53 is activated in response to DNA damage or a wide range of stress stimuli. There is considerable evidence indicating that p53 is also one of the central regulators in TNF-α apoptotic pathways (Yeung and Lau, 1998; Fernandez-Salas et al, 1999; Sata et al, 2001; Kawauchi et al, 2002; Boyle et al, 2003). For instance, TNF-α contributes to macrophage-induced smooth muscle apoptosis (Boyle et al, 2003) and p53 is activated in TNF-α-induced apoptosis of human promonocytic cells (Yeung and Lau, 1998). This indicates that p53 may be involved in apoptosis of endothelial cells during cerebral vasospasm after SAH. In the present study, we observed that TNF-α was congregated with high consistency in the fibroblasts in the adventitia, and in the endothelial intima, even though endothelial cells do not have direct contact with blood clot in subarachnoid space (Figures 4B, 4C). This may indicate that the expression of TNF-α may not be a direct result of blood clot action. In the PFT group, the expression of TNF-α in the fibroblasts in the adventitia remained, but the endothelial TNF-α expression was decreased. All these results suggest that endothelial cells generate TNF-α after SAH, because TNF-α could not pass the layers of smooth muscle cells that remained negative to TNF-α (Figures 4B, 4C). Even though p53 inhibitor may not interfere with the generation of endothelial TNF-α, PFT reduces p53 and apoptosis in endothelial cells, which may interrupt the signals for the circle of generation of TNF-α in the endothelial cells.
The death signaling pathways, involving p53, p53AIP1, and cytochrome c in endothelial cells at Day 7 of SAH, were identified by immunohistochemistry (Figures 4B, 4C). p53AIP1 (p53 regulated apoptosis inducing protein 1) expression is strictly controlled by p53 under specific conditions and is inducible by p53 (Oda et al, 2000). In addition, p53AIP1 is ectopically expressed within the mitochondria, reflecting a key role in the regulation of apoptosis (Li et al, 1999). Cytochrome c is a necessary factor for the activation of apoptosis, which translocates from the mitochondrial membrane to the cytosol, where it is required for the activation of caspase-3, and induction of caspase-dependent pathways (Liu et al, 1996). The mitochondria has been considered to be one of the most important organelles in the signaling process of apoptosis for its role in the regulation and amplification of apoptosis signals (Newmeyer and Ferguson-Miller, 2003). After PFT treatment, the immunohistochemical expression of p53, p53AIP, and cytochrome c in endothelial cells was attenuated (Figures 4D, 4H, 4L). This information indicates that, separate from the TNF-α and caspase-8 molecular pathway as shown previously by us (Zhou et al, 2004), TNF-α-induced apoptosis in cerebral endothelial cell involves, at least partially, the mitochondria pathway with p53 as one of the central regulators after SAH.
Role of p53 in Smooth Muscle Proliferation
Proliferation of smooth muscle cells has long been considered to be a key event in remodeling of the injured vascular wall, including cerebral vasospasm after SAH. Smooth muscle proliferation is a factor in the maintenance of vasospasm characterized by intimal thickening and wall stiffness (Borel et al, 2003). While the mechanisms regulating smooth muscle cell proliferation remain largely unknown, contraction, migration, and apoptosis may all be involved (Hetts, 1998; Hsieh et al, 2000). The balance between cell apoptosis and proliferation of the different cellular constituents of the vascular wall plays an important role in vascular remodeling. Recently, an in vitro study showed that apoptosis of endothelial cells induces the release of antiapoptotic mediator(s) active on smooth muscle cells, which inhibit apoptosis of smooth muscle cells (Raymond et al, 2004). Apoptosis in smooth muscle cells was found in atherosclerosis (Han et al, 1995; Isner et al, 1995), restenotic lesions (Perlman et al, 1997; Pollman et al, 1999), hypertension (Sharifi and Schiffrin, 1998; Diep et al, 1999), oxidative stress (Barbouti et al, 2002; Curtin et al, 2002), and stroke (Li et al, 2003). It was reported that apoptosis within the media, adventitia, and neointima peaked at 18 h to 7 days after percutaneous transluminal coronary angioplasty, and the PCNA staining peaked at 3 days after percutaneous transluminal coronary angioplasty (Malik et al, 1998). It was believed the apoptosis and proliferation occurred at the same time during 3 to 7 days.
It appears that the function of P53 is multiple; besides the regulation of apoptosis, it influences cell proliferation and cell cycle control mechanisms (Chene, 2001). There is considerable evidence indicating that p53 plays an important role in the proliferation of smooth muscle cells (Hsieh et al, 2000). For example, transfer of the wild-type p53 gene effectively inhibits smooth muscle proliferation in vitro and in vivo (Yonemitsu et al, 1998). p53 induces smooth muscle proliferation in restenotic human coronary arteries after percutaneous transluminal coronary angioplasty (Speir et al, 1994). p53 plays an important role in the endothelial nitric oxide synthase gene transfer for inhibition of smooth muscle proliferation (D'Souza et al, 2003).
We have previously observed apoptotic-like changes in smooth muscle cells in the spastic basilar artery by TUNEL staining (Zhou et al, 2004). In the present study, we reconfirmed apoptosis of smooth muscle cells in the basilar artery after SAH by TUNEL fluorescent staining (Figures 3F, 3G and 6A), immunohistochemistry staining of p53, p53AIP-1a, cytochrome c (Figures 5B, 5C, 5F, 5G, 5J, 5K), and by the Western blot analysis of protein expression of p53, Bax, and Bcl2 (Figures 7B, 7C). These results imply that apoptosis occurred in smooth muscle layers partly via mitochondrial apoptotic pathways. In addition, at Day 7 after SAH, both immunohistochemical staining and Western blot analysis show the overexpression of PCNA in samples from SAH and SAH+DMSO (Figures 5N, 5O, 6C and 7D). The treble fluorescence labeling imaging showed that TUNEL-positive nuclei were superimposed by the PCNA and p53 immunofluorescence, which strongly emphasized the occurrence of apoptosis and proliferation simultaneously at Day 7 after SAH. Pifithrin-α not only reduced endothelial apoptosis but also inhibited smooth muscle proliferation.
Role of P53 in Cerebral Vasospasm
Pifithrin-α is a novel p53 inhibitor and has potential in the treatment of neurological disorders, including stroke (Culmsee et al, 2001; Leker et al, 2004). Pifithrin-α is a small, stable and lipophilic molecule that predicts a high permeability at the blood–brain barrier, and allows systemic administration, an ideal approach in clinical settings. In the present study, PFT was injected into cisterna magna at concentration of 200 nmol/L, because higher concentrations (>500 nmol/L) were found to be toxic in cultured neurons (Culmsee et al, 2001). Cisterna magna injection allowed us to use a small amount of PFT to reach the therapeutic level. Pifithrin-α treatment resulted in the reduction of the expression of p53 and its downstream effectors, including p53AIP-1 and cytochrome c (Figures 5D, 5H, 5L and 7C) as well as PCNA (proliferation). In addition, PFT enhanced expression of Bcl2. These molecular changes may lead to the prevention of angiographic and histologic vasospasm. In conclusion, endothelial apoptosis and smooth muscle proliferation may contribute to the development of cerebral vasospasm. P53 may be an important regulator of these pathologic processes and inhibition of p53 may have potential in the treatment of vasospasm.