
Abstract
Interleukin-6 (IL-6) is pleiotropic cytokine involved in many central nervous system disorders including stroke, and elevated serum IL-6 has been found in acute stroke patients. IL-6 is implicated in the inflammation, which contributes to both injury and repair process after cerebral ischemia. However, IL-6 is one of the neurotrophic cytokines sharing a common receptor subunit, gp130, with other neurotrophic cytokines, such as leukemia inhibitory factor (LIF) and ciliary neurotrophic factor. The expression of IL-6 is most prominently identified in neurons in the peri-ischemic regions, and LIF expression shows a similar pattern. The direct injection of these cytokines into the brain after ischemia can reduce ischemic brain injury. The cytokine receptors are localized on the neuron surface, suggesting that neurons are the cytokine target. The major IL-6 downstream signaling pathway is JAK—STAT, and Stat3 activation occurs mainly in neurons during postischemic reperfusion. Further investigation is necessary to clarify the exact role of Stat3 signaling in neuroprotection. Taken together, the information suggests that IL-6 plays a double role in cerebral ischemia, as an inflammatory mediator during the acute phase and as a neurotrophic mediator between the subacute and prolonged phases.
Introduction
Interleukin-6 (IL-6) is a glycoprotein with a molecular mass of 20 to 30 kDa, depending on the cellular source and preparation, and is a cytokine with pleiotropic properties, playing a role in central host defense (Akira et al, 1993). Depending on the target cells, IL-6 can exert growth-inducing, growth-inhibitory, and differentiation-inducing effects. IL-6 is produced by many different cell types, including monocytes and macrophages, fibroblasts, keratinocytes, endothelial cells, mesangial cells, chondrocytes, osteoblasts, smooth muscle cells, T cells, B cells, granulocytes, mast cells, and certain tumor cells (Akira et al, 1993). Some culture cells have been found to release low levels of IL-6, which is greatly increased after stimulation (Van Wagoner and Benveniste, 1999).
The cytokine is also produced in the central nervous system (CNS) and is involved in the pathogenesis of many CNS disorders, such as inflammatory, autoimmune, and degenerative disorders, infection, neoplasms, and stroke (Van Wagoner and Benveniste, 1999). There have been increasingly findings regarding the role of IL-6 in clinical ischemic stroke and experimental ischemia models. Although these studies have suggested that IL-6 exerts not only inflammatory but also neurotrophic effects in cerebral ischemia, the precise roles of IL-6 in the pathophysiology of cerebral ischemia have not been fully elucidated. In other words, whether IL-6 works as a mediator of inflammatory damage or a neuroprotective agent in cerebral ischemia remains undetermined. The purpose of this review is to clarify the association between IL-6 and stroke and to discuss the roles of IL-6 in cerebral ischemia based on clinical and experimental data.
Association of interleukin-6 with occurrence of ischemic stroke
Atherosclerosis
From the standpoint of the risk factors for stroke, inflammatory processes are thought to be involved in the pathogenesis of atherosclerotic plaques and their thrombotic complications. Increased IL-6 expression has been found in atherosclerotic plaques, predominantly co-localized with macrophages (Schieffer et al, 2000). It is now thought that elevations in serum IL-6 correlate with an in-hospital and short-term adverse prognosis and may reflect not only a high prevalence of myocardial necrosis, ischemia-reperfusion damage, or severe coronary atherosclerosis but also a primary inflammatory instigator of coronary instability (Ridker et al, 2000; Libby et al, 2002). Moreover, higher serum IL-6 levels appear to be associated with unstable carotid plaques, suggesting a potential risk for stroke (Yamagami et al, 2004).
Interleukin-6 −174G/C Polymorphism
A polymorphism in the promoter region of the IL-6 gene is associated with the regulation of IL-6 gene expression and the inflammatory response. A G to C substitution at the −174 position in the IL-6 gene promoter was first reported in patients with juvenile chronic arthritis (Fishman et al, 1998). The IL-6 −174G/C polymorphism is also associated with carotid artery atherosclerosis and coronary heart disease (Rauramaa et al, 2000; Basso et al, 2002). As functional polymorphisms in inflammatory genes may influence the incidence and outcome of ischemic stroke, many authors have attempted to examine the association between the IL-6 −174G/C polymorphism and cerebral infarction (Jenny et al, 2002; Acalovschi et al, 2003; Balding et al, 2004; Flex et al, 2004; Chamorro et al, 2005; Grocott et al, 2005; Karahan et al, 2005; Weger et al, 2005; Lalouschek et al, 2006). Tso et al (2007) reviewed the published studies that independently examined the association between the −174 polymorphism and stroke: they concluded that there is a significant association with the occurrence of ischemic stroke, though some of the studies they reviewed did not support this conclusion. The effect of the −174G/C polymorphism on IL-6 expression may depend on the surrounding genetic variations, and the associations with a single polymorphism could be because of linkage disequilibrium with other genetic variants (Acalovschi et al, 2003). However, associations between the IL-6 promoter polymorphism and stroke are inherently difficult to interpret because IL-6 has a complex physiology, and genuine differences between stroke subtypes and the population studied have limitations in experimental design (Tso et al, 2007). Taken together, though the precise mechanisms of the effect of the IL-6 −174G/C polymorphism on the development of a stroke are unclear, this polymorphism seems to be closely associated with the occurrence of cerebral infarction.
Serum Interleukin-6 in Stroke Patients
An increase in serum IL-6 level has been reported in acute ischemic stroke patients by many investigators (Table 1; Fassbender et al, 1994; Beamer et al, 1995; Tarkowski et al, 1995; Ferrarese et al, 1999; Vila et al, 2000; Acalovschi et al, 2003; Smith et al, 2004; Waje-Andreassen et al, 2005). All of the authors showed that the serum IL-6 level was significantly increased in stroke patients as compared with normal controls. The increase in serum IL-6 started within 24 h and peaked between 2 and 4 days after the onset of stroke. Higher levels of serum IL-6 continued until 90 days after the stroke. Inflammatory biomarkers, including C-reactive protein, fibrinogen, IL-1 receptor antagonist, and tumor necrosis factor-α (TNF-α) were also increased in parallel with IL-6 elevation. Some researchers independently reported that the high level of serum IL-6 was correlated with infarct volume, early neurologic deterioration, body temperature, and a long-term poor outcome (Fassbender et al, 1994; Beamer et al, 1995; Tarkowski et al, 1995; Ferrarese et al, 1999; Vila et al, 2000). However, as there is no proof for the association between IL-6 elevation and pathologic process in acute stroke, the clinical markers of IL-6 for severe ischemic stroke are still controversial. In addition, it is widely accepted that IL-6 does not have a causative effect for stroke. Gladilin et al (2000) orally applied a neuroprotective antioxidant, ebselen, in rats subjected to focal cerebral ischemia, and found that ebselen decreased the plasma IL-6 level, possibly because of a protection of vessel walls and maintenance of the blood—brain barrier integrity and function.
Summary of published studies on the serum interleukin (IL)-6 levels in patients with cerebral infarction
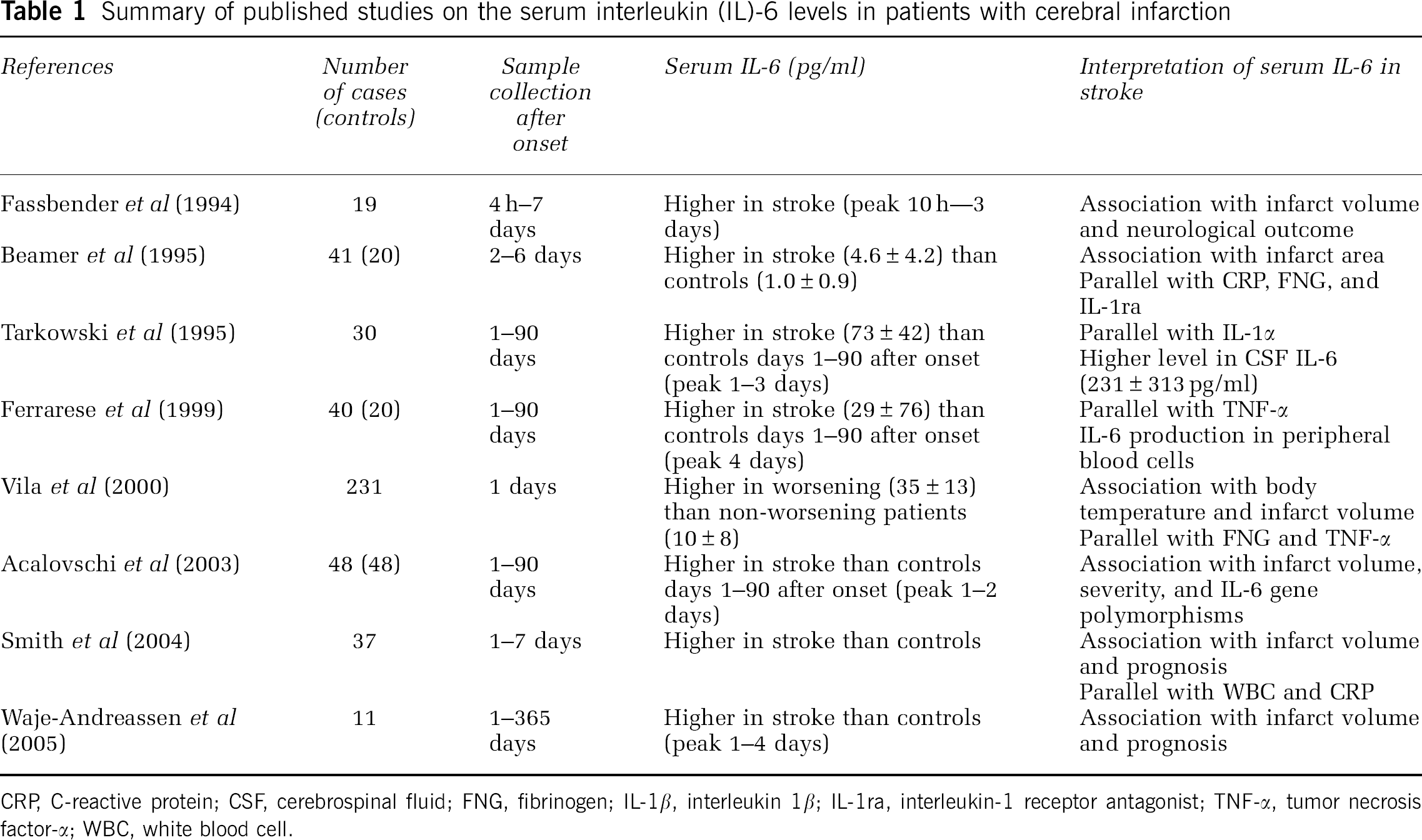
CRP, C-reactive protein; CSF, cerebrospinal fluid; FNG, fibrinogen; IL-1β, interleukin 1β; IL-1ra, interleukin-1 receptor antagonist; TNF-α, tumor necrosis factor-α; WBC, white blood cell.
Peripheral blood cells have been suggested to be a major source of serum IL-6 after stroke (Ferrarese et al, 1999). However, it is not conclusive, because a higher level of released IL-6 was detected after ex vivo stimulation using lipopolysaccharide.
Interleukin-6 is increased in the cerebrospinal fluid (CSF) of acute stroke patients (Beamer et al, 1995; Tarkowski et al, 1995). Systemically produced IL-6 could potentially be able to passively enter into the CSF component through the damaged blood—brain barrier. In this regard, Tarkowski et al (1995) reported significantly lower levels of serum IL-6 compared with the CSF levels during the first week after the onset of the stroke. This finding is strongly against the hypothesis of primary systemic IL-6 production with subsequent passage to the CSF. These findings suggest that the main source of IL-6 after a stroke is the brain.
Interleukin-6 as an inflammatory cytokine
Inflammation and Cerebral Ischemia
Ischemic brain injury results from a complex sequence of pathologic events that evolve over time and space. The major pathogenetic mechanisms of this cascade include excitotoxicity, peri-infarct depolarization, inflammation, oxidative damage, and programmed cell death. Among these mechanisms, inflammation is implicated as a general cardiovascular risk factor, a possible immediate trigger, and an exacerbating factor of the response to ischemic tissue injury (Dirnagl et al, 1999; Iadecola and Alexander, 2001; Muir et al, 2007).
Inflammatory Cytokines
The inflammatory response is characterized by the local expression of various inflammatory cytokines in the brain (DeGraba, 1998). It has been suggested that TNF-α and IL-1β, which are detected as early as 1 h after the onset of ischemia, induce neuronal injury. Interleukin-1 receptor antagonist and TNF-α blockers have been found to reduce infarct size in a rat model of focal cerebral ischemia and are thought to have neuroprotective effects (Iadecola and Alexander, 2001). However, Hallenbeck (2002) reviewed the role of TNF-α in cerebral ischemia, pointing out that the TNF ligand—receptor interaction may contribute to repair and recovery after stroke as an important modulator of inflammation.
Interleukin-6 is one of the CNS inflammatory cytokines and has been implicated in cellular responses. This cytokine orchestrates an inflammatory response between blood cells, the vascular endothelium, and resident cells of the brain parenchyma and can induce the synthesis of some chemokines and cell adhesion molecules that, together with blood—brain barrier leakage, can lead leukocyte infiltration (Lemoli et al, 1996; Prudhomme et al, 1996; Penkowa et al, 1999). In the brain parenchyma, IL-6 activates gliosis and leukocyte activation (Kharazmi et al, 1989; Chiang et al, 1994). It is speculated that upregulated IL-6 may be considered to act on the vascular endothelium to increase harmful mediators and mediate inflammatory cascades leading to the aggravation of cerebral ischemic damage (Huang et al, 2006). However, there is no direct evidence showing that IL-6 plays a detrimental function in acute phase of cerebral ischemia. Inflammatory IL-6 may be involved in not only ischemic brain injury, but also repair process after cerebral ischemia.
In the brain, inflammation acts as a double-edged sword. Brain inflammation spans a progression of events through repair and remodeling to neuronal plasticity and recovery of function (Barone and Feuerstein, 1999). Interleukin-6 is involved in the induction of acute reactions and controlling the level of acute inflammatory responses by decreasing the proinflammatory cytokines and increasing antiinflammatory molecules. In the prolonged phase, IL-6 is involved not only in eliciting an acute phase reaction, but also in the development of specific cellular and humoral immune response (Xing et al, 1998). In a contrast to negative effects of other cytokines including TNF-α and IL-1β, IL-6 can potentially exert beneficial or detrimental effects depending on the pathologic context.
Basic aspects of interleukin-6 as a neurotrophic cytokine
Interleukin-6 Type Cytokine
The family of IL-6 type cytokines consists of IL-6, leukemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF), IL-11, oncostatin-M, and cardiotrophin-1. A common signal transducing receptor subunit, gp130, acts in association with ligand-specific receptors for members of the IL-6 cytokine family (Heinrich et al, 2003). These cytokines show structural similarity and, more importantly, have shared biologic functions with IL-6 (Taga and Kishimoto, 1997). Based on this context, when the association between IL-6 and cerebral ischemia is discussed, it is also necessary to pay attention to LIF and CNTF. Besides their roles in inflammatory and immune responses, these cytokines play crucial roles in hematopoiesis, liver and embryonal development, and fertility (Taga and Kishimoto, 1997). In the CNS, these cytokines have neurotrophic effects, including neuronal regeneration, neuronal survival, and the differentiation of neuronal stem cells after various types of injuries. The cytokines signaling through gp130 are referred to as neurokines, and they are listed among the growth factors in the CNS (Londreth, 1999).
Receptors
Receptors involved in the recognition of IL-6 type cytokines can be subdivided into the nonsignaling ligand-specific α-receptors (IL-6Rα and CNTFRα) and the signal-transducing receptors (gp130 and LIFR). The latter are associated with Janus kinase (JAK) and become tyrosine-phosphorylated in response to cytokine stimulation. As indicated in Figure 1, IL-6 and CNTF first bind specifically to their respective α-receptor subunits and then recruit signal transducing receptor subunits to form active trimeric receptor complexes. Signals induced by IL-6 are transduced via gp130 homodimers, whereas signals induced by LIF and CNTF are transduced via heterodimer complexes of LIFR and gp130 (Heinrich et al, 2003). Ligand-induced receptor dimerization is thought to lead to their activation via inter- or intramolecular phosphorylation (Heinrich et al, 2003).
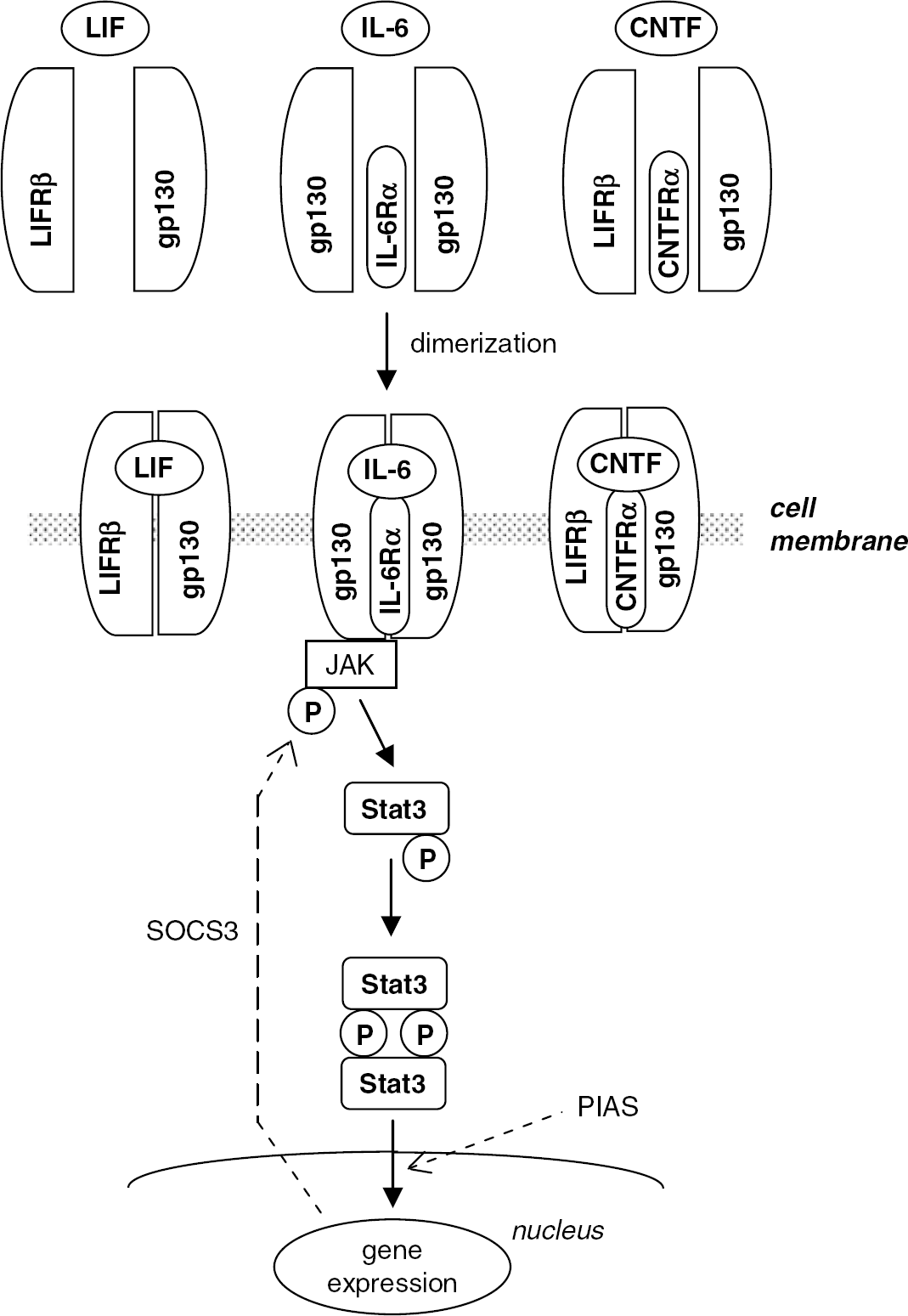
Interleukin (IL)-6 type cytokine receptor complexes and downstream JAK—STAT signaling. The IL-6 receptor is a gp130 homodimer with a ligand-specific α-subunit. The leukemia inhibitory factor (LIF) receptor is a heterodimer complex consisting of LIFR and gp130. The ciliary neurotrophic factor (CNTF) receptor is a heterodimer complex consisting of LIFR and gp130 with a ligand-specific α-subunit. Ligand-induced receptor dimerization initiates signaling. The activation of JAK leads to the phosphorylation of Stat3, resulting in the formation of Stat3 dimers, followed by dimer translocation to the nucleus, where they regulate the transcription of target genes. Negative modulators in the JAK—STAT pathway, such as SOCS (suppressor of cytokine signaling) and PIAS (protein inhibitor of activated stat), are shown as dotted lines.
Although gp130 is ubiquitously expressed, the number of cells that responds to a particular IL-6 type cytokine is limited because the expression of the other receptor subunits, especially α-receptors, is more restricted and tightly regulated. Soluble forms of IL-6α (sIL-6Rα) and gp130 (sgp130) are both present in the human serum. Soluble IL-6Rα can complex with IL-6 and bind and signal through gp130, thus serving as an agonist of IL-6-induced responses. However, sgp130 has potential antagonistic activity against IL-6/sIL-6 complexes (Heinrich et al, 2003).
JAK—STAT Signaling
The intracellular signal transduction induced by IL-6 involves the activation of JAK tyrosine kinase family members, leading to the activation of transcription factors of the signal transducers and activators of transcription (STAT) family. The JAK—STAT pathway is the main intracellular signaling pathway of the IL-6 cytokine family, though other signal cascades are also activated, such as PI3/Akt, MEK/Erk1/2, and NF-κB are also activated (Heinrich et al, 2003). In this review, we focus on the JAK—STAT pathway.
The binding of cytokines to their receptors induces the phosphorylation of the receptor-associated JAK family, which in turn leads to the phosphorylation of the downstream STAT family of transcription factors. A STAT proteins with a molecular weight of 92 kDa, Stat3, is an important element in the JAK—STAT pathway (Cattaneo et al, 1999). The phosphorylation of Stat3 at Tyr705 in response to gp130-stimulating cytokines leads to the formation of Stat3 dimers, followed by the translocation of these dimers to the nucleus, where they regulate the transcription of target genes.
There are important regulatory and negative modulators in the JAK—STAT pathway, such as suppressors of cytokine signaling (SOCS) and protein inhibitor of activated STAT (PIAS). Among SOCS proteins, SOCS3 is functionally the most potent in inhibiting the signal of IL-6 type cytokines. The SOCS3 protein blocks the phosphorylation status of JAK by binding to phosphotyrosine motifs within activated gp130. However, PIAS specifically binds to phosphorylated Stat3 and suppresses the DNA-binding activity of Stat3 (Heinrich et al, 2003). PIAS3 is essentially known to be specific for inhibition of Stat3-mediated gene expression mainly in nuclear (Rödel et al, 2000).
Expression of interleukin-6 type cytokine in cerebral ischemia models
Interleukin-6
Many investigators have studied IL-6 expression after cerebral ischemia in rodents (Table 2; Wang et al, 1995; Saito et al, 1996; Loddick et al, 1998; Hill et al, 1999; Orzylowska et al, 1999; Suzuki et al, 1999a, 1999b; Ali et al, 2000; Block et al, 2000; Legos et al, 2000). To further investigate the cellular localization and temporal profile of IL-6 expression, immunohistochemistry can detect the origin of released cytokines. Therefore, we investigated IL-6 expression in a rat model of focal ischemia model between 0.5 h and 7 days of reperfusion after 90-mins middle cerebral artery occlusion (MCAO; Suzuki et al, 1999a and unpublished data). Sham-operated rats showed no IL-6 expression. In the ischemic brain, IL-6 protein was principally localized in the neurons of the cerebral cortex. The neuronal expression of IL-6 started 3.5 h after ischemia, peaked after 24 h of reperfusion, and persisted for 7 days. The IL-6 immunoreactivity was most upregulated in the peri-ischemic regions (Figure 2A). In contrast, neuronal IL-6 expression was suppressed in the infarct region (Figure 2B). Accordingly, the IL-6 immunoreactivity clearly showed a boundary zone between the infarct and peri-ischemic regions after 24 h of reperfusion (Figure 2C). In other areas, not supplied by the occluded artery, including the contralateral cortex, the hippocampus, and the substantia nigra pars reticulata, IL-6 was also clearly detected in neurons (Figure 2D; Suzuki et al, 1999a; Dihne et al, 2001). It has also been reported that IL-6 expression was first detected in a focal ischemia model of spontaneously hypertensive rats at 2 h and peaked at 24 h after ischemia, but expression became undetectable from the fifth day onward (Legos et al, 2000). In experimental axonal injury, IL-6 was synthesized in the cortical, thalamic, and hippocampal neurons and was released into the CSF and serum (Hans et al, 1999). Other brain and nerve injury models have also indicated that neurons are important origins of IL-6 (Lemke et al, 1998; Murphy et al, 1999). Moreover, the autopsy of a patient with cerebral infarction also showed IL-6 positive cerebral neurons near the infarct area (Suzuki et al, 2002).
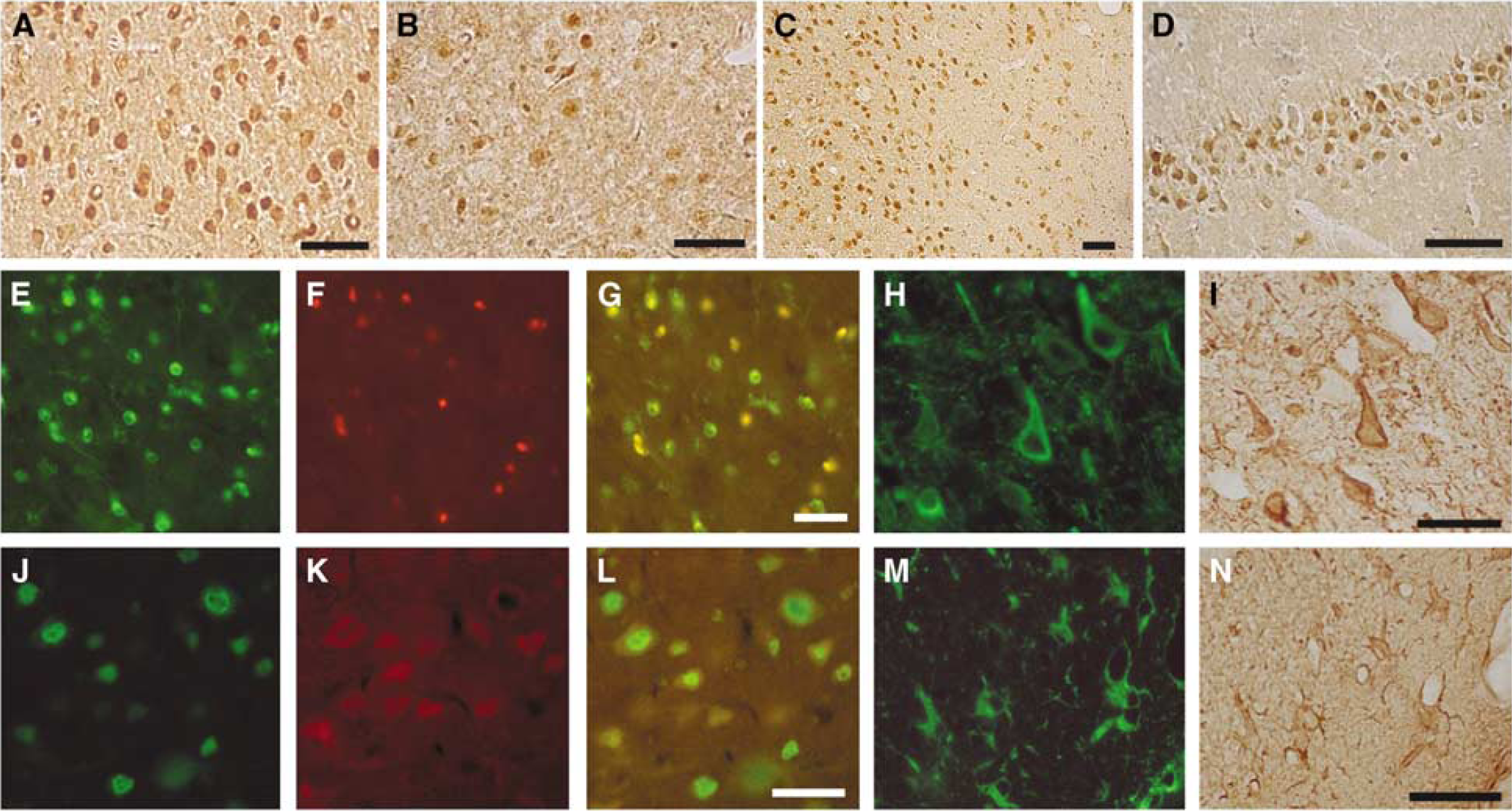
Interleukin (IL)-6 and leukemia inhibitory factor (LIF) immunoreactivity. Photomicrographs were obtained from consecutive frozen coronal sections prepared from ischemic rat brain after perfusion fixation and stained with specific antibodies. (
Summary of published studies on the expression interleukin (IL)-6 in experimental cerebral ischemia
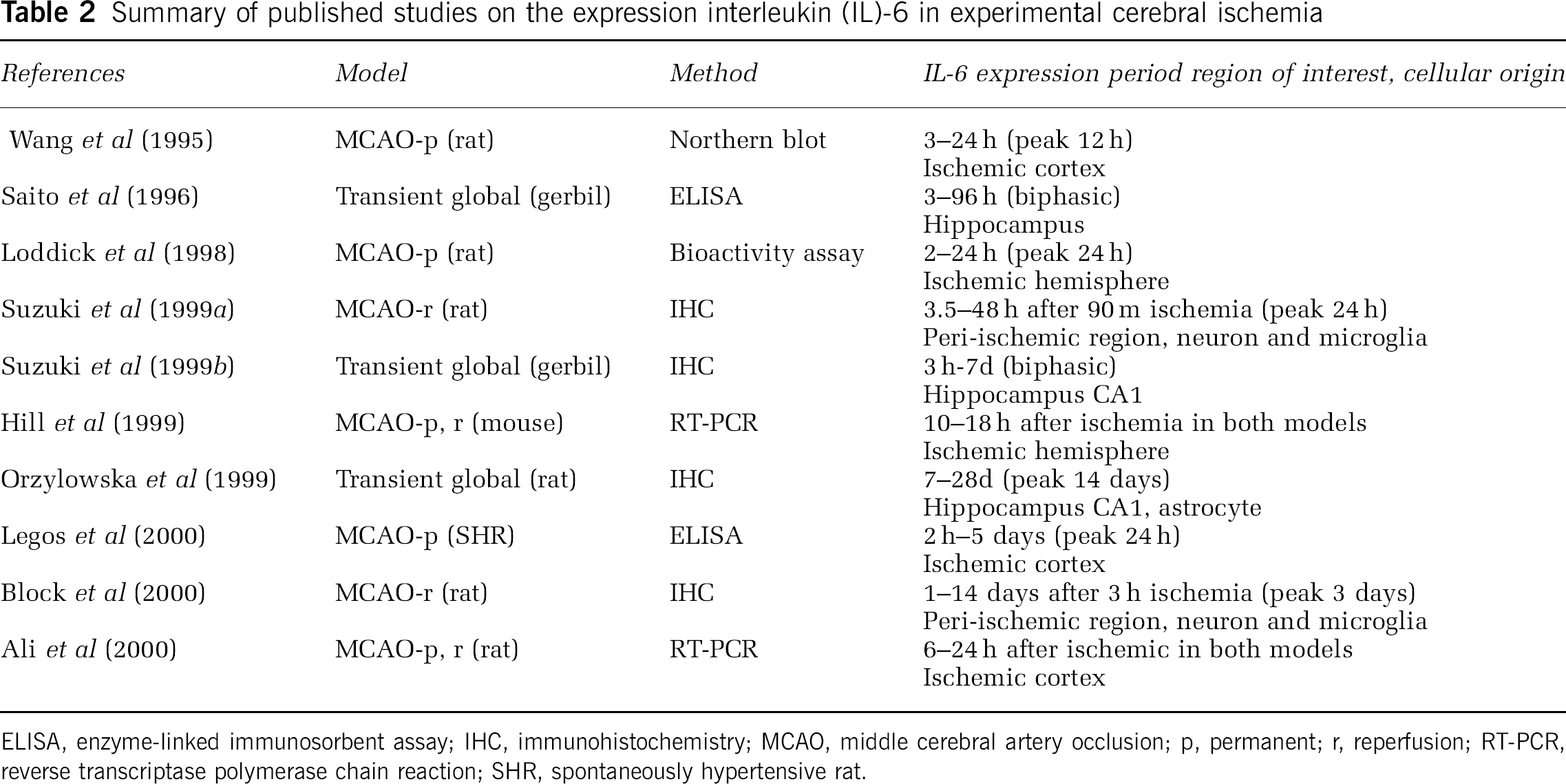
ELISA, enzyme-linked immunosorbent assay; IHC, immunohistochemistry; MCAO, middle cerebral artery occlusion; p, permanent; r, reperfusion; RT-PCR, reverse transcriptase polymerase chain reaction; SHR, spontaneously hypertensive rat.
Glial cells are another source of IL-6. The microglial reaction to ischemia is rapid, and several morphologic types of reactive microglia have been detected with various cell markers, such as B4-isolectin from Griffonia simplicifolia (GSAI-B4), ionized calcium-binding adaptor molecule 1 (Iba1), and ED1 (Ito et al, 2001). We have observed that round-type reactive microglia or blood-borne macrophages express IL-6 in the ischemic region, which peaked at 24 h after cerebral ischemia (Figure 2E—G; Suzuki et al, 1999a).
The third origin of IL-6 after cerebral ischemia is the vascular endothelium. We detected IL-6 immunoreactivity in the endothelium of the ischemic region 48 h after MCAO. Another in vivo experiment also revealed that, in response to noxious stimuli including ischemia, endothelial cells are a potential source of IL-6 (Reyes et al, 1999). The other possible source of IL-6 is oligodendrocytes. Based on our study in an experimental MCAO model (Tanaka et al, 2001), IL-6 is partially detected in oligodendrocytes in the medial region of the corpus callosum on the ischemic side, which corresponds to the peri-ischemic region of the white matter, after 3.5 h of reperfusion after cerebral ischemia. Finally, in vitro studies have shown that reactive astrocytes are also important sources of IL-6 (Van Wagoner and Benveniste, 1999). Prolonged induction of IL-6 in astrocytes has been reported in the hippocampus after transient global ischemia (Orzylowska et al, 1999). Although our experimental ischemia study revealed glial fibrillary acidic protein-positive astrocytes after 96 h of reperfusion after 90-min MCAO, astrocyte IL-6 immunoreactivity seemed weak compared with that of microglia.
Leukemia Inhibitory Factor and Ciliary Neurotrophic Factor
Although the induction of LIF after CNS injury has not been fully elucidated, LIF can be detected in neurons (Lemke et al, 1997). We first showed that LIF expression is upregulated after cerebral ischemia, and that neurons are the major source of LIF (Suzuki et al, 2000a). The LIF protein appeared in neurons later than IL-6 and, after focal ischemia, was expressed in the cortical and hippocampal neurons after 12 h of reperfusion onward (Figure 2H and 2I). Neuronal postischemia LIF expression peaked at 24 h and lasted for 7 days of reperfusion. The expression was most increased in the peri-ischemic region, and some neurons simultaneously expressed both IL-6 and LIF (Figure 2J—L; Suzuki et al, 2000a). The postmortem examination of patients with cerebral infarction has also shown that LIF localizes in the cortical neurons (Slevin et al, 2008). The LIF protein was also detected after ischemia in activated astrocytes after 96 h of reperfusion (Figure 2M and 2N), but not in reactive microglia or monocytes and macrophages (Suzuki et al, 2000a). In a model of CNS injury by contusion, LIF was detected in reactive astrocytes (Banner et al, 1997).
The expression of CNTF has only been detected in reactive astrocytes and not in neurons after cerebral ischemia. The expression in reactive astrocytes was present after 12 h of reperfusion after cerebral ischemia and gradually increased with a peak at 14 days (Lin et al, 1998; Park et al, 2000). These findings are common in focal cerebral ischemia and transient global ischemia models, but this CNTF expression pattern is apparently different from those of IL-6 and LIF (Lin et al, 1998; Park et al, 2000).
In conclusion, IL-6 is potentially expressed in various cell types, although the cellular sources of LIF and CNTF are restricted. In contrast to IL-6, astrocytes are major sources of LIF and CNTF. Unfortunately, the expressions of other IL-6 type cytokines, such as IL-11, oncostatin-M, and cardiotrophin-1, have not yet been studied in experimental cerebral ischemia.
Regulation of Neuronal Interleukin-6 Expression
In vitro studies on astrocytes have revealed that IL-6 induction is regulated by many factors, such as other cytokines, various neurotransmitters, and second messengers (Van Wagoner and Benveniste, 1999). Among these factors, increase in extracellular adenosine is suggested to increase primary astrocyte IL-6 secretion and gene transcription in ischemia (Schwaninger et al, 1997). However, the mechanism of neuronal IL-6 expression after cerebral ischemia has not been fully studied yet.
Membrane depolarization followed by the rapid translocation of Ca2+ from the extracellular to the intracellular space is one of the major events in the acute phase of cerebral ischemia (Dirnagl et al, 1999). Sallmann et al (2000) investigated the regulation of IL-6 expression in the PC-12 cell line. They clearly showed that neuronal IL-6 expression is regulated by membrane depolarization and inhibited by decreased extracellular Ca2+ levels. Additionally, IL-6 mRNA expression is induced by glutamatergic receptor activation (Minami et al, 1991; Schiefer et al, 1998). In an in vivo cerebral ischemia model, neuronal IL-6 expression was significantly suppressed by a specific blocker of voltage-sensitive Ca2+ blocker (Suzuki et al, 2000b). Spreading depression or ischemia-induced depolarization propagates from the ischemic cortex to the contralateral cortex, and it may be one of the most important factors for gene induction in remote regions (Sharp et al, 2000). Thus, neuronal IL-6 expression in the cerebral cortex on the nonischemic side is considered to be triggered by membrane depolarization followed by a massive influx of Ca2+. Ali et al (2000) showed that the calcium ions influx evoked by over-activation of N-methyl
Roles of interleukin-6 type cytokine in cerebral ischemia
Interleukin-6
Many in vivo and in vitro studies have shown that IL-6 exerts a neuroprotective effect in several types of brain injury (Hama et al, 1991; Maeda et al, 1994; Yamada and Hatanaka, 1994; Hirota et al, 1996). Interleukin-6 improves the survival of CNS neurons and reduces excitotoxic neuronal damage against NMDA-mediated injury (Toulmond et al, 1992; Carlson et al, 1999; Ali et al, 2000) and protects neurons against apoptosis (Shioda et al, 1998).
In the case of cerebral ischemia, Matsuda et al (1996) continuously injected recombinant IL-6 for 7 days into the lateral ventricle of gerbils subjected to transient forebrain ischemia, and they found that the IL-6 injection prevented learning disabilities and delayed neuronal loss. Loddick et al (1998) examined the effect of intracerebroventricular recombinant IL-6 injection in rats after permanent MCAO. Interleukin-6 dramatically reduced ischemic brain damage 24 h after MCAO compared with animals injected with saline (Loddick et al, 1998). These results from different experimental models clearly show the neuroprotective effects of IL-6 on cerebral ischemia, though the injection of a recombinant protein does not provide an accurate method for investigating the role of endogenous IL-6 (Matsuda et al, 1996; Loddick et al, 1998). In line with these findings, Yamashita et al (2005) intraperitoneally injected the anti-mouse IL-6 receptor monoclonal antibody (IL-6RA) in mice immediately after MCAO and found that it induced an increase in the number of apoptotic cells and resulted in an enlarged infarct size. In a traumatic brain injury model, IL-6 has been shown to promote posttraumatic healing in the CNS by repairing endothelial cells, which suggests that IL-6 may enhance angiogenesis or revascularization after ischemic injury (Swartz et al, 2001). In vivo studies have shown that the upregulation of IL-6 after induced ischemia represents an endogenous neuroprotective mechanism against NMDA receptor-mediated injury (Ali et al, 2000). It is now widely accepted that IL-6 induction after cerebral ischemia has neuroprotective effects.
However, Clark et al (2000) investigated whether mice lacking IL-6 were protected from acute ischemic injury and found that there were no differences in the infarct size or neurologic function between IL-6 knockout mice and normal littermates 24 h after cerebral ischemia. The authors speculated that IL-6 may not have a direct influence on acute ischemic injury. Interleukin-6 is a major regulator of body temperature (Chai et al, 1996). Herrmann et al (2003) speculated that this interpretation ignored postsurgical hypothermia in IL-6-deficient mice and reported that if body temperature is controlled by external warming after MCAO, IL-6-deficient mice show reduced survival, worse neurologic status, and larger infarcts than control animals.
Leukemia Inhibitory Factor and Ciliary Neurotrophic Factor
In contrast to the ample evidence for the neuroprotective effects of IL-6 from various studies, only one published study has examined the role of LIF in cerebral ischemia (Suzuki et al, 2005). We injected recombinant LIF stereotaxically into the cerebral cortex, adjacent to the inner boundary zone of the infarct area, soon after permanent MCAO in the rat. The LIF clearly reduced the neurologic deficits and infarct volume 24 h after permanent MCAO (Suzuki et al, 2005). In in vivo models of CNS injury, LIF enhanced the expression of neurotrophin-3 and corticospinal axonal growth after spinal cord injury (Blesch et al, 1999) and reduced immune-mediated demyelination by enhancing oligodendrocyte survival (Butzkueven et al, 2002). In addition, LIF is closely associated with CNS injury-induced neurogenesis and promotes the differentiation of neuronal progenitor cells (Hilton, 1992; Shimazaki et al, 2001; Hatta et al, 2002; Bauer et al, 2003). Taken together, these studies suggest that LIF may exert neuroprotective and neurogenesis effects after cerebral ischemia.
Various researchers have shown that CNTF is neuroprotective, and has been known to be one of the neurotrophic factors, in cerebral ischemia (Kumon et al, 1996; Ogata et al, 1996; Hermann et al, 2001). A continuous infusion of CNTF into the cerebral ventricles or an adenovirus-mediated CNTF treatment has been shown to reduce neuronal injury after experimental ischemia (Kumon et al, 1996; Hermann et al, 2001). However, the protective effect of CNTF in cerebral ischemia is likely to be weak compared with that of nerve growth factor or brain-derived neurotrophic factor (Ogata et al, 1996).
In conclusion, IL-6, LIF, and CNTF can be regarded as neurotrophic factors in cerebral ischemia, and they share some common functional properties. These proteins stimulate the same intracellular signaling pathways via a common receptor protein, gp130, though their temporal and cellular expressions show different kinetic patterns after cerebral ischemia.
Alteration of interleukin-6 type cytokine receptors and signal molecules
Receptors
To clarify the exact roles of cytokines in cerebral ischemia, it is necessary to examine the localization and alteration of the cytokine receptor components after cerebral ischemia. Receptor proteins of the IL-6 cytokine family, including gp130, IL-6R, LIFR, and CNTFR, are distributed in the neurons under normal conditions (Watanabe et al, 1996; Yamakuni et al, 1996; Morikawa et al, 2000; Park et al, 2000). These receptor proteins are decreased in parallel with the neuronal damage in cerebral ischemia. Surviving neurons continue to express these receptor proteins and remain as the possible targets for these cytokines. However, the receptor expression levels seem to be unchanged, in contrast to the marked upregulation of the cytokines themselves (Vollenweider et al, 2003; Suzuki et al, 2005).
After cerebral ischemia, glial cells and the endothelium possibly express receptors for the IL-6 cytokine family, though a varied expression of gp130, IL-6R, LIFR, and CNTFR has been reported by different investigators. Based on recent reports, it is now thought that the gp130 protein is widely distributed in various types of cells in the ischemic rat brain, whereas the α-subunits are likely to be limited to neurons (Watanabe et al, 1996; Morikawa et al, 2000; Park et al, 2000; Vollenweider et al, 2003). Other investigators have reported that reactive astrocytes express gp130 and CNTFR after ischemic insults, but not IL-6R (Choi et al, 2003; Kim et al, 2004; Vollenweider et al, 2003). Microglia, oligodendrocytes, and endothelial cells appear to express only gp130 (Park et al, 2000; Choi et al, 2003; Vollenweider et al, 2003; Kim et al, 2004; Suzuki et al, 2005). It is well known that glial cell cultures respond to IL-6 treatment. As the function of IL-6R is to efficiently induce dimerization in response to respective ligands (Taga and Kishimoto, 1997), some of IL-6 signaling is likely dependent on gp130 dimerization.
The above-mentioned findings are summarized in Figure 3. Although the alterations in these receptor components depend on the CNS injury model, the main source of receptor proteins, under both normal and ischemic conditions, is neurons. Together, these results indicate that cerebral neurons produce IL-6 and LIF in response to ischemic stress and these cytokines act as a neuroprotective mediators in a paracrine or autocrine manner.
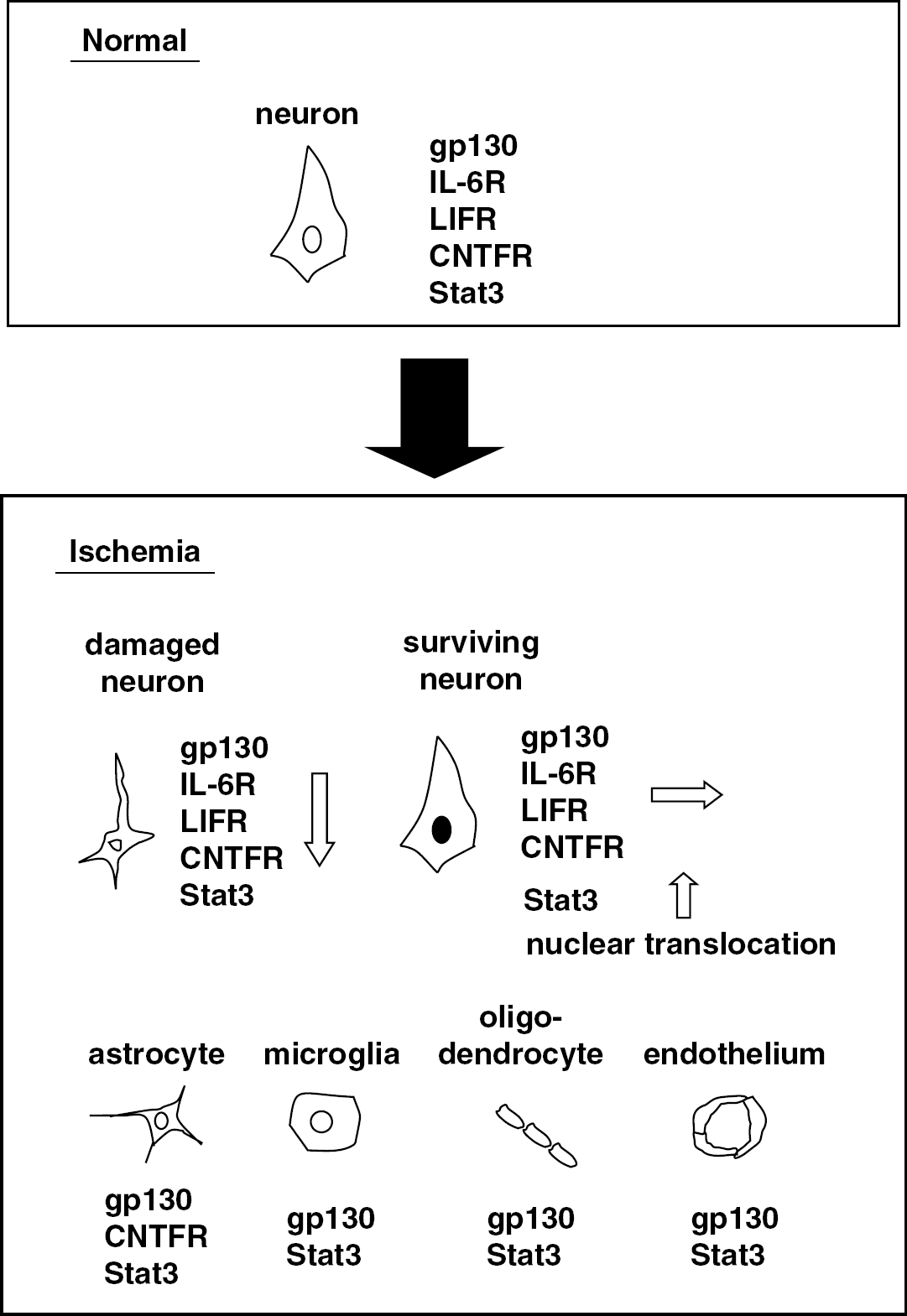
Cellular origins of interleukin (IL)-6 cytokine receptors and Stat3. A schema shows the alterations of IL-6 cytokine receptor components including gp130, IL-6R, leukemia inhibitory factor receptor (LIFR) and ciliary neurotrophic factor receptor (CNTFR), and Stat3 under normal conditions and in ischemia. In normal conditions, the receptors are localized in neurons. In ischemia, receptor expression is decreased in damaged neurons. In surviving neurons, however, Stat3 is only upregulated with nuclear translocation. Astrocytes, microglia, oligodendrocytes, and the endothelium express gp130 and Stat3.
Stat3
Among the various molecules involved in the JAK—STAT pathway, Stat3 is the most important and has attracted much attention. The wide distribution of Stat3 protein in normal rat neurons has been reported (Stromberg et al, 2000). In parallel with a decline in receptor proteins, the neuronal expression of Stat3 gradually decreases in the ischemic region (Suzuki et al, 2001). However, Stat3 is upregulated in the surviving neurons, especially in the penumbra. The protein is detected in both the cytoplasm and the nucleus of most neurons, indicating that the translocation of Stat3 occurs soon after ischemia as the initial step of activated signal transduction (Suzuki et al, 2001).
The Stat3 protein can be detected in various types of cells after cerebral ischemia: activated microglia, astrocytes, and oligodendrocytes are all potential sources of Stat3 (Planas et al, 1996; Choi et al, 2003; Kim et al, 2004). Moreover, the translocation of Stat3 into the nuclei of reactive microglia and astrocytes has been reported to occur after transient cerebral ischemia, suggesting the activation of downstream signal pathways (Justicia et al, 2000). Stat3 is also induced in the endothelium of the ischemic core 48 h after MCAO (Suzuki et al, 2001). The alteration of Stat3 localization after cerebral ischemia is illustrated in Figure 3. However, it should be noted that Stat3 is not specific for the signal transduction of the IL-6 cytokine family. The Stat3 protein can be activated by a variety of signals, including other growth factors, cytokines, and oxidative stress (Heinrich et al, 2003).
Phosphorylation of Stat3
Under normal conditions, Stat3 is not phosphorylated. After cerebral ischemia, Stat3 phosphorylation becomes significantly apparent in neurons (Figure 4A; Suzuki et al, 2001; Wen et al, 2001; Dziennis et al, 2007). We have observed that Stat3 is markedly phosphorylated 3.5 h after focal ischemia in the peri-ischemic region, where the normal neuron morphology was maintained. The Stat3 phosphorylation after MCAO peaked after 24 h of reperfusion and continued until day 7, which corresponded to the temporal profile of IL-6 and LIF expression. These data suggest that, after cerebral ischemia, these neurokines play crucial roles in stimulating the JAK—STAT pathway via gp130 in an autocrine and paracrine manner. In another injury model, Stat3 phosphorylation was also observed in neurons (Haas et al, 1999).
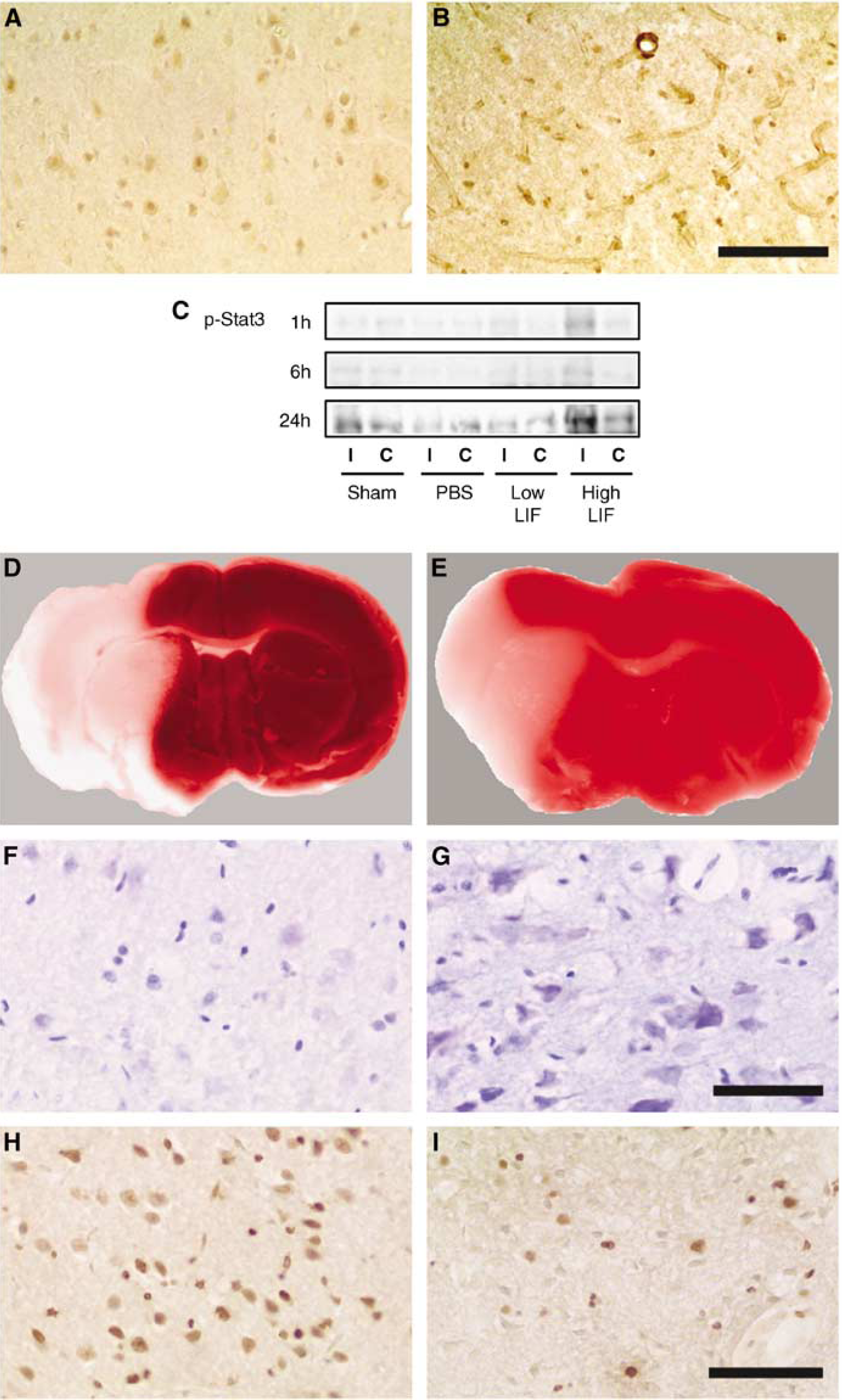
Cellular origins and roles of Stat3 phosphorylation. (
Another source of phosphorylated Stat3 is endothelial cells. In contrast to neurons, the immunoreactivity of phosphorylated endothelial Stat3 seems to be localized in the cytoplasm. Further investigations using specific vessel markers are necessary. Interestingly, endothelial Stat3 is selectively phosphorylated in the ischemic core 48 h after cerebral ischemia (Figure 4B), though a severe suppression of neuronal IL-6 and LIF expression has been observed (Suzuki et al, 2001). This finding may be closely associated with angiogenesis by blood-derived endothelial progenitor cells that activate IL-6 production in the endothelium (Fan et al, 2008). However, the reason why the kinetic pattern of Stat3 phosphorylation is different from that of IL-6 and LIF may be because of the effect of other cytokines, such as granulocyte colony-stimulating factor (Schabitz et al, 2003; Komine-Kobayashi et al, 2006).
Although reactive astrocytes, macrophages, and microglia are considered to be the main source of phosphorylated Stat3 after cerebral ischemia (Choi et al, 2003; Satriotomo et al, 2006), other studies have failed to detect glial Stat3 phosphorylation (Suzuki et al, 2001; Wen et al, 2001). Accordingly, varied cellular origins of Stat3 phosphorylation have been reported by different investigators. This discrepancy may be explained by differences in experimental protocols. The other possible reason is the effect of the JAK—STAT pathway regulatory system, such as SOCS3 and PIAS.
Regulatory System
Among the inhibitory molecules in the JAK—STAT pathway, the increased expression of SOCS3 mRNA has been found after transient MCAO in rats (Raghavendra Rao et al, 2002). In the mouse model of focal ischemia, SOCS3 mRNA was upregulated after ischemia, but this upregulation was markedly blocked by IL-6RA (Yamashita et al, 2005). However, the chronologic alteration and the cellular source of the SOCS3 protein and another inhibitory molecule, PIAS, have not yet been determined in cerebral ischemia. The molecular mechanisms underlying the regulation of the JAK—STAT pathway are very complex and still not fully understood, whereas there is no doubt about their major roles in the regulation of cytokine functions (Planas et al, 2006).
Roles of Stat3 phosphorylation in cerebral ischemia
In general, transcription factors play a pivotal role in controlling inflammatory genes after cerebral ischemia. The activation of hypoxia inducible factor-1, cAMP response element binding protein (CREB), c-fos, peroxisome proliferator-activated receptors (PPAR) and/or p53 is known to prevent ischemic neuronal damage, whereas the induction of inositol requiring (IRE)-1, activating transcription factor-2, Stat3, NF-κB, early growth response-1 (Egr1) and/or ccaat enhancer binding protein (C/EBP)-β promotes inflammation and neuronal death after cerebral ischemia (Tanaka, 2001; Yi et al, 2007). Satriotomo et al (2006) examined the functional significance of Stat3 activation in a rat model of focal ischemia by intraventricular infusion of specific JAK—STAT inhibitors. Blocking Stat3 phosphorylation significantly decreased the infarct volume. In regard to inflammation, Stat3 phosphorylation in macrophages or microglia may contribute to neuronal damage after cerebral ischemia (Satriotomo et al, 2006). In addition, Wen et al (2001) reported that Stat3 phosphorylation in cerebral ischemia is involved in cell death. In regards to neurogenesis, the suppression of Stat3 directly induces neurogenesis in neuronal stem cells (Gu et al, 2005). It has also been reported that the gp130-mediated activation of the Stat3 signaling pathway may play a key role in the induction of astrogliosis (Sriram et al, 2004).
However, other data do not support this conclusion. In fact, several studies have clearly shown that Stat3 phosphorylation is associated with neuroprotection against cerebral ischemia. We observed that rapid enhancement of Stat3 phosphorylation was detected after the injection of high-dose recombinant LIF into the cerebral cortex on the ischemic side in rat focal ischemia (Figure 4C). The phosphorylation of Stat3 was associated with a decrease in ischemic brain damage, including a reduction in the area of the infarct and the number of TUNEL-positive cells (Figure 4D—I; Suzuki et al, 2005). In line with this finding, a significant reduction of Stat3 phosphorylation induced by IL-6RA administration in mice after MCAO was found to be associated with an enlarged infarct size and aggravated neurologic deficits (Yamashita et al, 2005). The role of Stat3 in cell survival seems to be linked to the transcriptional regulation of antiapoptotic regulatory proteins, such as the Bcl-2 family (Battle and Frank, 2002), strongly suggesting that the phosphorylation of Stat3 has antiapoptotic effects.
The neuroprotective effects of Stat3 phosphorylation are also supported by recent reports. Estradiol was found to increase Stat3 activation and reduce infarct volume in the female rat brain after cerebral ischemia, and reduced infarct volume, and its neuroprotective effects were abolished by a Stat3 inhibitor (Dziennis et al, 2007). In addition, secretoneurin promotes neuroprotection and neuronal plasticity via Stat3 phosphorylation by inhibiting caspase-3 activity and increasing the expression of antiapoptotic proteins in a murine model of stroke (Shyu et al, 2008). Moreover, Stat3 phosphorylation occurs in neuronal stem cell or progenitor cells within the subventricular zone by Notch1 induction after perinatal hypoxia/ischemia (Covey and Levison, 2007). The Stat3 signaling pathway is now known as one of the transcriptional regulators of neurogenesis (Scholzke and Schwaninger, 2007). Besides neurogenesis, angiogenesis is also enhanced by Stat3 phosphorylation in vivo and in vitro (Fan et al, 2008). The phosphorylation of Stat3 has been detected in endothelial progenitor cells and activates angiogenesis, resulting in increased blood flow and the alleviation of tissue injury (Fan et al, 2008).
Taken together, further investigation is necessary to clarify the exact role of Stat3 signaling in neuroprotection.
Conclusions
Both clinical and experimental studies have indicated that cerebral ischemia promotes the marked expression of IL-6 in the brain and in serum. IL-6 is thought to play ambivalent roles, depending on the stages of cerebral ischemia (Figure 5). During the acute phase, IL-6 mainly works as an inflammatory cytokine and may contribute to ischemic injury. However, IL-6 possibly has the important effects of repair and regeneration, which can only be successful if the ‘mopping’ function of the inflammatory cells takes place. In addition, during the subacute phase, IL-6 mainly works as a neuroprotective mediator with LIF and CNTF. The phosphorylation of Stat3 is activated by IL-6, LIF, or CNTF and contributes to tissue repair after cerebral ischemia by antiapoptosis activity.
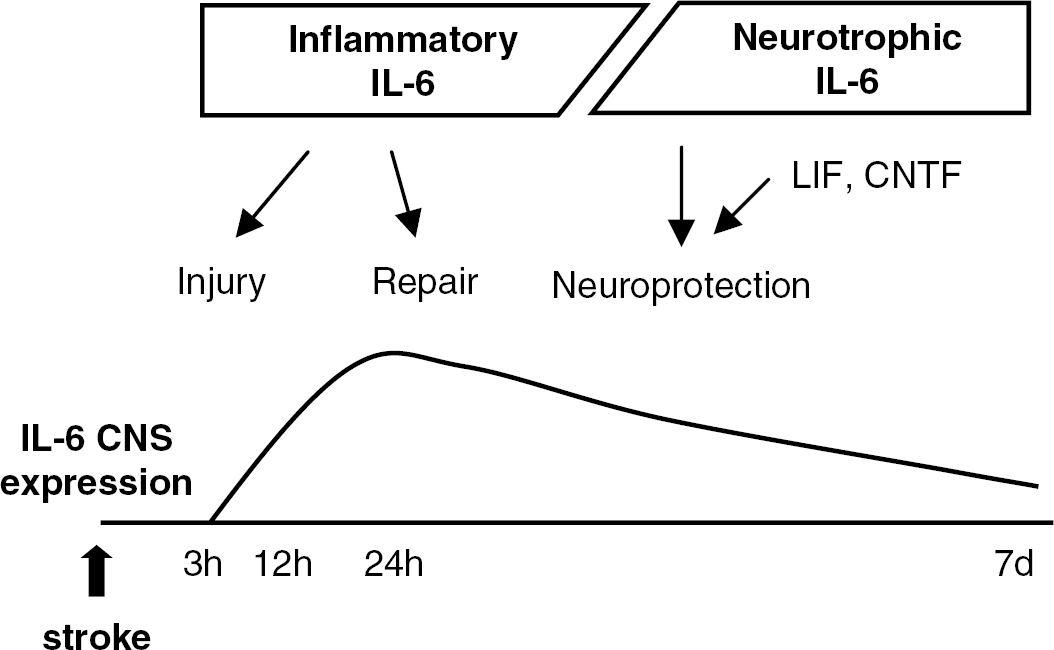
Ambivalent aspects of interleukin (IL)-6 after cerebral ischemia. In the central nervous system (CNS), IL-6 works as an inflammatory cytokine during the acute phase of cerebral ischemia and may contribute to both injury and repair. However, the peak in IL-6 expression is associated with neuroprotection in conjunction with leukemia inhibitory factor (LIF) and ciliary neurotrophic factor (CNTF).
Further studies should elucidate the precise role of IL-6 and Stat3 phosphorylation over a longer period, such as the first 2 to 3 months after cerebral ischemia using selective pharmacologic agents of this signaling.
Footnotes
Acknowledgements
The authors sincerely thank Dr Yasuo Fukuuchi, Dr Shigeru Nogawa, Dr Eiichiro Nagata, Dr Daisuke Ito, Dr Tomohisa Dembo, and Dr Arifumi Kosakai for their valuable collaboration and suggestions.
All authors have declared no conflict of interest.