
Abstract
Cytokine signaling through leukemia inhibitory factor receptor (LIFR)/gp130 is known to exert a neurotrophic action in the central nervous system, although the role of this signaling in cerebral ischemia remains unknown. We examined the effect of intracerebral injection of LIF after focal cerebral ischemia in rats. The animals underwent a sham operation (sham group) or middle cerebral artery occlusion (MCAO) followed by direct injection of either vehicle (phosphate-buffered saline, the PBS group) or recombinant LIF (10 ng in the low-LIF group and 100 ng in the high-LIF group) into the cerebral cortex adjacent to the inner boundary zone of the infarct area, and neurologic and histologic evaluations were conducted 24 h later. Expression of LIFR, gp130, and phosphorylated Stat3, Akt, and ERK1/2 was investigated by Western blot analysis and immunohistochemistry. The neurologic deficits and ischemic damage were significantly less severe in the high-LIF group than in the PBS group and the low-LIF group. Leukemia inhibitory factor receptor and gp130 were expressed in neurons, and the ischemic damage of these proteins was rescued in the high-LIF group. Early induction of phosphorylated Stat3 was significantly detected on the ischemic side in the high-LIF group after LIF injection. Exogenous LIF attenuates ischemic brain injury by activating cytokine signaling through LIFR/gp130.
Introduction
Leukemia inhibitory factor (LIF) exerts a broad range of effects on many types of cells and has a variety of potential functions in the developing and the mature nervous system (Hilton, 1992). Several in vitro studies have clearly shown that LIF promotes differentiation and survival of astrocytes, oligodendrocytes, and specific types of neurons (Guo et al, 1999; Nakashima et al, 1999), and LIF has also been reported to exert neurotrophic effects in some animal models of central nervous system (CNS) injury such as spinal cord injury, axotomy, and experimental autoimmune encephalomyelitis (EAE) (Tham et al, 1997; Blesch et al, 1999; Butzkueven et al, 2002). Based on these findings, LIF is listed among the growth factors in the CNS (Londreth, 1999).
A common signal transducing receptor component, gp130, acts in association with ligand-specific receptors for members of the interleukin-6 (IL-6) cytokine family, including IL-6, IL-11, LIF, ciliary neurotrophic factor (CNTF), oncostatin-M (OSM), cardiotrophin-1 (CT-1), and cardiotrophin-like cytokine (CLC) (Heinrich et al, 2003). Signals induced by LIF, CNTF, OSM, CT-1, and CLC are transduced via a heterodimeric complex of LIF receptor (LIFR) and gp130 (Heinrich et al, 2003), but the changes in cellular localization of these receptor components and cytokine signaling through LIFR/gp130 after CNS injury, including cerebral ischemia, have been poorly understood.
Recent studies have demonstrated that expression of LIF and CNTF is upregulated after cerebral ischemia (Suzuki et al, 2000; Lin et al, 1998), and although CNTF is known to be neuroprotective against cerebral ischemia (Hermann et al, 2001), the role of LIF and cytokine signaling through LIFR/gp130 has remained unexamined. The purpose of the present study was to elucidate the role of cytokine signaling through LIFR/gp130 induced by LIF injection in a rat model of focal cerebral ischemia.
Materials and methods
Animal Preparation
The protocol described here received prior approval from the Committee on Animal Experiment Guidelines of Keio University School of Medicine. Adult male Sprague—Dawley rats (Japan Laboratory Animals, Tokyo, Japan) weighing 270 to 330 g were anesthetized with an intraperitoneal injection of ketamine hydrochloride (100 mg/kg) and xylazine hydrochloride (15 mg/kg). A temperature probe (TD-300; Shibaura Electronics, Tokyo, Japan) was inserted into the rectum, and a heat lamp was used to maintain rectal temperature at 37°C to 37.5°C. Right middle cerebral artery occlusion (MCAO) was induced by the intraluminal filament technique of Belayev et al (1996) as previously reported (Suzuki et al, 1999). In brief, a 3-0 nylon monofilament suture (Matsuda Ikakogyo, Tokyo, Japan) whose distal segment had been coated with poly-
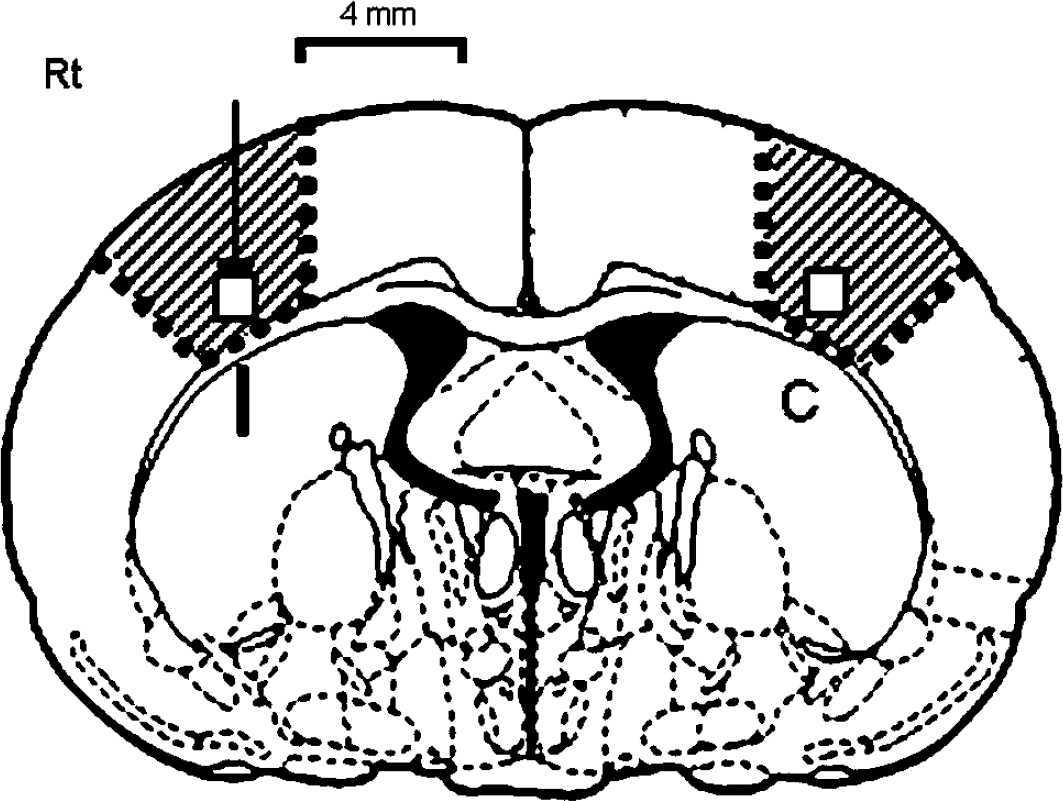
Schematic diagram of the rat coronal brain section (Paxinos and Watson, 1997) at the level of the globus pallidus (0.75 mm posterior to the bregma). The arrow indicates the site of leukemia inhibitory factor (LIF) injection, which corresponded to the region adjacent to the inner boundary zone of the infarct area. Squares indicate the region of interest in which the numbers of TUNEL and immunopositive cells were counted. All photomicrographs presented in Figures 2 and 3 were taken from this square on the ischemic side. Shaded areas indicate the regions from which protein samples were taken for Western blots. I: ischemic side, C: contralateral side.
(
(
Study Groups
The rats were randomly assigned to 4 groups: a sham group (n=18), a PBS group (n=22), a low-LIF group (n=20), and a high-LIF group (n=18). The sham group was subjected to the sham operation followed by PBS injection, and the PBS group was subjected to MCAO followed by PBS injection. The low-LIF and the high-LIF groups were subjected to MCAO followed by a 10 and 100 ng LIF injection, respectively. Animals (n=54) were allowed to survive until 24 h after the drug injection. The mortality rate was calculated after excluding deaths caused by anesthesia problems or subarachnoid hemorrhage during the surgical procedures. Before killing, rectal temperature was measured and the animals were examined for neurologic deficits. Remained animals (n=24) were killed at 1 or 6 h after the drug injection for experiments of signaling.
Neurologic Evaluation
Neurologic deficits were graded as severe, moderate, mild, or absent according to the method devised by Bederson et al (1986). In brief, animals that consistently circled were graded as 3. Rats that consistently showed reduced resistance to lateral push toward the left side with no circling movement were graded as 2. Rats with any amount of consistent forelimb flexion but no other abnormalities were graded as 1, and rats with no neurological deficit were graded as 0. Animals were evaluated for neurologic deficits by at least two persons masked to the experimental groups.
Triphenyltetrazolium Chloride Staining
Coronal brain slices were cut at the levels of 0.75 mm posterior to the bregma, and stained with 2% triphenyltetrazolium chloride (TTC) in each groups of animals (n=3 or 4 per group).
Infarct Area
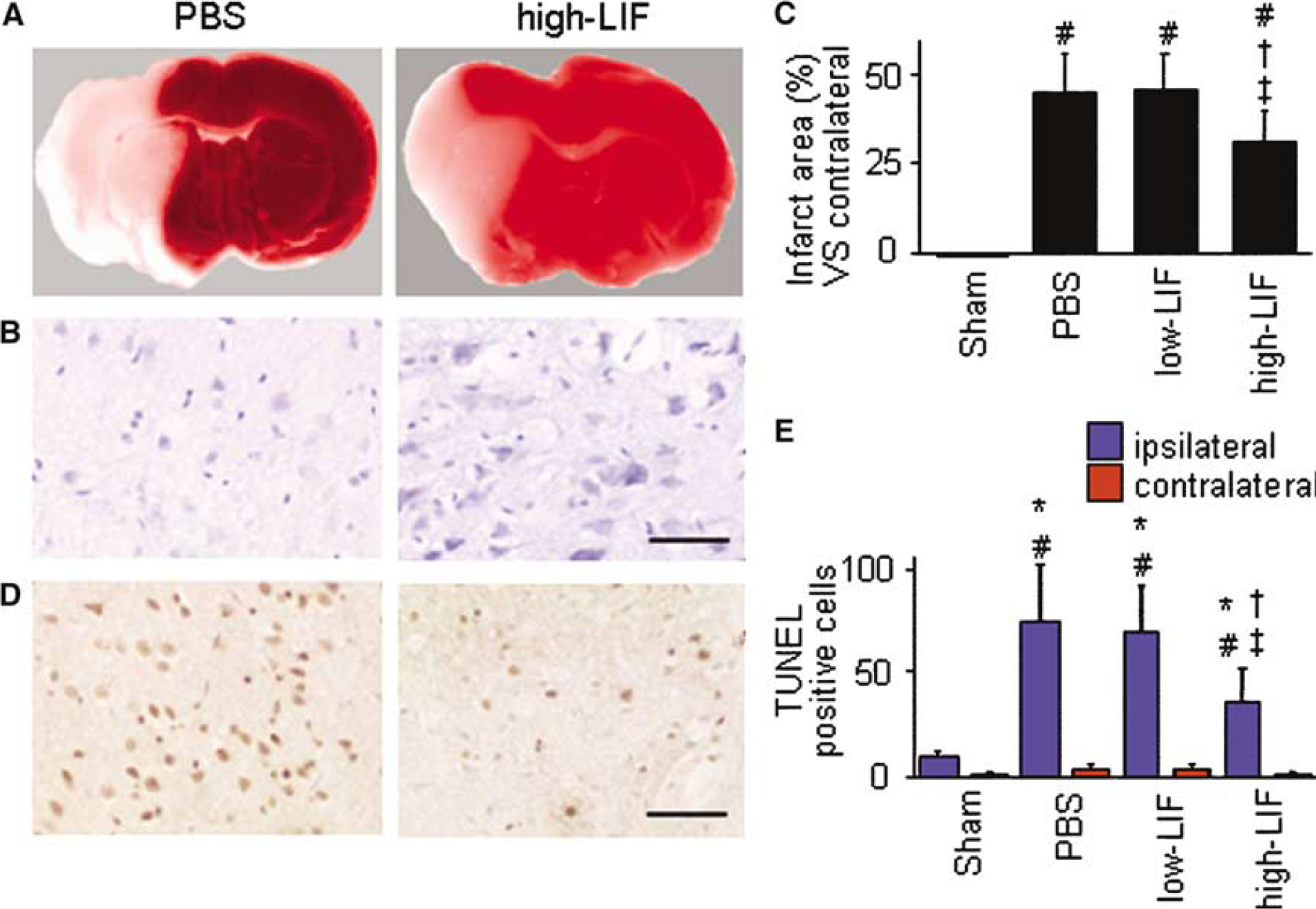
At 24 h after the drug injection, animals were perfused with 200 mL of PBS, followed by 200 mL of 4% freshly depolymerized paraformaldehyde in 0.1 mol/L phosphate buffer. The brains were removed and postfixed at 4°C before cryoprotection by sequential bathing in 10%, 20%, and 30% sucrose. They were then frozen in powdered dry-ice, and consecutive 20-μm thick coronal sections were prepared on a cryostat (Cryocut CM3050S, Leica Instruments GmbH, Nussloch, Germany). Coronal sections prepared from 0.5, 0.75, and 1.0 mm posterior to the bregma were stained with cresyl violet. To quantify the extent of the infarct area in the cerebral cortex, images of the sections were digitized by a computerized digital image analysis system (Inquiry, Loats Associates, Westminister, MD, USA) consisting of a charge-coupled device video camera (CCD72, Dege MTI, Michigan City, IN, USA) with Micro-Nikon lens (Nikon, Tokyo, Japan). An investigator masked to the experimental groups outlined the zone of infarction and the contour of the right and the left hemispheres. The infarct area in the cerebral cortex was calculated by subtracting the area of the normally stained ipsilateral cerebral cortex from the area of the contralateral cerebral cortex to reduce errors because of cerebral edema. The mean values of 3 different sections were reported as a percentage of the entire area of the contralateral cerebral cortex (n=8 per group).
TUNEL Staining
Sections were treated with proteinase K, and the fragmented DNA was visualized with biotinylated-dUTP and terminal transferase (TUNEL method) according to the manufacturer's protocol (Roche Diagnostics, Basel, Switzerland). Absolute TUNEL-positive cell counts (per 0.25 mm2) were made in the regions of interest with an ocular micrometer (Nikon) attached to a light microscope at × 200 magnification, as shown in Figure 1.
Western Blot Analysis
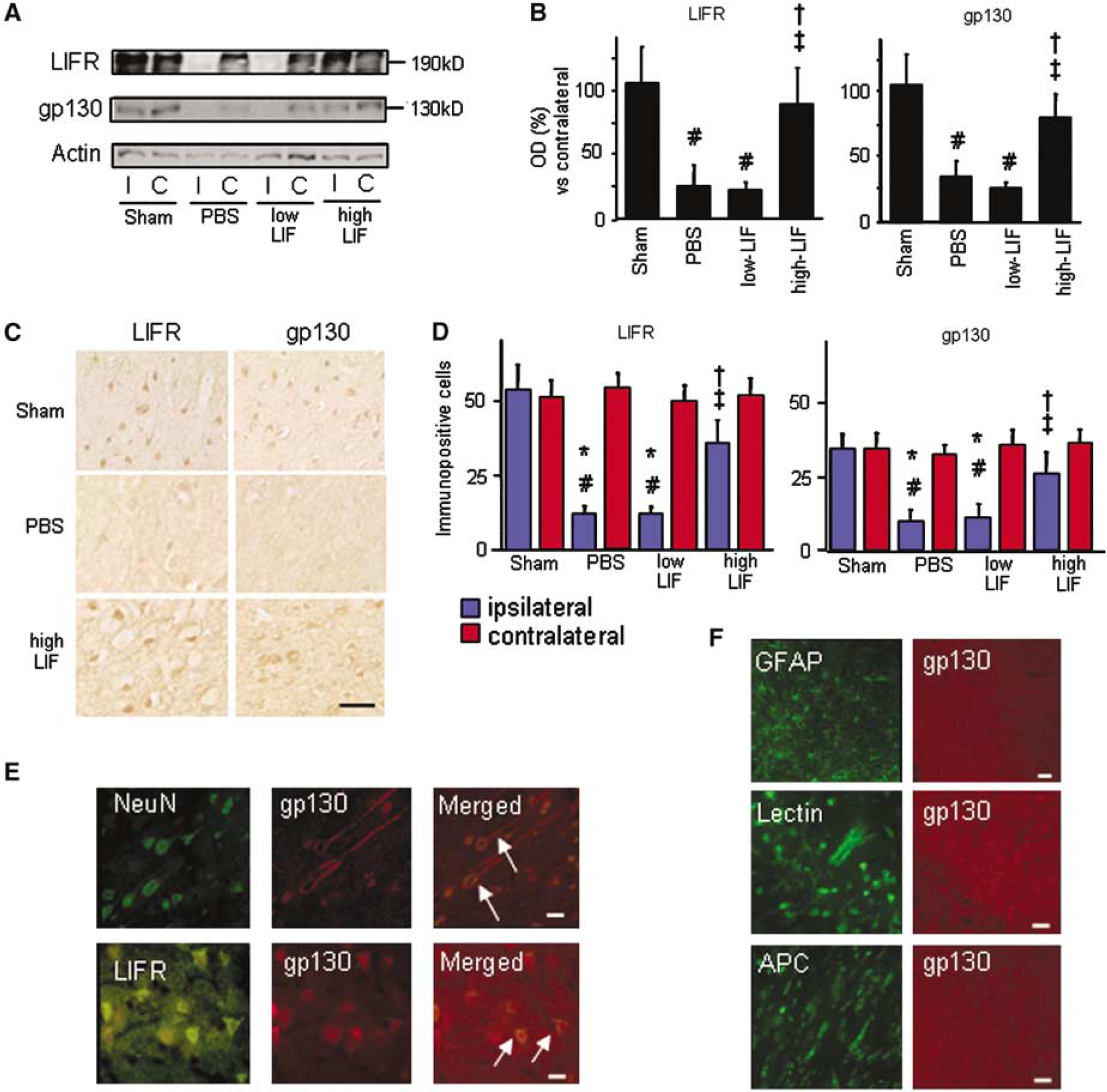
Specimens were collected from the cerebral cortex on both sides as shown in Figure 1. The method used for the Western blot analysis has been described in detail elsewhere (Suzuki et al, 1999). In brief, samples containing 20 μg of protein were subjected to 10% polyacrylamide gel electrophoresis. Polyclonal rabbit antibodies for LIFR (1:500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), gp130 (1:500 dilution; Santa Cruz), phosphorylated Stat3 (Tyr705) (1:250 dilution; Cell Signaling, Beverly, MA, USA), phosphorylated Akt (Ser473) (1:1,000 dilution; Cell Signaling), phosphorylated ERK1/2 (Thr202/Tyr204) (1:1,000 dilution; Cell Signaling), and actin (1:200 dilution; Santa Cruz) were used. Quantitative densitometric analysis of Western blots was performed with a computerized digital image analysis system (Inquiry). The optical density (OD) on the ischemic side was expressed as a percentage of the value on the contralateral side.
Immunohistochemistry
After blocking endogenous peroxidase and nonspecific binding, the sections were incubated overnight at 4°C with rabbit polyclonal anti-LIFR and anti-gp130 antibodies at 1:200 dilution. The slides were then reacted with an avidin-biotinylated enzyme complex system (Vectastain ABC Elite Kit, Vector Laboratories, Burlingame, CA, USA), and the immunoreactive product was visualized with diaminobenzidine (DAB). To assess the specificity of the immunoreactivity, the primary antibody was omitted to provide a nonspecific control. The numbers of LIFR- and gp130-immunoreactive cells were counted as shown in Figure 1. Double-staining immunohistochemistry studies were also performed with the following monoclonal antibodies: anti-NeuN antibody (Chemicon), anti-LIFR antibody (Santa Cruz), anti-GFAP antibody (Boehringer-Mannheim, Philadelphia, PA, USA), anti-adenomatous polyposis coli (APC) antibody (Oncogene, Boston, MA, USA), and isolectin-B4 from Griffonia simplicifolia seeds (lectin, Sigma) (Suzuki et al, 1999). After incubation with the primary antibodies for 16 h, the sections were incubated for 2 h with a fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG antibody (Amersham, Buckinghamshire, UK) and a Texas Red-conjugated anti-rabbit IgG antibody (Amersham), and the sections were examined with a fluorescence microscope (Eclipse E-800, Nikon).
Statistical Analysis
All data are reported as means±s.d. Neurologic deficits were analyzed with the Mann—Whitney U-test. Infarct area, densitometric analysis of Western blots, and cell counts for TUNEL and immunohistochemical staining were analyzed with Student's t-test with Bonferroni corrections for multiple simultaneous comparisons. A value of P<0.05 was considered statistically significant.
Results
Neurologic Evaluation
The mortality rates, rectal temperatures, and neurologic deficits are summarized in Table 1. All rats in the sham group and the high-LIF group survived until 24 h after the injection, but 25% of the animals in the PBS group and 14% in the low-LIF group died of massive cerebral infarction. Statistic differences on the mortality rate were not obtained. Rectal temperature was lower in the sham group than in the other three groups subjected to MCAO. None of the animals in the sham group exhibited neurologic deficits. The neurologic deficits in the high-LIF group were significantly less severe than in the PBS group (P=0.02) and the low-LIF group (P=0.006).
Effect of LIF assessed 24 h after the injection
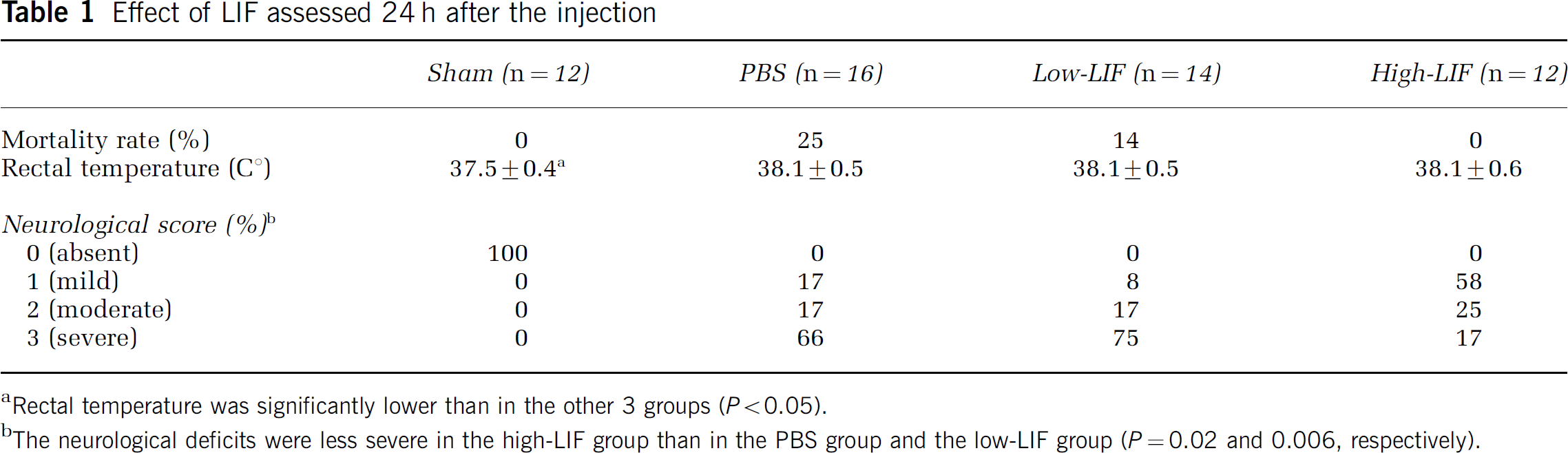
Rectal temperature was significantly lower than in the other 3 groups (P<0.05).
The neurological deficits were less severe in the high-LIF group than in the PBS group and the low-LIF group (P=0.02 and 0.006, respectively).
Histologic Evaluation
Representative coronal brain slices stained with TTC are shown in Figure 2A. Triphenyltetrazolium chloride staining clearly outlined a massive infarct region in the PBS group and the low-LIF group, whereas the infarct area in the cerebral cortex was clearly smaller in the high-LIF group. Cresyl violet staining revealed severe ischemic damage in the PBS group and the low-LIF group, while there was less severe in the high-LIF group (Figure 2B). As shown in Figure 2C, the infarct area in the cerebral cortex in the high-LIF group was significantly less than in the PBS group (P=0.03) and the low-LIF group (P=0.02).
The results of TUNEL staining are shown in Figures 2D and 2E. TUNEL-positive cells were detected around the injection site in all groups, but very few TUNEL-positive cells were detected in the contralateral cortex. The number of TUNEL-positive cells on the ischemic side in the high-LIF group were significantly lower than in the PBS group (P=0.01) and the low-LIF group (P=0.01).
Leukemia Inhibitory Factor Receptor and gp130 Expression
The results of the western blot analysis with anti-LIFR and anti-gp130 antibodies are shown in Figures 3A and 3B. Equal expression of LIFR and gp130 protein was detected in the cerebral cortex on both sides in the sham group. Densitometric analysis revealed that expression of both LIFR and gp130 protein on the ischemic side was significantly lower in the PBS group and the low-LIF group than in the sham group. By contrast, expression of both proteins was well preserved in the high-LIF group.
The results of immunohistochemical studies are shown in Figures 3C and 3D. LIFR immunoreactivity was clearly detected in the soma and neurite of the cerebral cortical neurons on both sides in the sham group and on the contralateral side in the other three groups. Immunoreactivity for gp130, however, was localized in the soma of neurons. There were fewer LIFR- and gp130-immunopositive neurons on the ischemic side in the PBS group and the low-LIF group, and these findings were consistent with the Western blot data. The numbers of LIFR- and gp130-immunopositive cells on the ischemic side were significantly higher in the high-LIF group than in the PBS group and the low-LIF group. No immunoreactivity was detected when the primary antibodies were omitted. Double-staining immunohistochemistry showed that the gp130-immunopositive cells were exclusively neurons (Figure 3E) and that some neurons expressed both LIFR and gp130 (Figure 3E). Glial cells, including astrocytes, microglia, and oligodendrocytes, had proliferated in the ischemic region at 24 h after the injection, but the distribution of the glial cells was different from that of the LIFR- and gp130-immunopositive cells (Figure 3F). These findings indicate that the main source of LIFR and gp130 was neurons.
Phosphorylation of Stat3, Akt, and Erk1/2
The temporal profile of the Western blot analyses showed that Stat3, Akt, and Erk1/2 were phosphorylated at 1 h after the drug injection (Figure 4A). Densitometric analysis revealed that early induction of phosphorylated Stat3 was significantly detected on the ischemic side in the high-LIF group at 1 and 6 h after LIF injection (Figure 4B). Although the phosphorylation of Akt and Erk1/2 was induced after cerebral ischemia, densitometric analysis revealed that an increase in phosphorylation of these proteins in the high-LIF group at 1 and 6 h after LIF injection was small and not statistically significant compared with the PBS group and the low-LIF group.
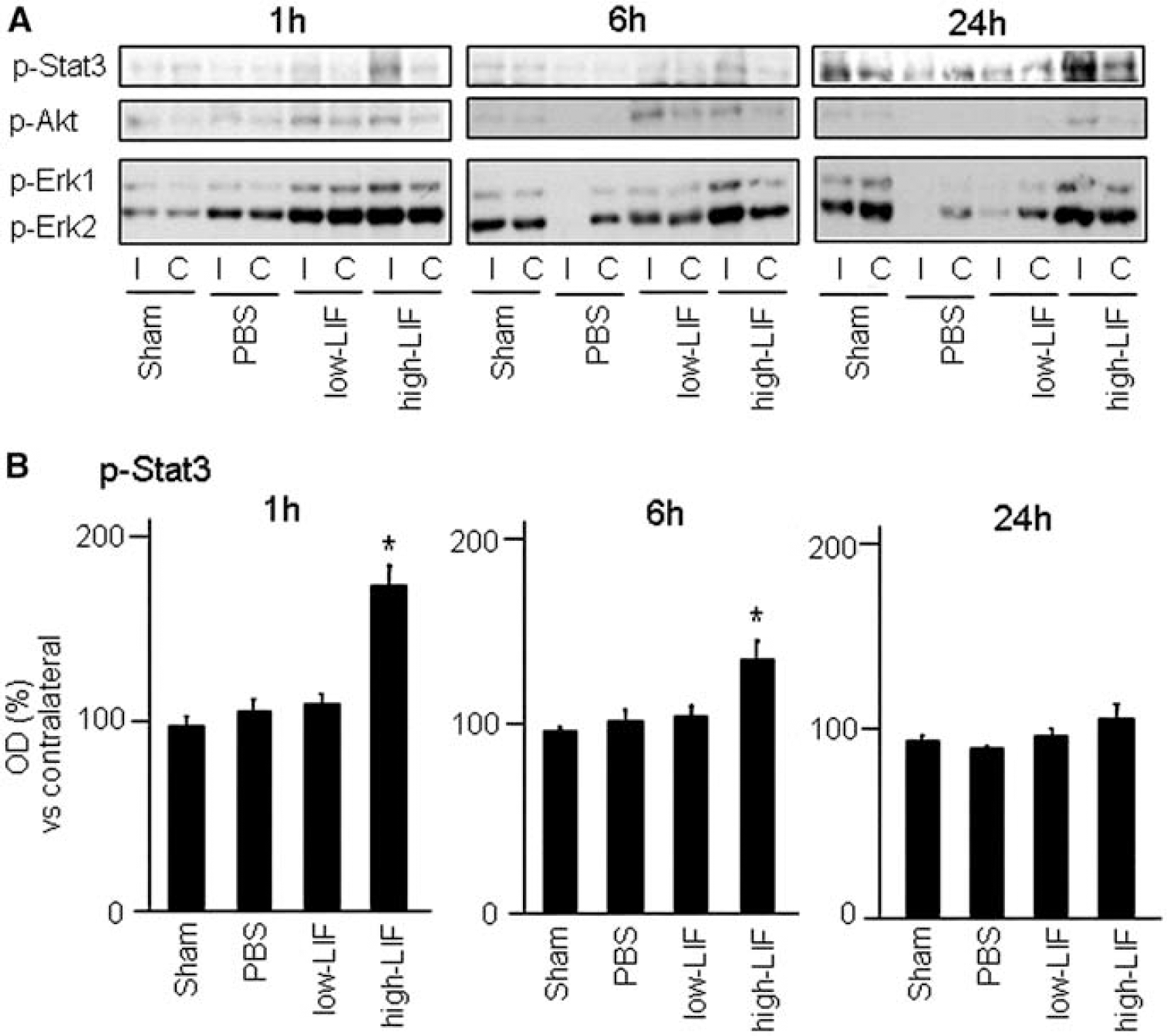
(
Discussion
We selected the region adjacent to the inner boundary zone of the infarct area as the site of LIF injection for the following reasons. First, we had already showed that endogenous LIF is expressed in the cerebral cortical neurons in the peri-ischemia area, and its expression peaked at 24 h after MCAO (Suzuki et al, 2000). By contrast, expression of endogenous LIF was suppressed in the region adjacent to the inner boundary zone of infarct area. Second, this region corresponds to the ischemic penumbra that can be salvaged by pharmacological agents (Memezawa et al, 1992).
Leukemia inhibitory factor receptor and gp130 have been shown to be located in the cerebral and cerebellar neurons in normal rat brain (Watanabe et al, 1996; Yamakuni et al, 1996; Morikawa et al, 2000), although the alterations in these receptor components under pathological conditions was varied by investigators (Haas et al, 1999; Getchell et al, 2002; Choi et al, 2003). Getchell et al (2002) reported upregulation of LIFR mRNA in the olfactory receptor neurons after olfactory bulb ablation. Haas et al (1999) found that gp130 mRNA levels were unaltered in the rat facial nucleus after axotomy, but Choi et al (2003) reported that gp130 mRNA was upregulated after cerebral ischemia. Leukemia inhibitory factor receptor has also been reported to be increased in the spinal cord in EAE, but gp130 is not increased (Butzkueven et al, 2002). Taken together, we concluded that the alterations in these receptor components depended on the model of the CNS injury. In the clinical study, it was reported that the serum concentration of soluble gp130 was significantly reduced the first week after stroke (Acalovschi et al, 2003). We showed that the main source of LIFR and gp130 under both normal and ischemic conditions is neuron, and that the loss of these proteins was rescued by LIF injection.
Much attention had been paid to the role of LIFR/gp130 in the developing and mature CNS, and cytokine signaling through LIFR/gp130 has been reported to be necessary for the maintenance of forebrain neuronal stem cells and the survival of postmitotic neurons during development (Shimazaki et al, 2001). Leukemia inhibitory factor receptor/gp130 signaling also reduced immune-mediated demyelination by enhancing oligodendrocyte survival (Butzkueven et al, 2002), and either LIFR- or gp130-deficient mice exhibited significant loss of motor neurons (Nakashima et al, 1999; Li et al, 1995). Based on these findings, we speculated that the LIFR/gp130-mediated signaling induced by LIF injection had a significant neuroprotective effect on the cerebral ischemia in the present study.
We propose the presumed downstream signal pathways through LIFR/gp130, which are activated by LIF (Figure 5). It is now acceptable that these signaling pathways are closely associated with the cell survival and anti-apoptosis (Alonzi et al, 2001; Park et al, 2003; Heinrich et al, 2003). Among them, JAK-STAT is the most important pathway, and is strongly regulated by Tyr705 phosphorylation of Stat3 (Heinrich et al, 2003). In the present study, we demonstrated that recombinant LIF induced especially Stat3 phosphorylation, and the JAK-STAT was the main pathway activated after cerebral ischemia. In addition, PI3K/Akt and MEK/Erk1/2 pathways are also known to be activated after LIF injection (Alonzi et al, 2001; Park et al, 2003), but their activation was less than that of JAK-STAT.
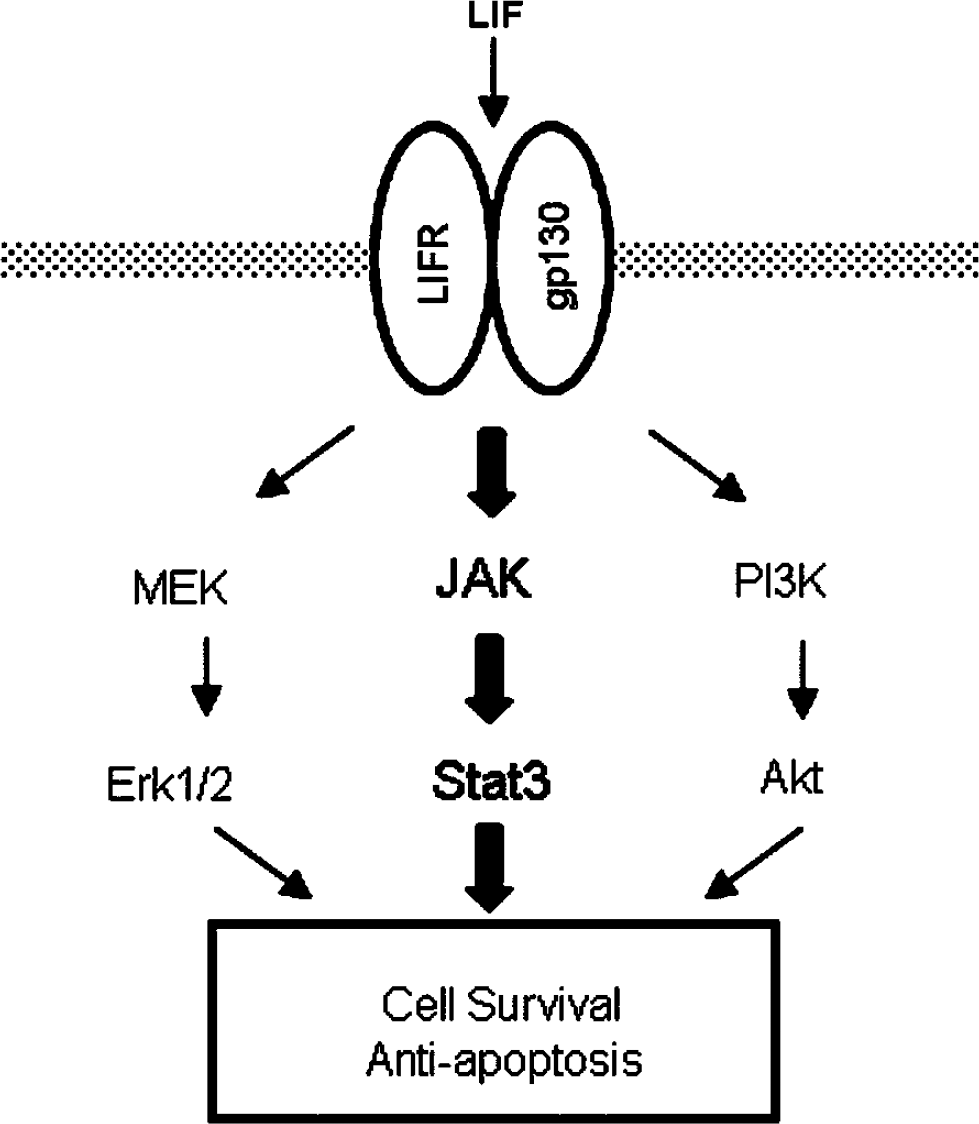
A presumed diagram of signaling pathways through Leukemia inhibitory factor receptor/gp130, which are activated by leukemia inhibitory factor (LIF).
Although we and other investigators have reported enhanced phosphorylation of Stat3 after cerebral ischemia (Justicia et al, 2000; Suzuki et al, 2001; Wen et al, 2001), the exact role of Stat3 phosphorylation has not been elucidated. Wen et al (2001) observed that phosphorylated Stat3-positive neurons were also TUNEL-positive and speculated that this signal plays a crucial role in ischemia-induced neuron death. By contrast, we found that LIF injection induced rapid enhancement of Stat3 phosphorylation, indicating that LIF may be associated with an important neuroprotective role against cerebral ischemia. In this regard, we had already shown that Stat3 phosphorylation is predominantly detected in surviving neurons after transient MCAO (Suzuki et al, 2001). In addition, IL-6 was proven to have neuroprotective effects against cerebral ischemia (Loddick et al, 1998; Herrmann et al, 2003). We recently found that the administration of anti-IL-6 receptor antibody to mice after MCAO enlarged the infarct size and aggravated the neurological deficits together with a significant reduction in Stat3 phosphorylation (Yamashita et al, submitted). Based on these findings, we concluded that Stat3 phosphorylation after cerebral ischemia was closely related to neuroprotection. Because we demonstrated that LIF could decrease the number of TUNEL-positive cells, the most important neuroprotective mechanism of Stat3 phosphorylation was antiapoptosis. In fact, it was reported that the role of Stat3 on cell survival has been linked to the transcriptional regulation of apoptotic regulatory proteins such as Bcl-2 family (Battle and Frank, 2002).
The present study showed that proliferating glial cells, including astrocytes, microglia, and oligodendroglia, are not the main source of LIFR and gp130. One of the reasons why gp130 was not detected in glial cells may be because of the sensitivity of polyclonal antibody used in this study. Unfortunately, it was an only commercially available antibody for immunohistochemistry. The survival and differentiation of astrocytes and oligodendroglia have been shown to be potentiated by LIFR/gp130 signaling, and astrocyte differentiation is dramatically impaired in either gp130- or LIFR-deficient mice (Nakashima et al, 1999; Butzkueven et al, 2002). In fact, activation of the JAK-STAT pathway involving Stat3 was observed in reactive astrocytes after cerebral ischemia (Choi et al, 2003; Justicia et al, 2000), and we have showed expression of endogenous LIF in astrocytes at 96 h of reperfusion after transient MCAO (Suzuki et al, 2000). Taken together, we think that an experimental protocol with a longer period after cerebral ischemia will be necessary to determine the exact role of signaling through LIFR/gp130 in glial cells.
In summary, we have demonstrated that recombinant LIF can attenuate ischemic brain injury in rats by activating cytokine signaling through LIFR/gp130.