
Abstract
Sublethal insults can induce a transient tolerance toward subsequent lethal ischemia, a phenomenon termed ischemic preconditioning (IPC). In the myocardium, nitric oxide derived from ‘inducible’ nitric oxide synthase (iNOS or NOS II) plays a critical role in the expression of IPC produced by sublethal ischemia. Here, we investigated whether iNOS is involved in IPC in brain. Ischemic preconditioning was produced in mice by three episodes of 1-min bilateral common carotid artery (BCCA) occlusion, each followed by 5 mins of reperfusion. After 24 h, mice underwent middle cerebral artery (MCA) occlusion for 20 mins. Intraischemic cerebral blood flow was monitored during both in BCCA and MCA occlusion (MCAO) by laser-Doppler flowmetry. Mice were killed 3 days after MCAO, and infarct volume was determined in thionine-stained sections. Infarct volume was significantly reduced 24 h after IPC (70%; P<0.05). Treatment with the iNOS inhibitor aminoguanidine (400 mg/kg), abolished the IPC-induced protection. Furthermore, IPC failed to induce ischemic tolerance in iNOS-null mice. In wild-type mice, IPC increased the resistance to Ca2+-mediated depolarization in isolated brain mitochondria. However, in iNOS-null mice IPC failed to induce such resistance. We conclude that iNOS is required for the full expression of IPC and that such effect is coupled to an increased resistance of mitochondria to injury. Thus, iNOS-derived nitric oxide, in addition to its deleterious effects on the late stages of ischemic brain damage, can also be beneficial by promoting ischemic tolerance through signaling, ultimately resulting in mitochondrial protection.
Introduction
Activation of injury cascades by a sublethal stimulus induces tolerance toward a subsequent lethal ischemic insult, a phenomenon termed ischemic preconditioning (IPC) (Dirnagl et al, 2003). In many organs, IPC occurs in two temporally distinct types: early and delayed. In early preconditioning the tolerance develops within minutes to hours after the inducing stimulus, while in late preconditioning within hours to days (Bolli, 2001). In brain, both forms of preconditioning have been described, but delayed preconditioning has been studied more extensively (Kirino, 2002). A wide variety of stimuli can induce ischemic tolerance, including pharmacological agents, sublethal ischemia, oxidative stress, hyperbaric oxygenation, inflammation, seizures, spreading depression and heat stress (Dirnagl et al, 2003; Kirino, 2002). Although the cellular and molecular bases of the phenomenon have not been fully elucidated, there is evidence that preconditioning stimuli increase mitochondrial resistance to stress (Argaud et al, 2004; Dave et al, 2001; Zhan et al, 2002; Zhang et al, 2003). However, the factors that trigger such increased mitochondrial resistance remain unknown.
Nitric oxide (NO), a free radical well known for its participation in ischemic brain injury, has also been implicated in the mechanisms of ischemic tolerance (Kirino, 2002; Nandagopal et al, 2001). The endothelial isoform of NO synthase (eNOS) contributes to the induction of tolerance in neonatal hypoxic–ischemic brain injury (Gidday et al, 1999), while eNOS and neuronal NOS (nNOS) have been implicated in the development of early preconditioning in a model of focal cerebral ischemia (Atochin et al, 2003). Much less is known on the role of ‘inducible’ NOS (iNOS or NOSII). In the heart, iNOS plays a crucial role in the expression of IPC (Dawn and Bolli, 2002). However, in brain it is unknown whether iNOS is involved in the expression of ischemic tolerance triggered by prior sublethal ischemia.
In this study, we used pharmacological and genetic approaches to investigate whether iNOS is required for cerebral ischemic tolerance in a model of preconditioning induced by sublethal ischemia. We found that iNOS is critical for the expression of tolerance to focal cerebral ischemia induced by brief episodes of bilateral carotid occlusion, and for the attendant increase in mitochondrial resistance to Ca2+ overload underlying the tolerance. The findings suggest that iNOS, depending on the setting of its action, can either be deleterious to the postischemic brain or can activate neuroprotective pathways, resulting in increased mitochondrial resistance to injury.
Materials and methods
Animals
All experimental procedures were approved by the institutional animal care and use committee. Experiments were performed in male C57BL/6J mice (age 2 to 3 months; weight 20 to 22 g) and male iNOS-null mice obtained from an in-house colony (Iadecola et al, 1997). The iNOS-null mice were backcrossed into the C57BL/6J strain for more than 10 generations. Procedures for breeding and genotyping iNOS-null mice were described previously (Iadecola et al, 1997).
Ischemic Preconditioning by Bilateral Carotid Occlusion
Mice were anesthetized with a mixture of isoflurane (1.5% to 2%), oxygen, and nitrogen. A flexible fiber-optic probe was glued to the parietal bone (−2 mm AP, 5 mm ML to bregma) and connected to a laser-Doppler flowmeter (Periflux System 5010, Perimed, Sweden). Cerebral blood flow (CBF) was continuously recorded using a computer-based data acquisition system (Perisoft). Through a midline incision of the neck, 4–0 surgical threads were loosely placed around common carotid arteries. Preconditioning was induced by three episodes of 1-min occlusion of both common carotid arteries, each followed by 5 mins of reperfusion. Wounds were closed and animals were returned to their cage. In sham-operated mice, the carotid arteries were exposed, surrounded by the thread but not occluded. Some mice were treated with the iNOS inhibitor aminoguanidine (AG; 400 mg/kg, i.p.; Sigma). We have previously shown that this dose of AG inhibits iNOS selectively (Iadecola et al, 1995a; Sugimoto and Iadecola, 2002).
Transient Middle Cerebral Artery Occlusion
At 3 to 72 h after the preconditioning stimulus, mice were reanesthetized and the fiber-optic flow probe was reattached to the right parietal bone for continuous measurement of CBF (see previous section). The right middle cerebral artery (MCA) was transiently occluded by an intravascular filament as described previously (Park et al, 2004a). Briefly, a heat-blunted black monofilament surgical suture (6–0) was inserted via the external carotid artery, advanced into the internal carotid artery and wedged into the circle of Willis to obstruct the origin of the MCA. The filament was left in place for 20 mins and then withdrawn to reestablish CBF. Only animals that exhibited a reduction in CBF >85% during MCA occlusion (MCAO) and in which CBF recovered by >90% after 10 mins of reperfusion were included for the study. Rectal temperature was kept at 37.0°C±0.5°C by heating a pad thermostatically regulated by a rectal probe, both during surgery and in the recovery period until animals regained consciousness.
Infarct Volume Measurement
Procedures for measurement of infarct volume have been described in detail previously (Iadecola et al, 1997; Nogawa et al, 1997) and are briefly summarized. Mice were killed 3 days after MCAO. Brains were removed, frozen and sectioned (30 μm thicknesses) in a cryostat. Brain sections were collected serially at 600 μm intervals, and stained with thionine. Infarct volume was determined using an image analyzer (MCID, Imaging Research Inc.). To eliminate the contribution of postischemic edema to the volume of injury, infarct volume measurements were corrected for swelling according to the method of Lin et al (1993).
Inducible Nitric Oxide Synthase Immunocytochemistry
Immunocytochemical procedures were identical to those described previously (Iadecola et al, 1996; Nogawa et al, 1997). Sections (7 μm) from formalin-fixed, paraffin-embedded brains were incubated overnight (4°C) with an iNOS polyclonal antibody (Upstate Biotechnology Incorporated; dilution 1: 5000) or with antibodies to the peroxynitrite marker 3-nitrotyrosine (Upstate Biotechnology Incorporated; dilution 1: 2000). Sections were washed and incubated with the secondary antibody (Vector) for 30 mins. In certain conditions associated with inflammation, tyrosine nitration can also be promoted by nitrite (Eiserich et al, 1998). However, in the absence of florid inflammation, this is unlikely to be a relevant factor affecting the specificity of 3-nitrotyrosine as a marker of peroxynitrite. The immunocomplex was visualized using the ABC complex (Vectastain Elite Kit, Vector). The specificity of the immunolabel was tested by preadsorption of the antigen and by removing the primary antibody as described previously (Iadecola et al, 1995b; Park et al, 2004b).
Mitochondria Isolation and Membrane Potential Measurement
Mitochondria from mouse brain cortex were isolated using a differential centrifugation method adapted from Anderson and Sims (2000). Briefly, sham or ischemic preconditioned mice were anesthetized and brains were quickly removed from the skull and placed into ice-cold isolation buffer (225 mmol/L mannitol, 75 mmol/L sucrose, 10 mmol/L HEPES-KOH, pH 7.4, 1 mmol/L EGTA, 0.1% BSA). Dissected cortices from 2 to 3 mice were pooled, homogenized in 7 mL isolation buffer, centrifuged for 5 mins at 2000 g at 4°C. The supernatant was centrifuged subsequently at 12,000 g for 10 mins. The resulting pellets were resuspended in 4 mL of isolation buffer, overlaid onto a 20% to 40% discontinuous Percoll gradient, and the tubes were centrifuged in a Beckman SW41 rotor at 40,000 g for 45 mins. The dark gray band was collected, washed in mitochondria isolation buffer, and centrifuged 8,000 g for 5 mins. The final pellet was resuspended in isolation buffer and the protein concentration of the purified mitochondria was measured using a DC protein assay kit (Bio-Rad). Mitochondria membrane potential was measured according to the procedures described by Fiskum et al (2000). Purified mitochondria were suspended in a quartz cuvette (0.2 mg/mL) containing measuring buffer (210 mmol/L mannitol, 70 mmol/L sucrose, 5 mmol/L KH2PO4, 10 mmol/L HEPES pH 7.4, 2 μmol/L rotenone, 10 μmol/L safranine O, and 5 mmol/L succinate), and the cuvette was place in a fluorescence spectrophotometer (Hitachi F, 2000). The intensity of fluorescence was measured at set excitation/emission wavelengths (485/585 nm). The depolarization of the mitochondrial membrane was achieved by adding CaCl2 into the mitochondria solution (50 μmol/L).
Statistical Analysis
Data are presented as mean±s.d. Comparisons between two groups were statistically evaluated by the Student's t-test. Multiple comparisons were evaluated by the analysis of variance and Fisher's PLSD test. Differences were considered significant at P<0.05.
Results
Temporal Profile of Protection by Ischemic Tolerance
First, we sought to define the time point after the preconditioning stimulus at which the maximal protection could be obtained. C57BL/6J mice were subjected to three brief episodes of bilateral common carotid occlusion (preconditioning stimulus) (Figure 1A), and 3, 24, or 72 h later underwent MCAO for 20 mins (Figure 1B). Infarct volume was assessed 72 h after MCAO. Cerebral blood flow during occlusion and reperfusion did not differ among the groups of mice both during the preconditioning stimulus and MCAO (Figures 2A and 2B). However, infarct volume was reduced (−70%) in only in animals in which the MCA was occluded 24 h after preconditioning, but not 3 or 72 h (Figures 2C and 2D). Although longer durations of preconditioning have been reported in other models, the duration of the protection varies depending on the preconditioning stimulus and nature of the ischemic insult (Atochin et al, 2003; Stagliano et al, 1999; Stenzel-Poore et al, 2004; Wu et al, 2001; Yanamoto et al, 2004).
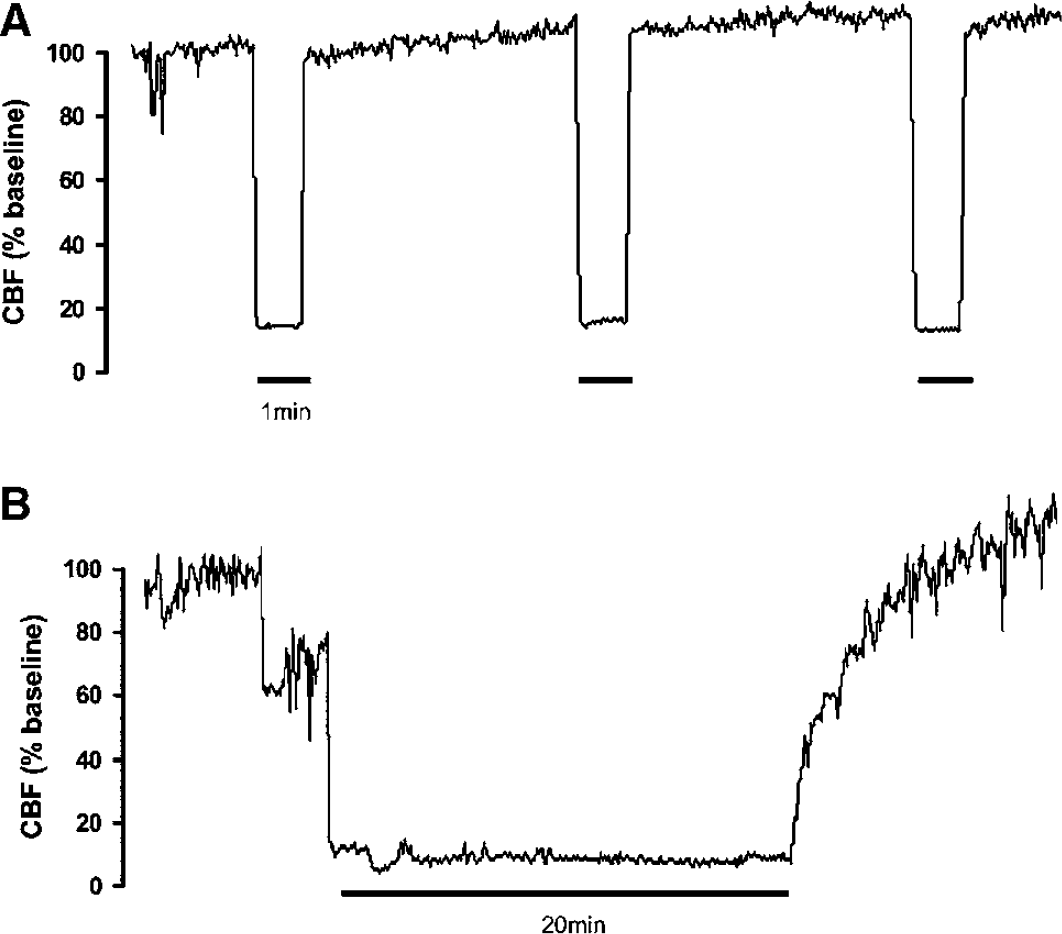
Laser–Doppler CBF recordings during the preconditioning stimulus and MCAO. (
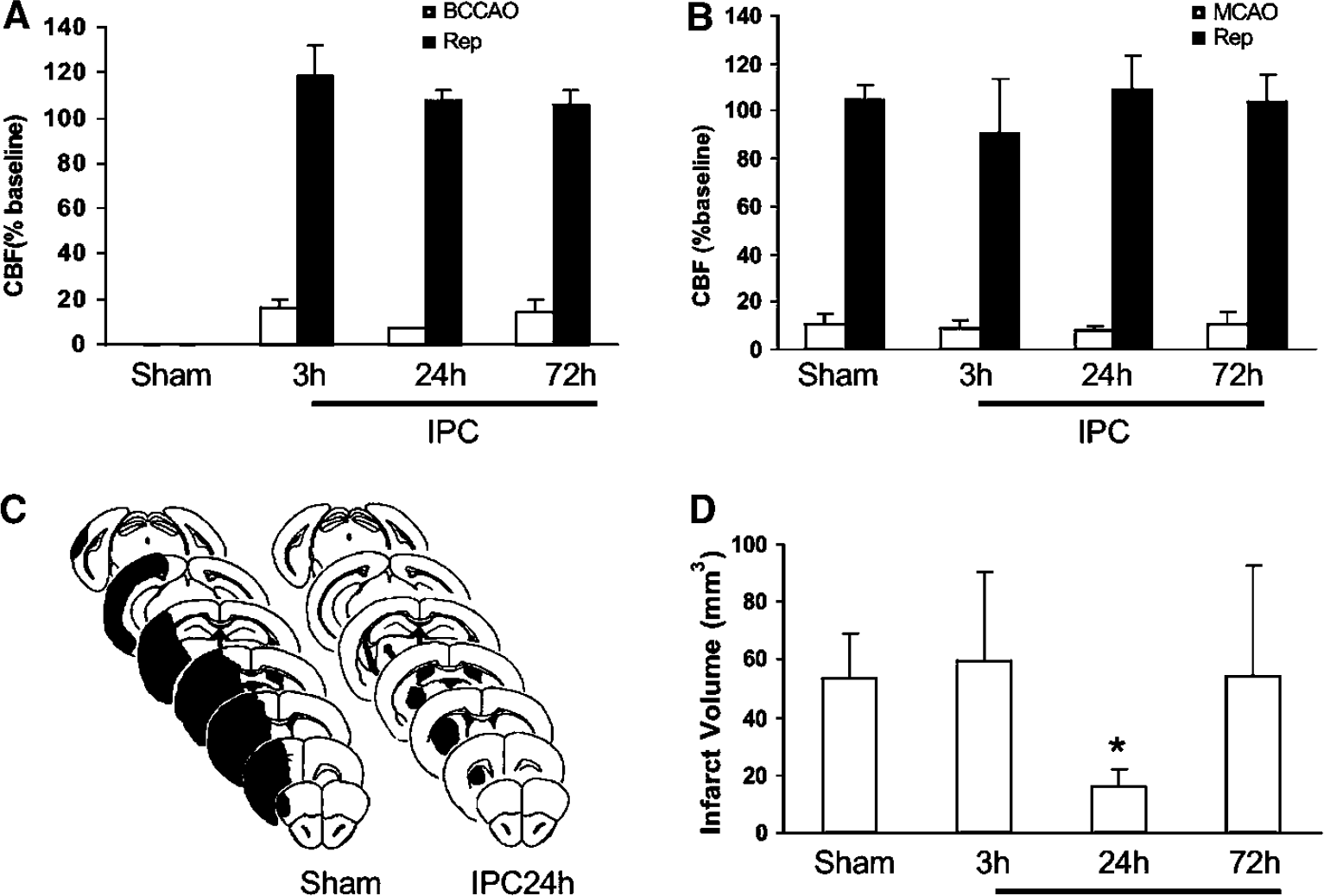
Time window of neuroprotection after preconditioning (IPC). (
Aminoguanidine Prevents Ischemic Preconditioning
We then examined whether iNOS is involved in the protection exerted by IPC. Mice were treated with the iNOS inhibitor AG (400 mg/kg, i.p.) or vehicle 10 mins and 6 h after preconditioning, and underwent MCAO 24 h later. Although CBF during occlusion and reperfusion did not differ between AG-treated and vehicle-treated mice (Figures 3A and 3B), the protection conferred by IPC was not observed in mice receiving AG (Figure 3C). Similarly, treatment with AG without IPC did not reduce infarct volume (Figure 3C).
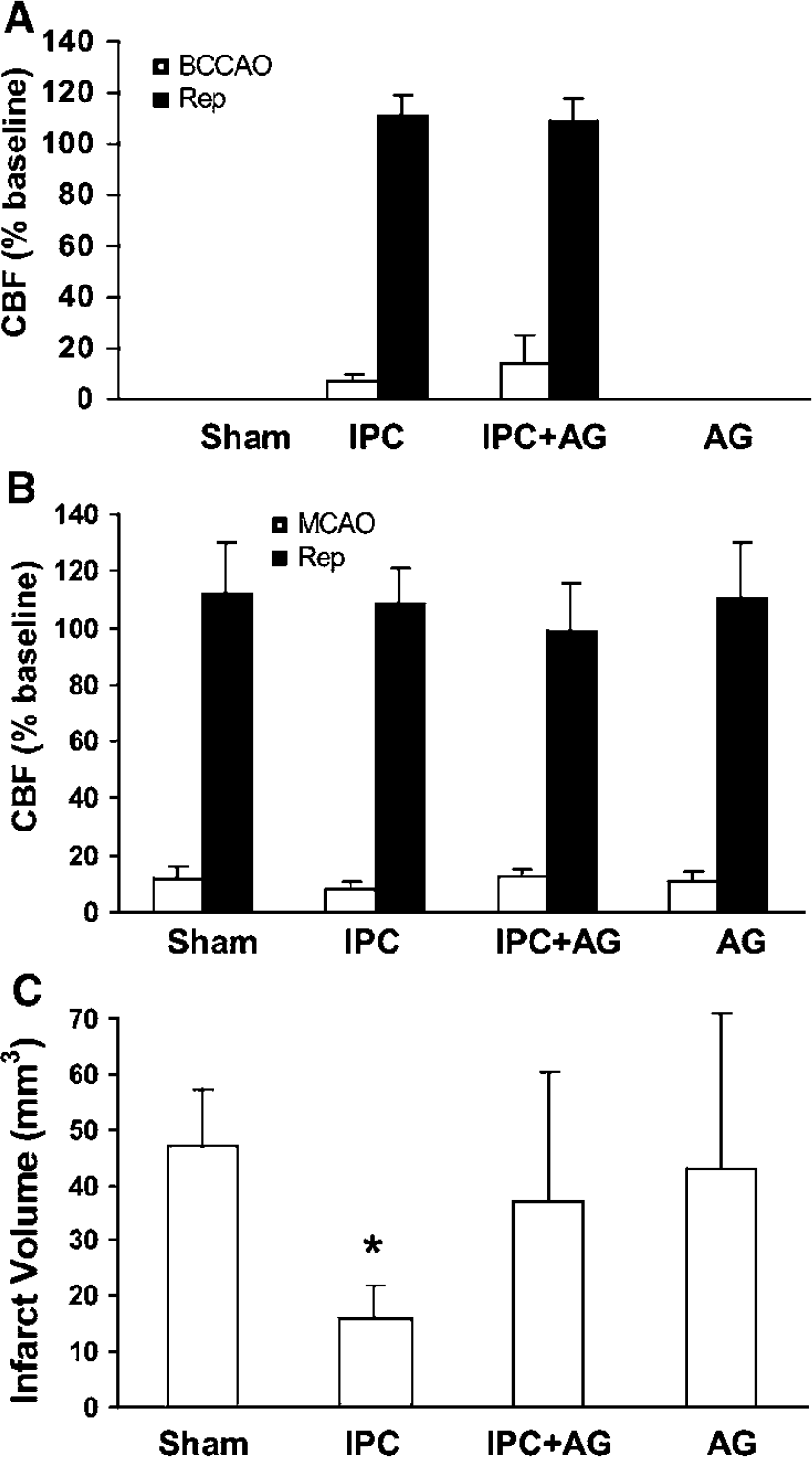
Effect of AG on the IPC induced by bilateral carotid artery occlusion (BCCAO). (
Ischemic Preconditioning Induces Inducible Nitric Oxide Synthase Immunoreactivity in Cerebral Blood Vessels
To determine whether the preconditioning stimulus induced iNOS expression, we used immunocytochemistry. At 24 h after bilateral carotid occlusion, iNOS immunoreactivity was observed in cerebral blood vessels of both cerebral hemispheres, including the cerebral cortex and striatum (Figure 4). Positive vessels had the morphological characteristics of arterioles (diameter 20 to 30 μm, thick wall) (Figure 4A) and venules (40 to 50 μm, thin wall) (Figure 4B). We also examined the immunoreactivity for 3-nitrotyrosine, a marker of peroxynitrite, the reaction product of NO and superoxide (Ischiropoulos, 2003). The 3-nitrotyrosine immunolabel was also located in vessels with a distribution similar to that of iNOS (Figure 4E). Inducible nitric oxide synthase or 3-nitrotyrosine immunoreactivity was not observed in sham-operated controls (Figures 4D and 4F), or after removal of the primary antibody or preadsorption of the antigen (not shown).
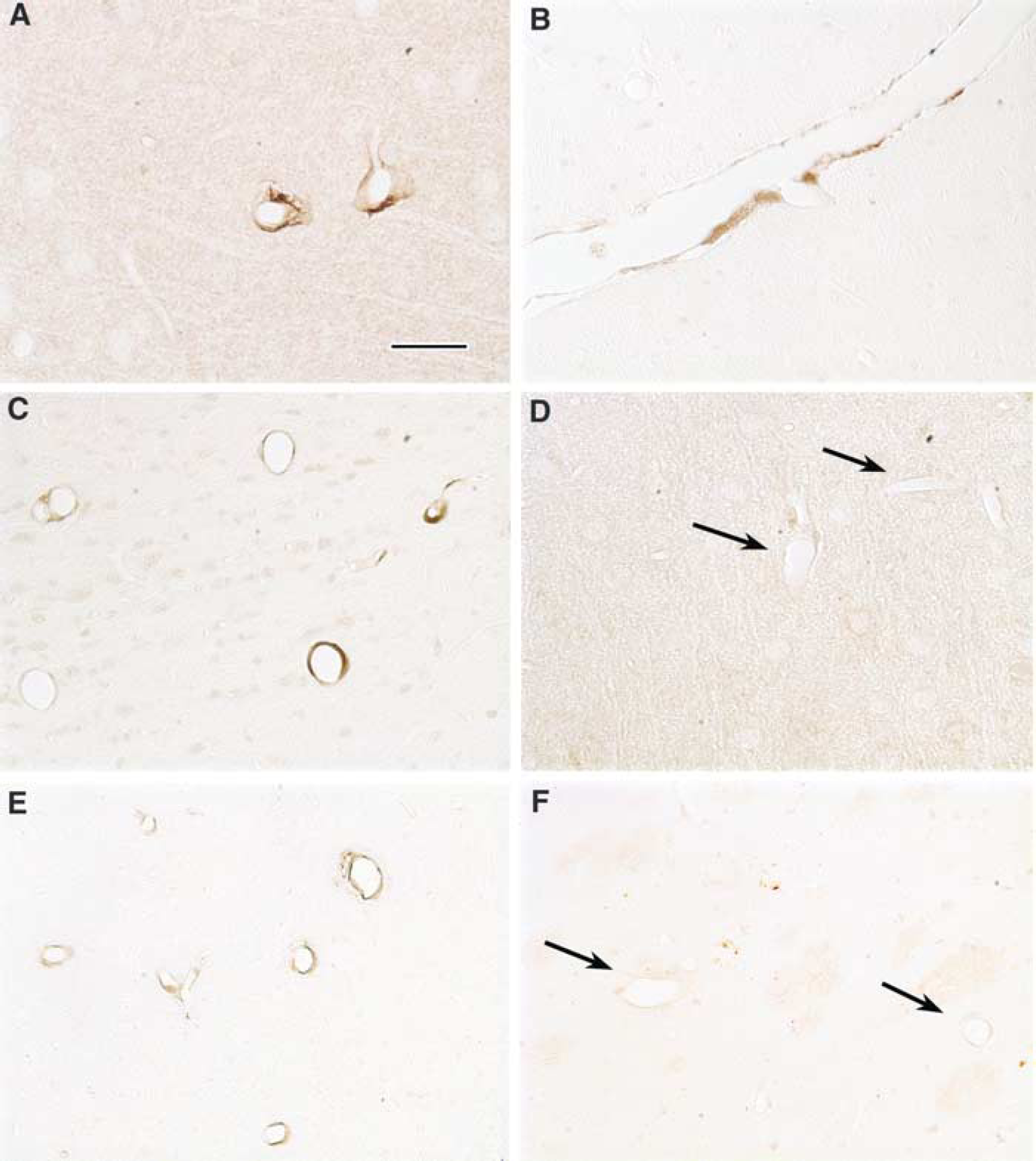
Inducible nitric oxide synthase and 3-nitrotyrosine immunoreactivity 24 h after IPC. Inducible nitric oxide synthase immunoreactivity is observed in 20- to 30-μm vessels with thick walls presumably arterioles (
Ischemic Preconditioning cannot be Induced in Inducible Nitric Oxide Synthase-null Mice
The role of iNOS was also studied in iNOS-null mice. Cerebral blood flow during the preconditioning stimuli and during MCAO–reperfusion did not differ between iNOS-null mice and wild-type littermates (Figures 5A and 5B). However, the preconditioning stimulus was effective in wild-type mice, but failed to produce neuroprotection in iNOS-null mice (Figure 5C).
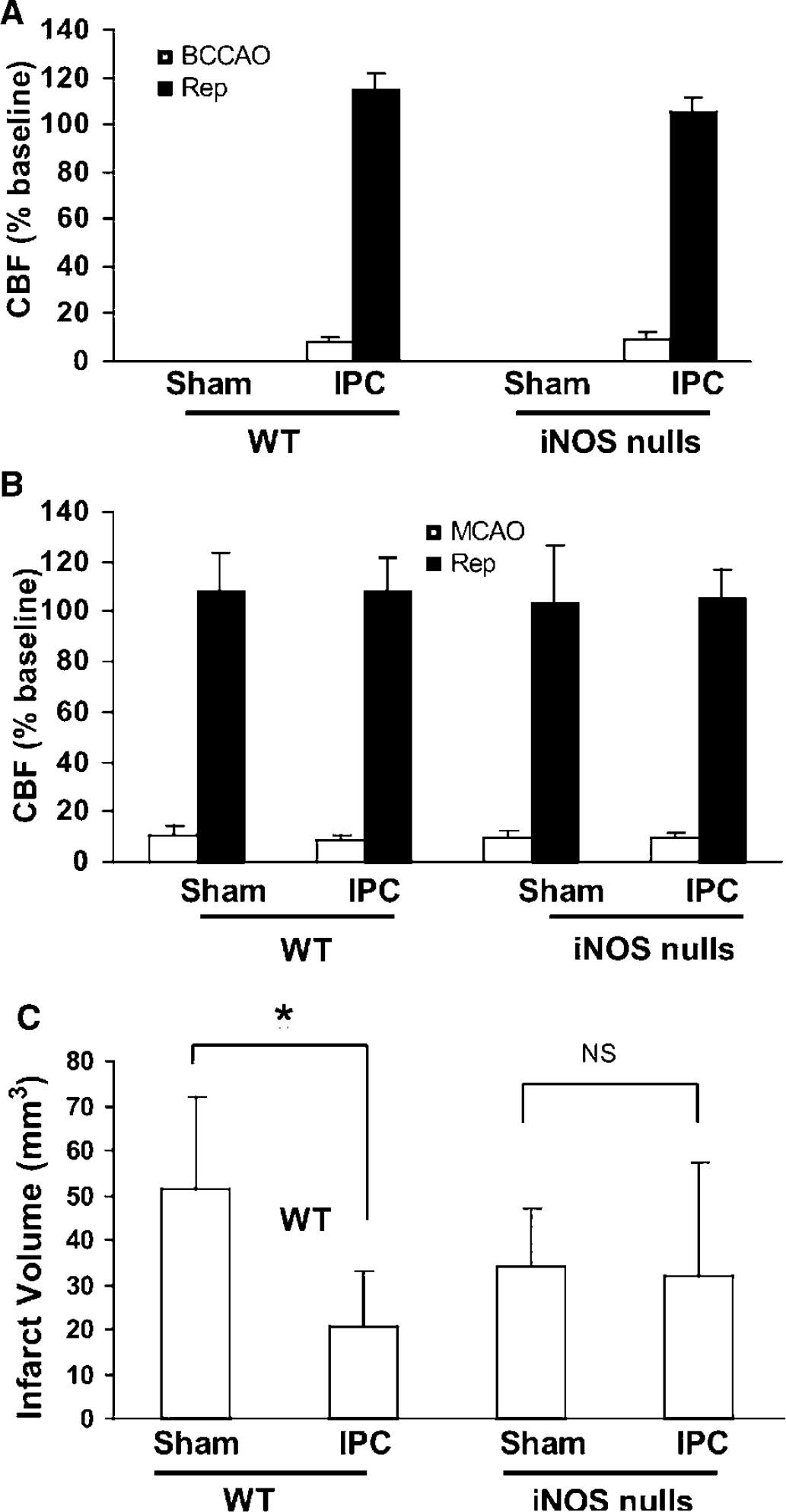
Effect of IPC induced by bilateral carotid artery occlusion (BCCAO) in iNOS-null mice. (
Ischemic Preconditioning does not Protect the Mitochondria of Inducible Nitric Oxide Synthase-null Mice
To investigate whether the role of iNOS in IPC involved the mitochondria, Ca2+-induced depolarization was examined in brain mitochondria isolated from wild-type and iNOS-null mice. Mitochondria from preconditioned wild-type mice maintained a normal membrane potential better than sham-operated mice, indicating increased resistance to injury (Figure 6A). However, mitochondria from preconditioned iNOS-null mice failed to exhibit such a resistance (Figure 6B). In the absence of preconditioning, the mitochondrial tolerance to Ca2+ did not differ between wild-type and iNOS-null mice (data not shown).
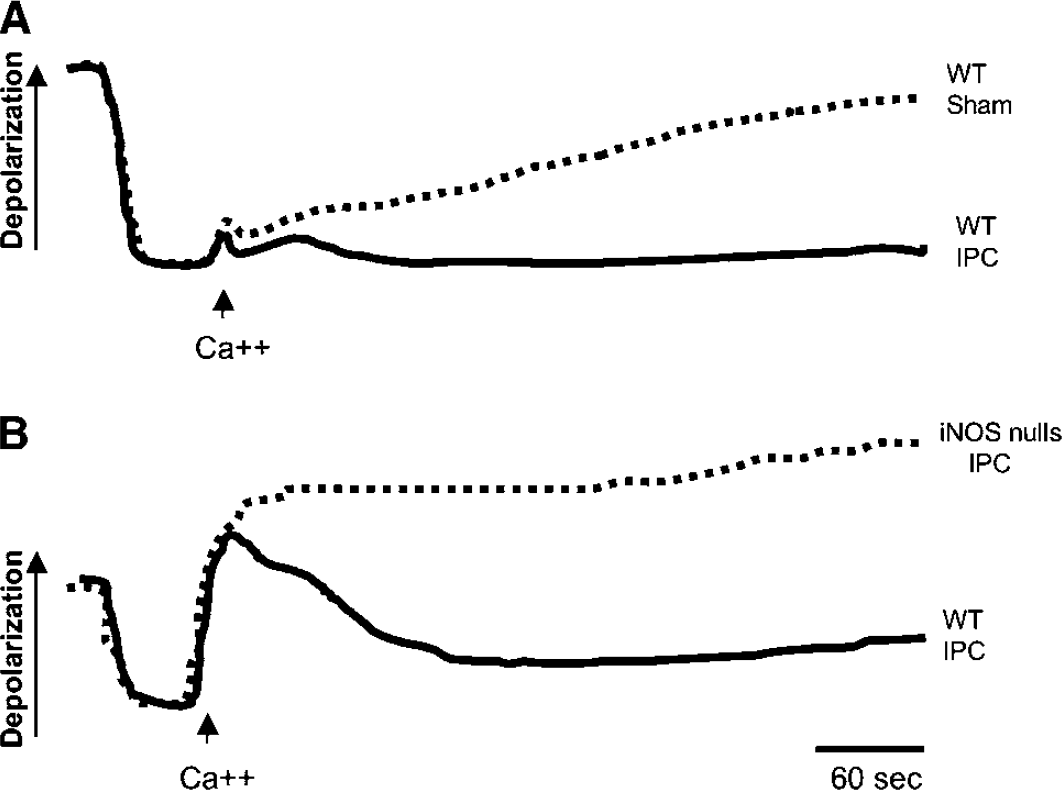
Effect of IPC on Ca2+-induced mitochondrial membrane depolarization in wild-type (WT) and iNOS-null mice. The time interval between IPC and testing was 24 h. (
LPS does not Induce Ischemic Tolerance in Aminoguanidine-treated Mice or in Inducible Nitric Oxide Synthase-null Mice
To determine whether iNOS is also involved in other forms of ischemic tolerance, we examined its role in the preconditioning induced by LPS (Chen et al, 1996; Dawson et al, 1999; Tasaki et al, 1997). Administration of LPS (0.5 mg/kg) reduced the infarct produced by occlusion of the MCA 24 h later by 40% (Figure 7). The reduction was not observed in wild-type mice treated with AG (400 mg/kg, i.p.; 10 mins and 6 h after LPS) (Figure 7A) or in iNOS-null mice (Figure 7B).
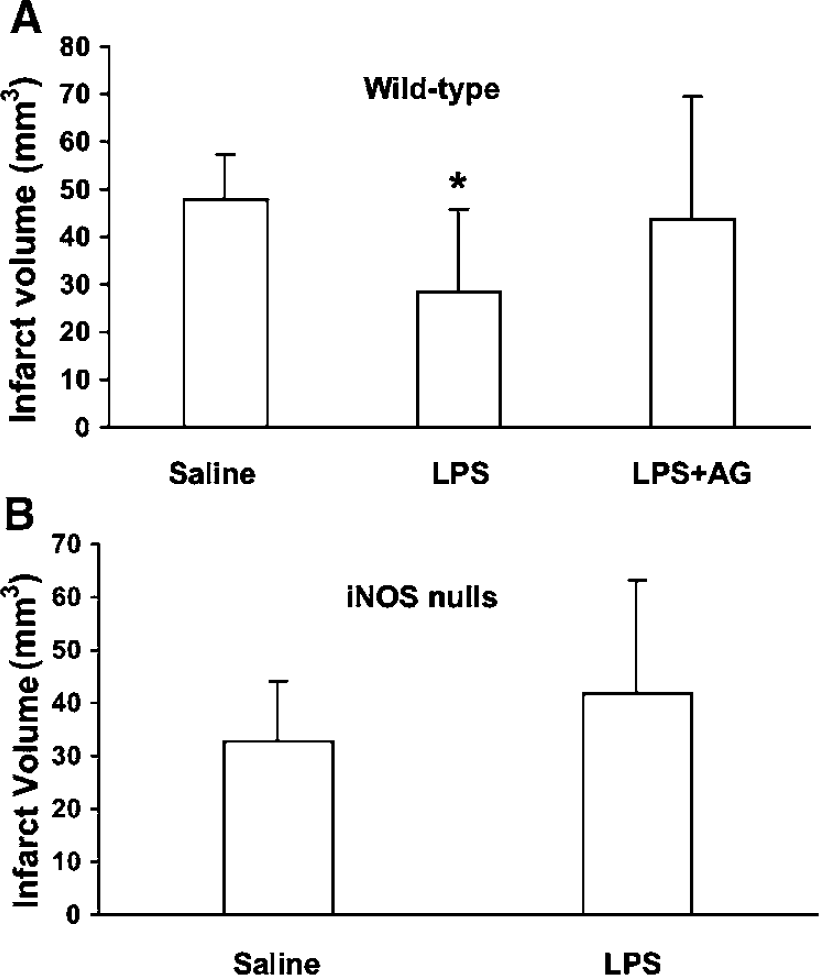
Role of iNOS in the preconditioning induced by LPS. The MCA was occluded 24 h after administration of LPS (0.5 mg/kg) or saline. (
Discussion
We have shown that three brief episodes of bilateral carotid artery occlusion result in a substantial reduction in brain damage in a model of transient focal cerebral ischemia (IPC). The protection is observed 24 h after the preconditioning stimulus and is associated with an increased resistance of neocortical mitochondria to Ca2+-mediated depolarization. We found that such ischemic tolerance is associated with iNOS immunoreactivity in cerebral blood vessels, is abolished by treatment with the iNOS inhibitor AG, and is not observed in iNOS-null mice. Furthermore, the increased mitochondrial resistance to Ca2+ conferred by preconditioning in wild-type mice was lost in iNOS-null mice. We also found that the tolerance conferred by treatment with LPS was not observed in mice treated with AG or in iNOS-null mice. Collectively, these findings show, for the first time, that iNOS is a critical factor in the development of ischemic tolerance produced either by transient ischemia or administration of LPS. Furthermore, they suggest that iNOS, in addition to its deleterious effects in the late stages of cerebral ischemia, can also be beneficial by promoting ischemic tolerance.
The findings of the present study cannot be attributed to differences in experimental conditions in the groups of mice studied. The temperature of the mice was monitored and carefully controlled. Furthermore, the CBF reductions produced by the preconditioning stimulus and by the MCAO were comparable in the different groups of mice. However, because CBF was assessed by LDF, a technique that measures relative flow, we cannot completely exclude the effects on resting CBF.
Inducible nitric oxide synthase-null mice have smaller infarcts before preconditioning. Therefore, it could be argued that the absence of ischemic tolerance in iNOS-null mice is because the infarct is already maximally reduced. While we cannot completely exclude this possibility, two observations suggest that this is unlikely to be the case. First, we have previously shown that the nNOS inhibitor 7-nitroindazole attenuates infarct volume in iNOS-null mice (Nagayama et al, 1999). Therefore, the infarct in iNOS-null mice is not maximally reduced and can be further attenuated. Second, in the experiments in which AG was used to inhibit iNOS, there was no difference in the baseline infarct before preconditioning; yet, ischemic tolerance could not be induced. Therefore, the concordance of the results obtained with AG and those in iNOS-null mice suggests that the reduction in baseline infarct in the null mice is unlikely to play a role in the lack of preconditioning. Although it is possible that AG acts through mechanisms independent of iNOS inhibition (Zimmerman et al, 1995), the observation that AG fails to reduce infarct volume in iNOS-null mice (Sugimoto and Iadecola, 2002) suggests that this is unlikely to be the case in the present study. Furthermore, the involvement of iNOS in IPC in brain is not unexpected. Inducible nitric oxide synthase has an analogous role in IPC in the heart (Bolli, 2001) and, in brain, this enzyme has been implicated in the preconditioning mediated by volatile anesthetics (Kapinya et al, 2002).
There is evidence that NO is involved in the phenomenon of ischemic tolerance (Keynes and Garthwaite, 2004; Nandagopal et al, 2001). In neuronal cultures, a brief nonlethal episode of oxygen glucose deprivation (OGD) confers tolerance toward longer OGD-causing lethal injury (Gonzalez-Zulueta et al, 2000). Treatment of the cultures with the NOS inhibitor nitro-L-arginine blocks the induction of tolerance, while administration of NO donors mimics the protective effect of preconditioning (Gonzalez-Zulueta et al, 2000). In vivo studies have shown that IPC is abolished in eNOS or nNOS null mice (Atochin et al, 2003), while hypoxic preconditioning is blocked by the NOS inhibitor nitro-L-arginine in a model of neonatal hypoxic–ischemic injury (Gidday et al, 1999). In the present study, we provided pharmacological and genetic evidence that iNOS is required for IPC. The finding that iNOS is responsible for ischemic tolerance induced by prior ischemia or by treatment with LPS suggests a critical role of iNOS in the preconditioning process, because it is also involved in the tolerance resulting from a preconditioning stimulus different from that inducing the subsequent injury (cross tolerance) (Kirino, 2002). This conclusion is supported also by a study in which AG was reported to block the relative tolerance to focal ischemia induced by prior isoflurane anesthesia (Kapinya et al, 2002).
The mechanisms by which iNOS mediates ischemic tolerance have not been elucidated. Our finding that iNOS is required for the increased mitochondrial tolerance to Ca2+ induced by preconditioning indicates that iNOS initiates signaling that leads to increased mitochondrial resistance. In the brain as in the heart, mitochondria play a crucial role in the expression of IPC (Kirino, 2002; Murphy, 2004). Thus, preconditioning has been reported to preserve mitochondrial function by opening the mitochondrial KATP channel, by inducing the expression of neuroprotective genes, delaying cytochrome c release and protecting from Ca2+-mediated mitochondrial membrane depolarization (Argaud et al, 2004; Dave et al, 2001; Zhan et al, 2002; Zhang et al, 2003). However, the precise signaling mechanisms by which iNOS-derived NO mediates the tolerance at the level of mitochondria are not known. Our finding that the preconditioning stimulus induces iNOS expression in cerebral blood vessels suggests that the process might be initiated by vascular signaling through the close association between cerebral blood vessels astroglia and neurons, the so-called neurovascular unit (Lo et al, 2003). Alternatively, vascular production of NO by iNOS, through its vasodilatatory and antiaggregant effects, could improve microvascular perfusion as previously described for LPS preconditioning (Dawson et al, 1999). Irrespective of the mechanisms of the effects and the signaling pathways involved, our findings clearly show that iNOS is required for the effect of preconditioning on mitochondrial Ca2+ buffering capacity that confers profound resistance to injury.
An important question concerns whether IPC occurs also in the human brain. In the cardiac literature, evidence from clinical trials suggests that patients suffering from angina before infarction have smaller cardiac lesions (Tomai et al, 1999). In addition, brief episodes of ischemia may protect the myocardium in high-risk procedures, such as angioplasty, involving interruption of coronary artery flow (Yellon and Downey, 2003). The evidence for IPC in the human brain is less firm than in the heart. Although some studies suggest that patients with stroke who had prior TIAs have milder neurological deficits at presentation and a better functional recovery (Moncayo et al, 2000; Sitzer et al, 2004; Wegener et al, 2004; Weih et al, 1999), the mechanisms of the effect remain controversial (Sitzer et al, 2004). In particular, the contributions of embolus size/composition and recanalization to the favorable outcome have not been ruled out. Nevertheless, these studies raise the possibility that preconditioning may also occur in the human brain and, as such, interventions based on the mechanisms of ischemic tolerance could be used in preventive or treatment strategies. One example of this approach is provided by erythropoietin, a hormone that contributes to IPC (see Dawson (2002) for a review) and has also been shown to be a promising drug for the treatment of acute stroke (Ehrenreich et al, 2002).
In conclusion, we have shown that the powerful tolerance to focal cerebral ischemia conferred by prior transient ischemia or administration of LPS is abolished by treatment with the iNOS inhibitor AG or in iNOS-null mice. The effect is associated with loss of tolerance to Ca2+-mediated mitochondrial injury induced by IPC. The findings suggest a central role of iNOS in the mechanisms of ischemic tolerance in brain and unveil a beneficial effect of iNOS-derived NO in the setting of cerebral ischemia.