
Abstract
Acute cerebral ischemia occurs after subarachnoid hemorrhage (SAH) because of increased intracranial pressure (ICP) and decreased cerebral perfusion pressure (CPP). The effect of hyperbaric oxygen (HBO) on physiological and clinical outcomes after SAH, as well as the expressions of hypoxia-inducible factor-1α (HIF-1α) and its target genes, such as BNIP3 and VEGF was evaluated. Eighty-five male SD rats (300 to 350 g) were randomly assigned to sham, SAH, and SAH+HBO groups. Subarachnoid hemorrhage was induced by endovascular perforation. Cortical cerebral blood flow (CBF), ICP, brain water content, brain swelling, neurologic function, and mortality were assessed. HBO (100% O2, 2.8 ATA for 2 h) was initiated at 1 h after SAH. Rats were sacrificed at 24 h to harvest tissues for Western blot or for histology. Apoptotic morphology accompanied by strong immunostaining of HIF-1α, VEGF, and BNIP3 were observed in the hippocampus and the cortex after SAH. Increased expressions of HIF-1α, VEGF, and BNIP3 were quantified by Western blot. HBO reduced the expressions of HIF-1α, VEGF, and BNIP3, diminished neuronal damage and improved CBF and neurologic function. HBO reduced early brain injury after SAH, probably by inhibition of HIF-1α and its target genes, which led to the decrease of apoptosis and preservation of the blood–brain barrier function.
Introduction
Subarachnoid hemorrhage (SAH) represents 5% of all strokes; however, it accounts for 27.3% years of potential life lost caused by all stroke events. Assuming that relatively younger people are affected by SAH, with premature death and overall higher mortality rates, the social impact of SAH is comparable to other stroke types (Johnston et al, 1998). Despite progress in diagnostics and treatment employing early neurosurgical intervention, case fatality is approximately 50% and 30% of survivors remain dependent on others (Van Gijn and Rinkel, 2001). One of the major factors for the devastating outcome of SAH is early brain injury, including brain edema, blood–brain barrier (BBB) disruption, elevation of intracranial pressure (ICP), and acute cerebral ischemia. However, the molecular mechanisms of early brain injury remain unclear and no effective treatment is currently available.
Hypoxia-inducible factor-1α (HIF-1α) is a key mediator of hypoxic responses and HIF-1α expression increases in the early period after global and focal cerebral ischemia and after intracerebral hemorrhage (Bergeron et al, 1999; Jiang et al, 2002; Pichiule et al, 2003). Hypoxia-inducible factor-1α activates its target genes including BNIP3, a proapoptotic member of the Bcl-2 family that participates in apoptosis, and vascular endothelial growth factor (VEGF), which is involved in the enhanced permeability of BBB, both of which probably contribute to brain injury. HBO has been used as an adjunct treatment of SAH to counter cerebral ischemia, particularly for cerebral vasospasm (Isakov et al, 1985; Kohshi et al, 1993), although physiologic and molecular mechanisms and efficacy of its action in the early brain injury period after SAH are not clear. We propose that HBO reduces the expression of HIF-1α and its target genes, especially VEGF and BNIP3, and decreases early brain injury after SAH. In parallel, effects of HBO on SAH-induced pathophysiologic and morphologic changes as well as on mortality and neurologic outcome were evaluated.
Materials and methods
Experimental Groups
Eighty-five SD male rats weighing 300 to 350 g were randomly assigned to the following groups: sham, SAH, and SAH+HBO. All brain samples were obtained at 24 h after SAH.
Subarachnoid Hemorrhage Modeling, Intracranial Pressure Monitoring, and Cerebral Blood Flow Measurement
We used an endovascular perforation rat model of SAH described by Bederson et al (1995) and modified by Schwartz et al (2000). Under 1% α-chloralose 60 mg/kg and 10% urethane 600 mg/kg anesthesia (intraperitoneally), animals were tracheotomized, intubated, and mechanically ventilated throughout the operative period. After intubation, rats were placed in the prone position to perform cannulation of the subarachnoid spinal space below cauda equina for monitoring ICP according to the method described recently in our laboratory (Kusaka et al, 2004). Next, the animals were placed in the stereotaxic frame (Stoelting), the skin of the head was incised, and the skull exposed. Using a microdrill cooled with 0.9% NaCl, skull bone on the right side was thinned to translucency. By not opening the skull for LDF monitoring, reliability of ICP, Cerebral blood flow (CBF), and brain swelling measurements was assured. In addition, communication of the brain with the hyperbaric environment was avoided. Blood flow in the thinned bone (less than 0.1 mm thick) and dura mater (several μm) is negligible. A plastic holder containing a small, straight LDF probe was attached to the bone with acrylic glue. Laser Doppler flow at the site (localized 5 mm lateral and 1 mm posterior to the bregma), within the territory supplied by the right middle cerebral artery (Schmid-Elsaesser et al, 1998), was recorded by laser Doppler monitor (Periflux System 5000, Perimed) generating the laser beam at a wavelength of 740 nm, penetrating brain tissue approximately 1 mm beneath the probe ending (Dirnagl et al, 1989). Contralateral hemisphere was chosen for CBF measurements tending to investigate CBF alterations related to ICP avoiding influence of brief ipsilateral CBF decrease after filament insertion (Bederson et al, 1995). Then animals were placed in the supine position, and a plastic catheter was inserted into the right femoral artery for blood pressure registration and blood sample withdrawal for blood gases measurements (Blood Gas and Electrolyte Analyzer ABL 77Sci, Radiometer Copenhagen). Animals were attached to the operating table, and placed on a heating blanket. During surgery, postoperative period, and after oxygenation, rectal temperature was maintained at a level of 37.0°C±0.5°C.
In rats subjected to SAH, 4–0 monofilament (obliquely cut) was inserted into the surgically obtained stump of the left external carotid artery at the common carotid artery and internal carotid artery clamped with small aneurysm clips, and advanced as described previously (Kusaka et al, 2004). At 1 h after surgery, blood samples were taken for analyses. All rats were monitored within 1 h after SAH, and then were returned to their cages or transferred to the HBO chamber.
HBO Treatment
HBO with 100% oxygen for 2 h at 2.8 ATA (26.5 psi) was initiated 1 h after SAH. Rats were placed in the small hyperbaric chamber (model 1300B, Sechrist Industries Inc., Anaheim, CA, USA) and the compression started at a rate of 5 psi/min. Oxygen flow was maintained at a rate of 22 L/min. Accumulation of CO2 was prevented by using a small container with calcium hydronate crystals.
Mortality and Neurologic Score
Overall mortality was calculated; then mortality distribution was calculated in the following time intervals after SAH: 0 to 1, 1 to 3, 3 to 8, and 8 to 24 h. We used a neurologic scoring system proposed by Garcia and modified in our laboratory (Garcia et al, 1995; Kusaka et al, 2004).
Brain Water Content
At 24 h after SAH, brains were harvested and quickly separated to left and right hemispheres, cerebellum, and the brain stem. Brain samples were weighed on a precise electronic balance (Denver Instruments, Denver, CO, USA) and placed in the oven at a temperature of 105°C for 48 h (Schwab et al, 1997). After 48 h, samples were weighed again and water content was calculated according to the following formula:
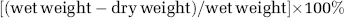
Brain Volume and Cerebral Blood Volume
Plastic 10 mL vials containing 1.7 mL. of saline were marked equally to the level of liquid by using ultrathin marker. After meticulous clot removal, the brain was put into the vial and the increased level of liquid was marked. Vials were not discarded after assay and brain volume was measured by titrated saline infusion within marked levels. Vertical position of vials upon measurements was maintained.
Parallel samples of brain and arterial blood were harvested for hemoglobin content assay according to the method described by Choudhri et al (1997). After having measured hemoglobin content in the given volume of blood, cerebral blood volume (CBV) was calculated based on measured brain hemoglobin content and compared with the measured volume of the entire brain.
Histology
Animals were sacrificed in deep anesthesia by perfusion through the left ventricle with 200 mL of ice-cold 0.1 mol/L PBS followed by 400 mL of 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (PBS) (pH 7.4). Brains were postfixed in the same fixative overnight. For IgG staining, brains were fixed by immersion for 7 days. After fixation, brains were cryopreserved. Coronal sections of 20 μm thick were cut by use of a cryostat (CM3050S, Leica) as described (Yin et al, 2003; Zhou et al, 2004).
Nissl Staining
Brain sections were air dried for 30 mins and hydrated in 0.1% cresyl violet for 5 mins. After rinsing with water they were dehydrated in increasing concentrations of ethanol and cleared in xylenes, then mounted with Permount, coverslipped and observed under a light microscope.
Immunohistochemistry
Brain sections were hydrated with 0.01 mol/L PBS with Tween 20 and subsequently treated with 3% hydrogen peroxide for 10 mins and blocking sera for 2 h at room temperature; then sections were incubated with primary antibodies overnight at 4°C. The following polyclonal antibodies (Santa Cruz Inc., Santa Cruz, CA, USA) were used: rabbit anti-HIF-1α (H206), rabbit anti-VEGF (147), and goat anti-Nip3 (C18) at a concentration 1:100 each. After rinsing with PBS (5 mins, 3 times), ABC staining system (Santa Cruz Inc.) was used as described (Zhou et al, 2004).
Double Fluorescence Labeling
Brain coronal sections were used for double fluorescent labeling using rabbit anti HIF-1α antibody diluted 1:100 and Texas red-conjugated goat anti-rabbit IgG antibody diluted 1:200 (Santa Cruz Inc.) in combination with TUNEL fluorescence kit (green, In situ Cell Death Detection Kit, Fluorescein, Roche Inc., Mannheim, Germany). Incubation with primary antibodies was held overnight at 4°C. Secondary antibodies conjugated with fluorescent dyes were incubated in the dark for 2 h at room temperature. The method for TUNEL staining was described previously (Sun et al, 2004). Sections were cover-slipped with 30% glycerin and observed under fluorescent microscope operating with digital camera (OLYMPUS BX51). Texas red and FITC were excited at 595 to 605 and 488 nm, respectively. Digitalized microphotographs of immunofluorescent sections were saved to HDD of PC computer. Merged images were generated by means of Image ProPlus software.
IgG Staining
Brain immunolocalization of IgG was studied according to methods described previously (Richmon et al, 1998). Sections were incubated overnight with anti-rat IgG biotin-conjugated antibody 1:100 (Santa Cruz Inc.) at 4°C and subsequently treated with ABC staining kit.
Western Blot
Animals in deep anesthesia were perfused through the left ventricle with 200 mL ice-cold 0.1 mol/L PBS. Brains were kept at −80°C until analysis. Western blot analysis was performed as described previously with some modifications (Sun et al, 2004). Briefly, brain samples were homogenized in extract buffer (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 1% Triton X-100, 10%glycerol, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L NaF, 20 mmol/L Na4P2OP2, 2 mmol/L Na3VO4, 0.1% SDS, 0.5% deoxycholate, 1 mmol/L PMSF, 10 nmol/L eupepsin, 10 nmol/L pepstatin A, pH 7.4), and centrifuged at 14 000g for 15 mins at 4°C (Marathon 21000R centrifuge, Fisher Scientific, Pittsburg, PA, USA). Protein content was measured by using DC protein assay (Bio-Rad, Hercules, CA, USA). Protein samples 30 μL each were suspended in sample buffer (125 mmol/L Tris, 4% SDS, 20% glycerol, 10% B-merkaptomethanol, 0.025% bromophenol blue, pH 6.8) denatured at 95°C for 2 mins and electrophoresed in 7% (for HIF-1α) or 12% (for VEGF and BNIP3) dodecyl sulfate-polyacrylamide gel at 125 V (Mini Protean 3, BioRad). Proteins from gels were transferred at 100 V for 75 mins onto polyvinylidene difluoride (PVDF) membrane previously blocked with 5% dry nonfat milk in TBS. Membranes were incubated overnight at 4°C with the same primary antibodies as for immunohistochemistry—raised against HIF-1α, VEGF, and BNIP3, diluted 1:200 with 1% nonfat milk in Tween-TBS (TBST). Probing the same membranes with goat anti β-actin antibody 1:1000 (Santa Cruz, Inc.) served as loading control. After washing with TBS, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies diluted 1:1000 for 30 mins at 37°C. Analysis of protein content was based on detection of chemiluminescence developed by ECL kit (BioRad) in ChemiDoc chamber (Bio Rad). Protein bands were quantified by densitometry (Quantity One Software, Bio Rad).
Data Analysis
Data are expressed as mean±s.d. Statistical significance was verified by analysis of variance performed in one-way ANOVA followed by Tukey test for multiple comparisons. Follow-up analyses of blood pressure, ICP, CPP, and CBF were performed by repeated measures ANOVA. Significance of differences in neurologic scores was analyzed by Kruskal–Wallis one-way ANOVA followed by multiple comparison procedures by Dunn's method. Differences in mortality between groups were tested using Fisher exact test. Probability value of P<0.05 was considered statistically significant.
Results
Physiologic Variables
A significant increase in mean arterial blood pressure, by 36.76% in the SAH group and by 29.22% in SAH assigned for the HBO group, was observed acutely after SAH (Table 1). Mean arterial blood pressure reached its peak close to 150 mm Hg in both experimental groups 5 mins after perforation of the middle cerebral artery. Blood pressure level returned to control values within the next 10 mins, stabilized and on the first postoperative day there were no differences between experimental groups. Mean values of pO2, pCO2, and pH, measured at the end of surgery are presented in Table 1. No significant changes within these variables occurred.
Mean arterial blood pressure, pH, pO2, and pCO2 in three experimental groups
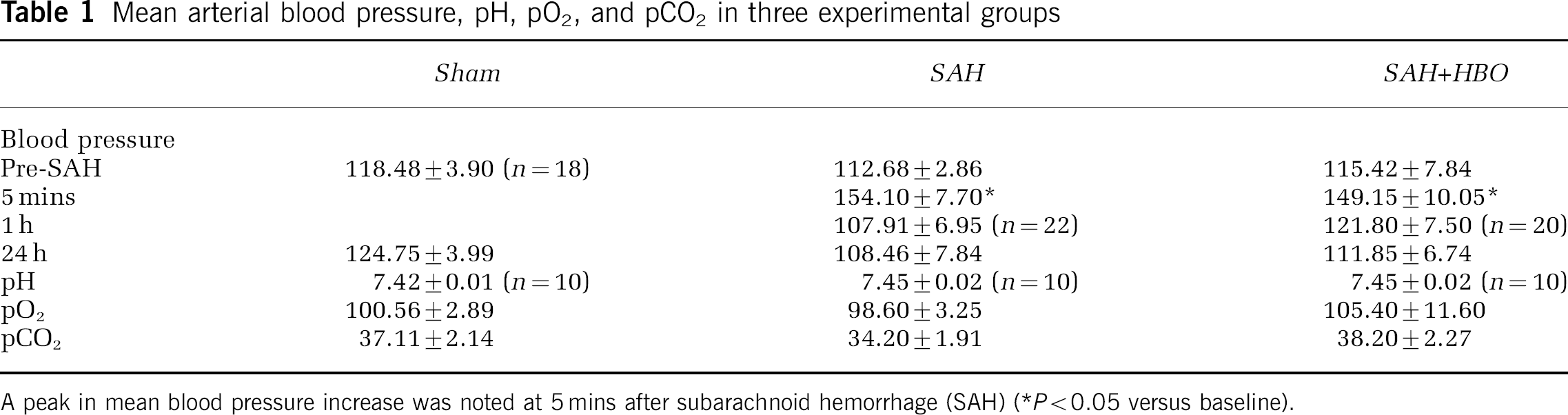
A peak in mean blood pressure increase was noted at 5 mins after subarachnoid hemorrhage (SAH) (*P<0.05 versus baseline).
Mortality
In SAH group, 20 of 42 animals died, which yielded 47.62% mortality rate (Figure 1A). Treatment with HBO reduced mortality rate to 20% (5 of 25). Statistical analysis revealed significant difference between mortality rates in the two experimental groups at a level of P=0.036 (Fisher exact test). No animals died in the sham group. Mortality within 24 h after SAH was artificially divided into the following time intervals: 0 to 1, 1 to 3, 3 to 8 and 8 to 24 h to obtain temporal distribution of deaths in not treated versus HBO-treated groups of rats (Figure 1B). In the aforementioned time periods 10%, 20%, 25%, 45% versus 20%, 0%, 20%, 60% of total deaths occurred in SAH and SAH+HBO groups, respectively. This analysis indicates that no rat died in the HBO chamber during treatment at 1 to 3 h after SAH as against 20% death without HBO treatment.
(
Neurologic Score
Neurologic score was 17.05±0.21 points in control group versus maximum 18 points obtainable. A slightly reduced exploring behavior contributed to this insignificant difference. A significant reduction of neurologic score to 5.05±0.52 was found in animals 24 h after SAH (Figure 1C). Treatment with HBO alleviated diminution in neurologic score (P<0.05 versus SAH), even though 9.10±0.98 point level calculated in this group was significantly lower than control value (P<0.05 versus control, ANOVA).
Intracranial Pressure, CPP, Cerebral Blood Flow
Pre-SAH values of ICP were 7.3±1.18 mmHg in no treatment group and 7.83±1.47 mmHg in animals assigned to HBO (Figure 2A). Intracranial pressure rose significantly after SAH and reached 75.18±14.83 mm Hg in the SAH group and 80.39±10.43 mmHg in SAH assigned to the HBO group at the first minute (Figure 2A). After 5 mins, ICP dropped 56.24±17.63 and 56.10±15.23 mmHg, respectively, then gradually declined, and at the end of the first hour after SAH, showed 28.07±3.92 and 30.72±4.39 mm Hg values in SAH and SAH assigned to HBO groups, respectively. A tendency towards lower ICP values occurred in animals treated with HBO immediately after treatment (3 h after SAH): 22.41±2.35 mm Hg (SAH+HBO) versus 29.51±3.71 mm Hg in untreated rats (SAH). At 24 h after SAH, untreated rats showed significantly elevated ICP, whereas ICP in rats with HBO treatment was close to control values (10.28±1.09). Statistical analysis by ANOVA also revealed significant difference (P<0.05) in ICP between two groups with SAH at 24 h after bleeding.
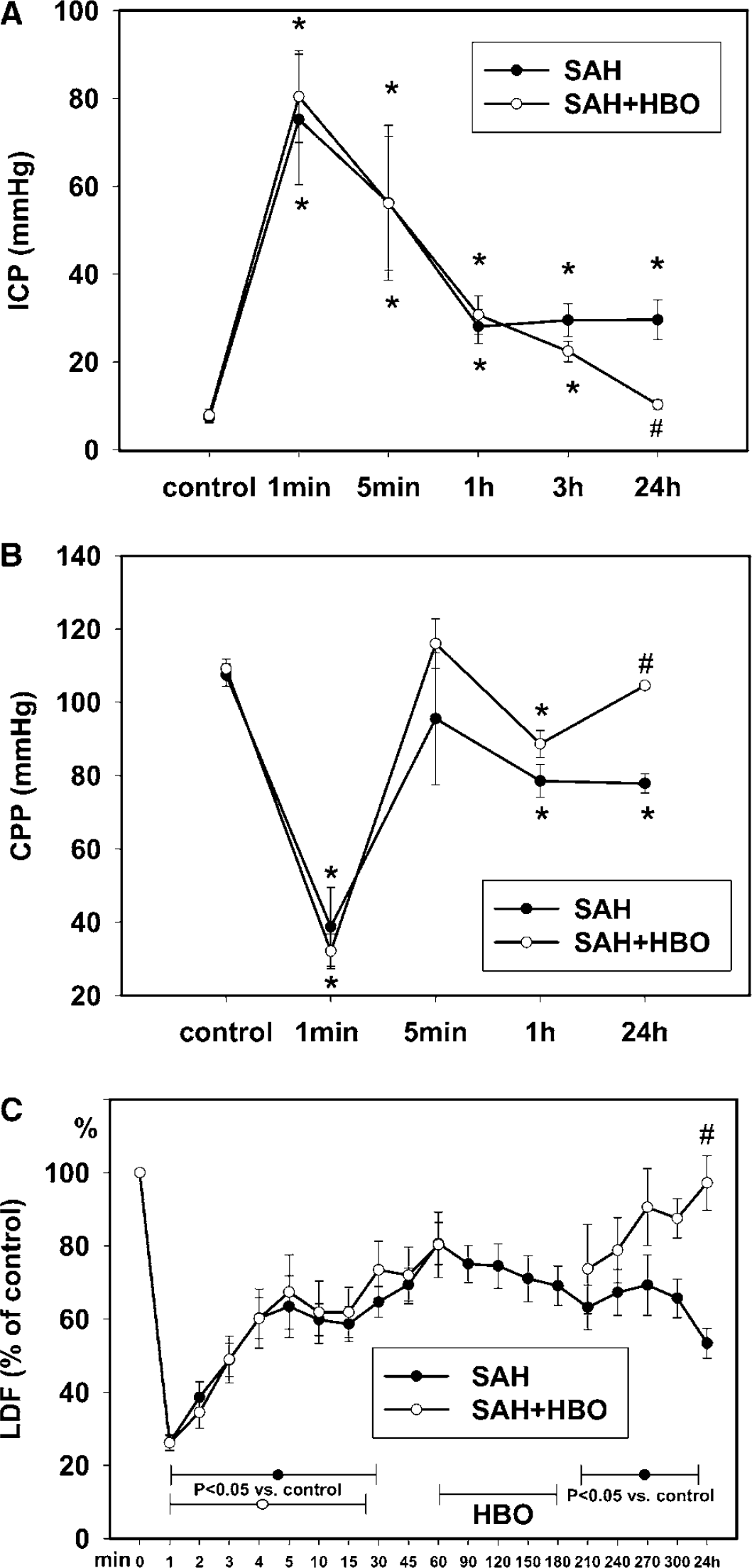
(
Baseline values for CPP were calculated at a level of 107.38±3.02 and 109.13±2.67 mm Hg in SAH and SAH assigned to HBO rats, respectively. CPP decreased and reached its nadir at 1 min after SAH: 38.71±10.76 mm Hg in the SAH group and 32.09±4.73 mm Hg in SAH assigned to the HBO group (Figure 2B). Subsequently, a recovery was observed with a peak CPP value occurring at 5 mins (95.48±18.09 mm Hg versus 115.99±6.77 mm Hg) followed by gradual decrease to 78.49±4.48 mm Hg (SAH) and 88.60±3.69 mm Hg (SAH assigned to HBO) at 1 h after SAH. At 24 h after SAH, CPP in untreated rats stabilized at a level of 77.79±2.62 mm Hg, significantly lower than basal value, however after HBO complete recovery was observed (104.55±0.99 mm Hg, 95.80% of baseline). At 21 h after HBO treatment, CPP was significantly higher as compared with untreated animals (P<0.05; ANOVA).
LDF (CBF) dropped very rapidly, reaching its nadir within 1 min after puncture − 26.26%±2.23% of control in the SAH group and 26.13%±2.03% of control in SAH assigned to the HBO group (Figure 2C). Values of LDF continued decreasing during 1 min and then an increase at a slow rate began. Cortical microflow remained significantly lowered, below baseline in both groups during the first 20 mins after SAH (P<0.05 versus sham; ANOVA). At 1 h after hemorrhage, LDF showed approximately 80% restoration of control level in both groups. From this time point, LDF started to decrease in no treatment animals and displayed statistical significance for a value of 63.2%±6.09% of control, noted at 210 mins after SAH. LDF stabilized at a level of 53.36%±4.08% of control at 24 h after induction of SAH. In rats treated with HBO, cortical microflow decreased slightly immediately after treatment (by 8.28%) as compared with pretreatment values; however, it started to increase in later time intervals. At 24 h after SAH, a complete recovery of LDF values was observed in the HBO group: 97.21±7.46%, a value significantly higher than in the no treatment group.
Brain Swelling
Brain swelling is represented by wet brain weight, brain volume (size), and cerebral blood volume (hemoglobin content). Both brain wet weight and brain volume were significantly increased, by 8.79% and 16.56%, as compared with the sham group at 24 h following SAH (Figures 3A and 3B). HBO attenuated increases in wet brain weight and brain volume down to the control level (3.84% and 0.72% of control, respectively).
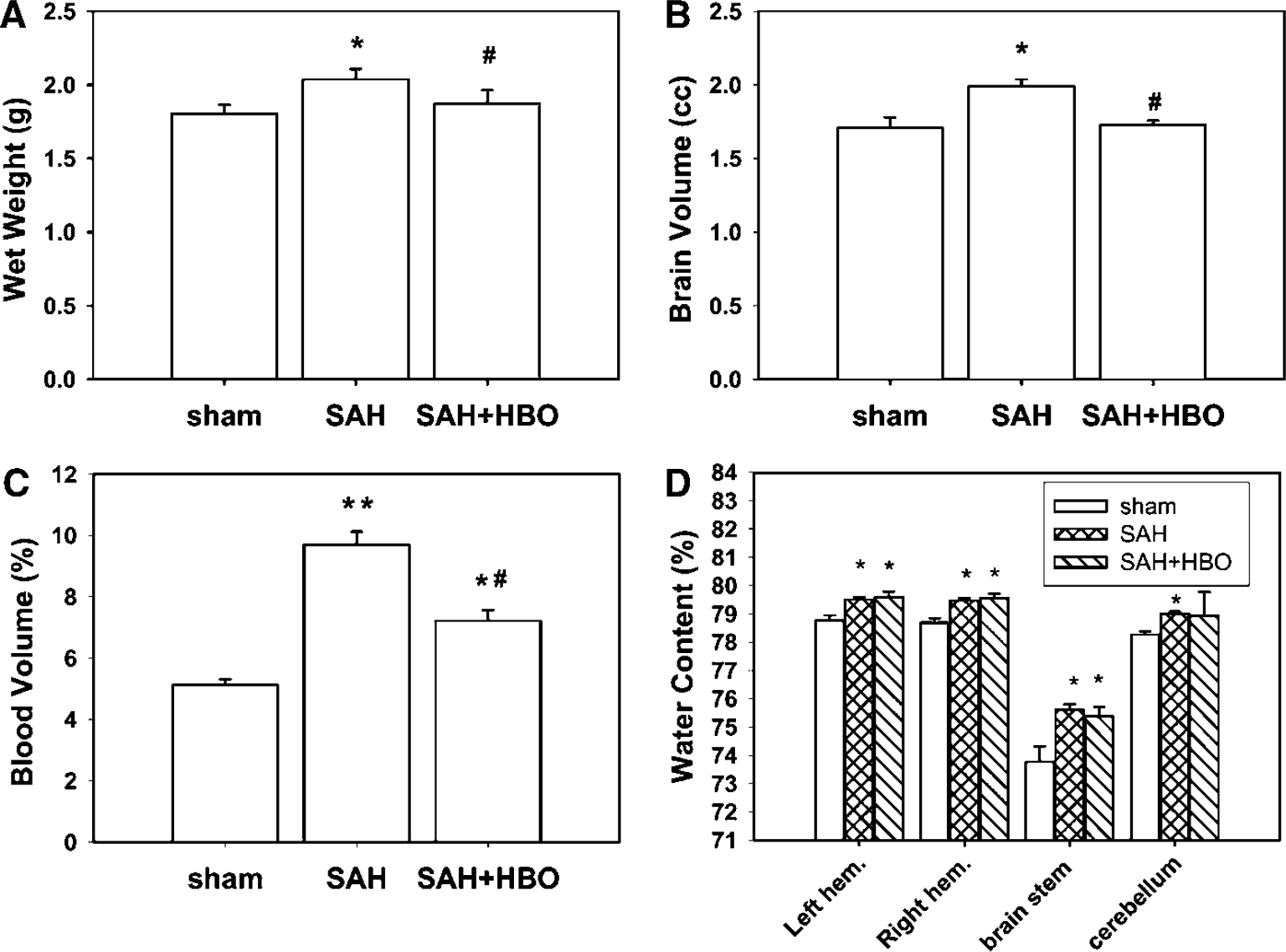
(
A marked increase in cerebral blood volume, by 89.54%, was noted after SAH (P<0.001 versus sham, ANOVA, Figure 3C). Cerebral blood volume in the HBO group was significantly lower than in animals with not treated SAH, although significantly elevated, by 34.52%, as compared with control (P<0.05).
Brain Water Content
Mean brain water content values in sham-operated animals were 78.76%±0.19% in the left hemisphere, 78.68%±0.17% in the right hemisphere, 73.77%±0.55% in the brain stem, and 78.26%±0.11% in the cerebellum (Figure 3D). In the left hemisphere, water content significantly increased to 79.51%±0.09% and 79.60%±0.19% in the SAH and SAH+HBO groups, respectively. Significant increases in brain water content were observed in no treatment and HBO groups also in the right hemisphere, 79.47%±0.09% and 79.56%±0.16%, and in the brain stem, 75.60%±0.20% and 75.37±0.34, respectively. In the cerebellum, water content increased significantly only in the SAH group. However, any significant effect of HBO on water content was not observed in the examined brain regions.
Morphologic Changes
Results of Nissl staining of the hippocampus and cerebral cortex in three experimental groups are presented in Figure 4. Light microscopic examination revealed extensive neuronal changes in the CA1 sector of the hippocampus 24 h after SAH. They comprised pyknotic nuclei, dark, shrunken cytoplasm and twisted axonal processes of pyramidal neurons (Figure 4C). In the cerebral cortex, the above changes, together with small foci of neuronal loss, were seen (Figure 4A). HBO prevented appearance of morphologic alterations in the hippocampus (Figure 4D). In the cerebral cortex, ischemic cell changes were much less abundant, represented by sparse shrunken neurons. Surviving cells presented some darkening of the cytoplasm (Figure 4B).
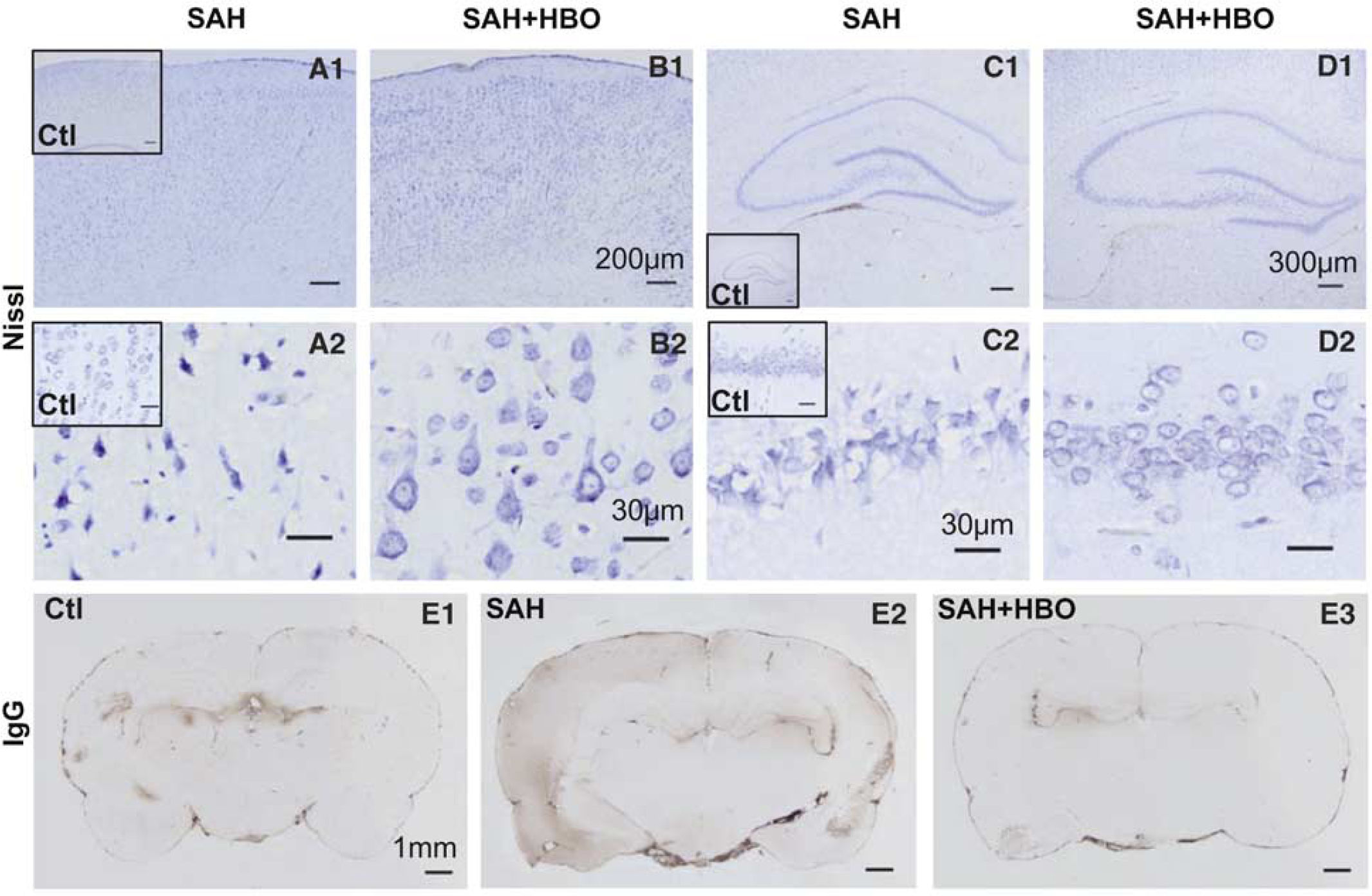
Nissl staining was performed on brain sections obtained from the sham, no treatment subarachnoid hemorrhage (SAH), and HBO-treated SAH groups. Neurons in the cerebral cortex (
Blood–Brain Barrier Permeability
In control brain sections, IgG staining revealed BBB leaks limited to the periventricular area, which has physiologic permeability (Figure 4E1). At 24 h after SAH, a diffuse leakage of IgG protein within the left hemisphere was present. The most intense staining was found in the upper layers of cerebral cortex, especially of cingular and basal areas (Figure 4E2). To a smaller extent, IgG immunoreactive areas were found in the right hemisphere; however, in the right hippocampus the immunostaining signal seemed to be more intense than contralaterally. HBO reduced the extent of IgG staining in the SAH brain (Figure 4E3). Areas of increased BBB permeability were limited to the left-sided hippocampus and periventricular region.
Hypoxia-Inducible Factor-1α Immunostaining
The cerebral cortex and the hippocampus of sham-operated rats showed very weak HIF-1α immunostaining, limited to nuclei in single cells (insert, Figures 5A and 5C). Extensive foci of positive HIF-1α staining were present in the cerebral cortex after SAH (Figure 5A). Brain samples obtained from animals at 24 h after SAH showed strong HIF-1α staining in the nuclei of neurons forming all hippocampal zones and DG (Figure 5C). In the CA1 sector of the hippocampus, strong HIF-1α immunoreactivity in pyramidal neurons was colocalized with morphologic features of cell damage represented by condensed nuclei and shrunken cytoplasm. Cerebellar Purkinje cells and neurons within gray matter formations of the brain stem also showed HIF-1α-positive staining (data not shown). Marked attenuation of HIF-1α immunoreactivity was observed in brain sections from animals treated with HBO. Decrease in the extent of HIF-1α immunostaining correlated with the presence of well-preserved neurons in the cerebral cortex and diminution of neuronal changes in the hippocampus (Figures 5B and 5D).
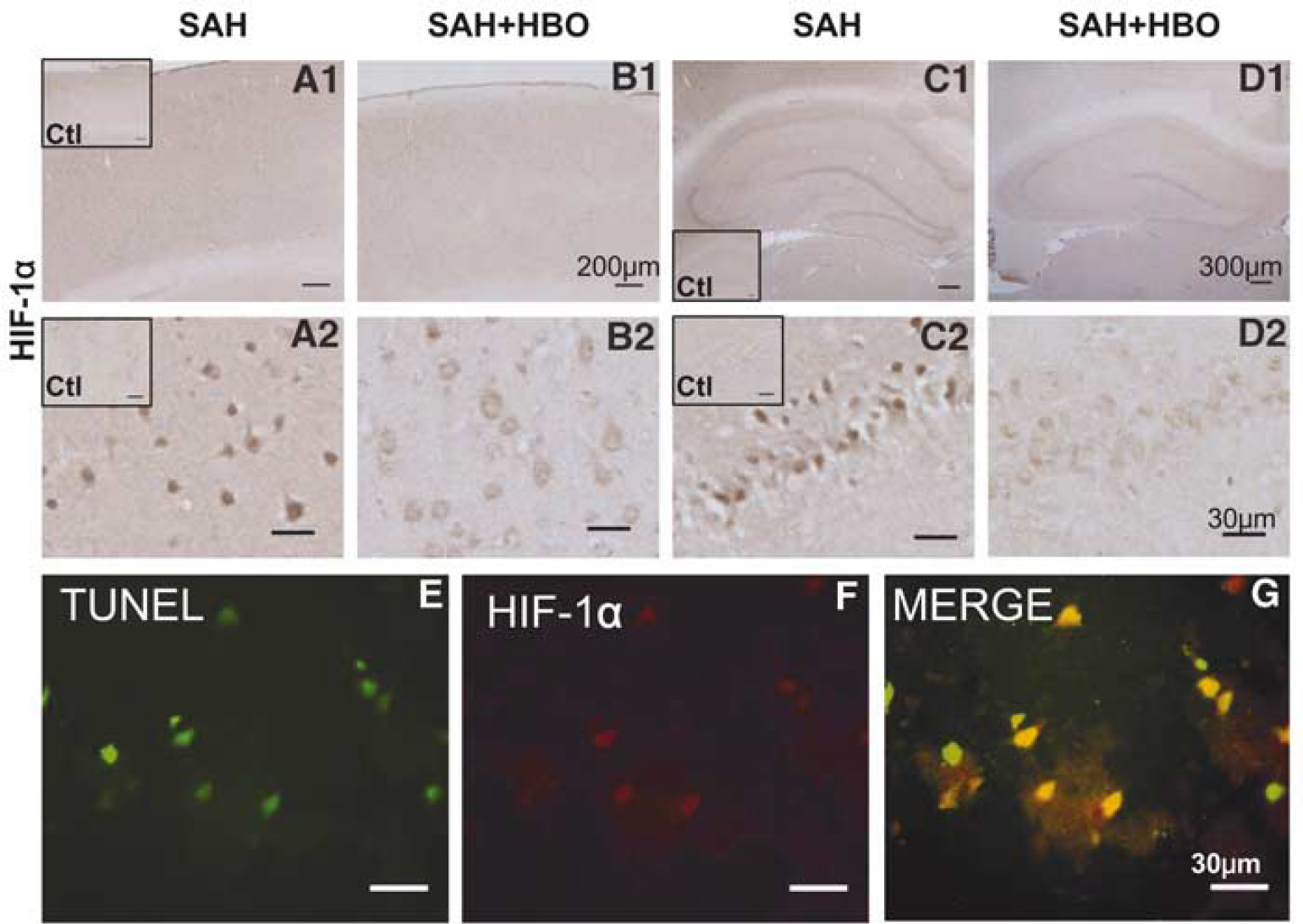
(
Hypoxia-Inducible Factor-1α and TUNEL Staining
We applied double fluorescent staining to delineate patterning of neuronal death occurring within 24 h after SAH, and to assess HIF-1α contribution to this process. Limited foci of TUNEL-positive cells were scattered within the cerebral cortex of both hemispheres (Figure 5E). Most of the neurons displaying TUNEL positive nuclei were costained with HIF-1α. Merging images, acquired at different excitation waves, resulted in the development of yellow color in the cell nucleus, indicating nuclear colocalization of apoptotic alterations and HIF-1α protein expression (Figure 5G).
VEGF and BNIP3 Staining
Spatial pattern of VEGF expression was similar, however, localization within the cell appeared to be different from HIF-1α. On HIF-1α immunochemical staining, immunoreactivity was confined to the cell nucleus, whereas staining with anti-VEGF antibody was localized dispersedly within the cell body (Figures 6A and 6C). Immunoreactive cells were found mostly in the brain structures localized ipsilaterally to the ruptured artery, that is, left cortex and left hippocampus. Similar to HIF-1α staining, immunopositive cerebellar Purkinje cells and brain stem neurons were found (data not shown). HBO treatment prevented staining of VEGF in the hippocampus and reduced VEGF staining in the cerebral cortex (Figures 6B and 6D). Besides, nearly negative staining was found in the cerebellar neurons and within the brain stem (data not shown).
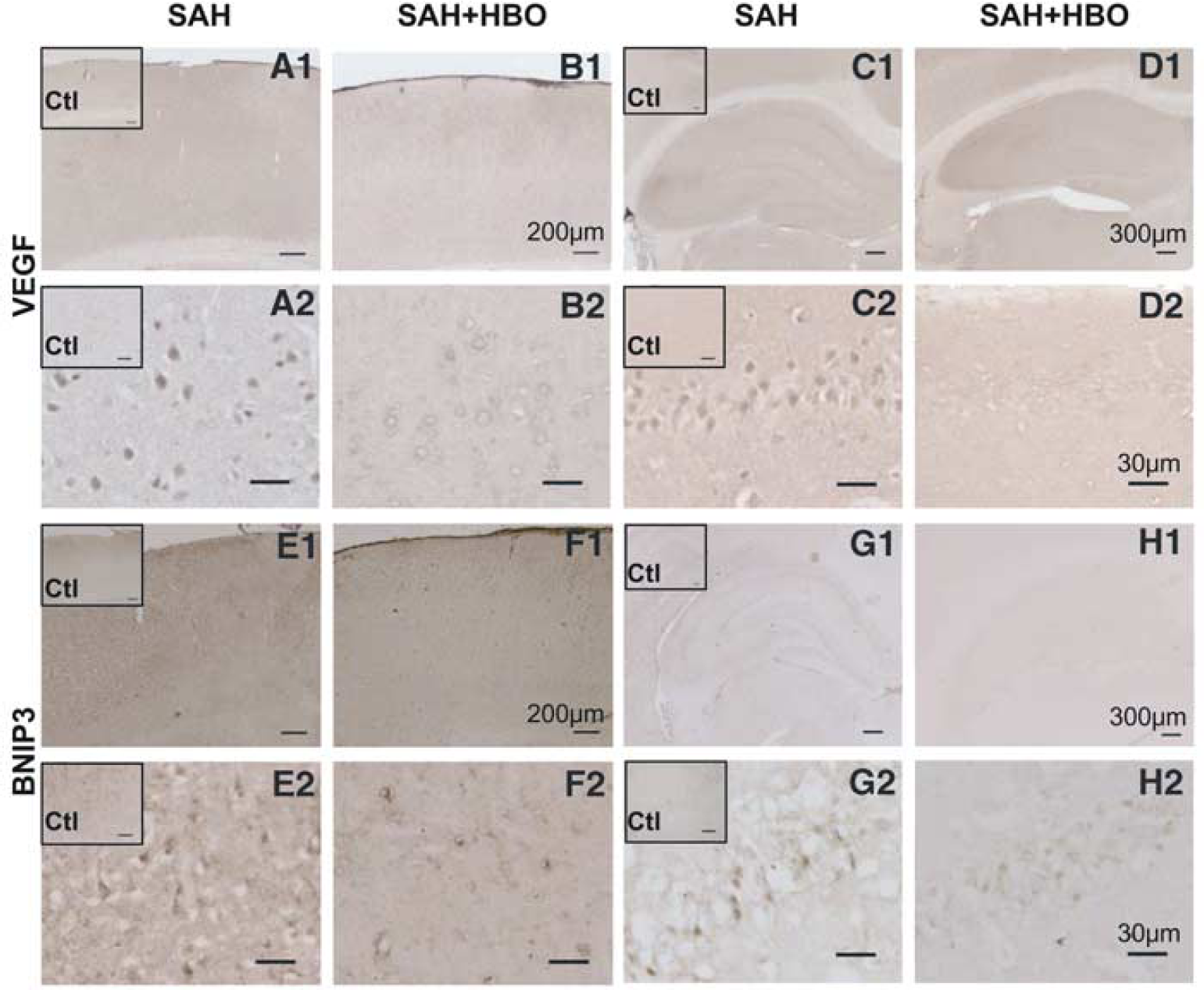
Positive immunohistochemical staining for VEGF was observed both in the hippocampus, mostly in CA1 sector, and in the cerebral cortex after subarachnoid hemorrhage (SAH) (
Light microscopic examination revealed that immunostaining of BNIP3 was localized mostly in the profoundly injured nuclei of pyramidal neurons in the CA1 sector of the hippocampus (Figure 6G). Analogous staining in the cerebral cortex brought very limited results; only sparse neuronal foci showed weak BNIP3 immunoreactivity (Figure 6E).
Hypoxia-Inducible Factor-1α, VEGF, BNIP3 Expression
Accumulation of HIF-1α was further verified by Western blot analysis. Weak HIF-1α protein band (120 kDa) was detected in the sham-operated rats, in contrast with HIF-1α accumulation at 24 h after SAH. The highest increase in HIF-1α protein content was noted in the brain stem (2.02-fold increase; P<0.01 versus sham, Figure 7B). Comparable (approximately 2-fold) increases were demonstrated in the hippocampus and cerebellum (P<0.05 versus sham, Figures 7A and 7C). Treatment with HBO resulted in significant diminution of HIF-1α protein content (P<0.05 versus SAH group), especially in the hippocampus. Small and insignificant increases in HIF-1α protein content were present in the cerebellum (21.98%) and in the brain stem (15.36%) despite HBO treatment. In the cerebral cortex, increases in HIF-1α protein contents appeared to be insignificant.
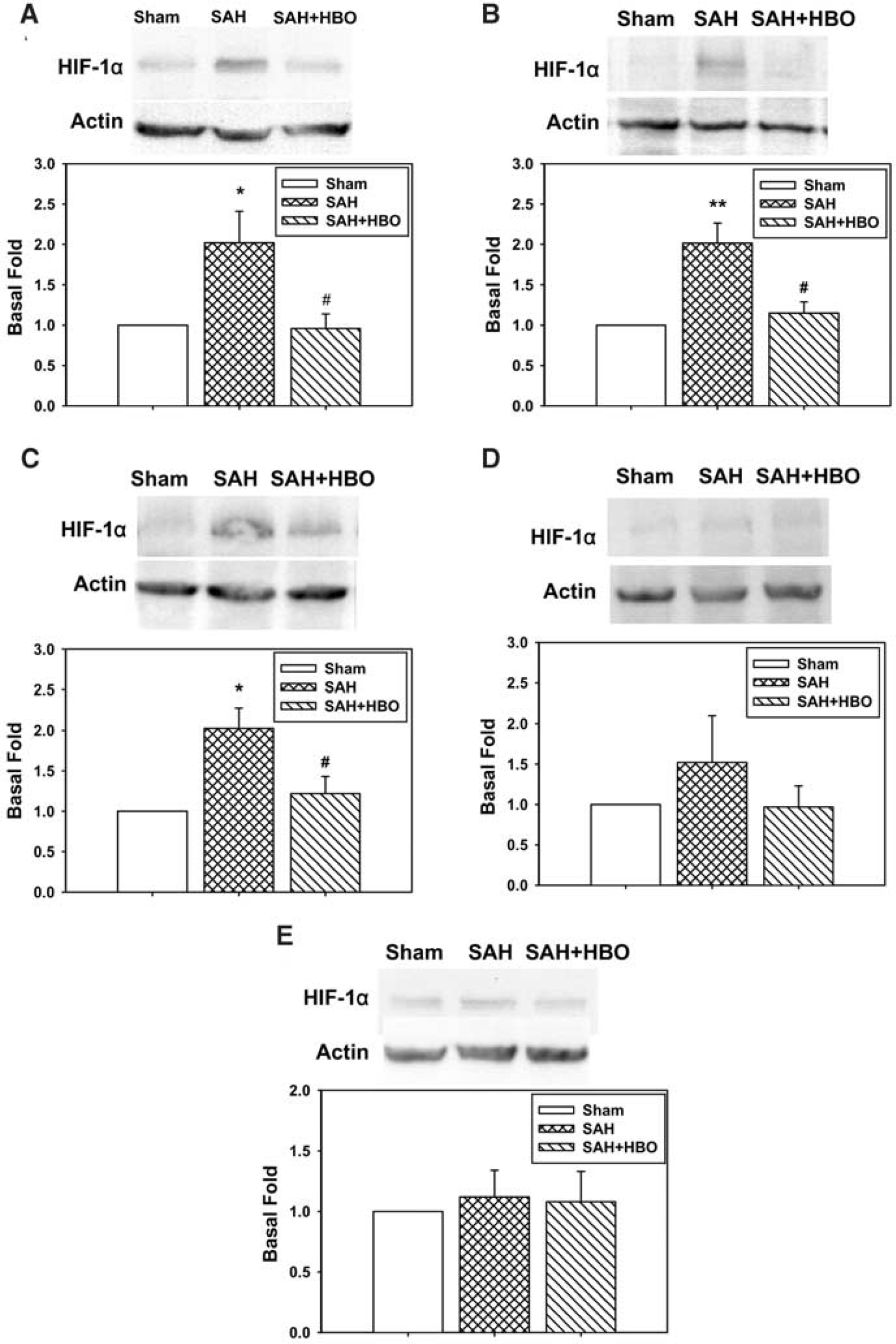
Western blot analysis of hypoxia-inducible factor-1α (HIF-1α) protein expression in the hippocampus, brain stem, cerebellum, and cerebral cortex. The top panels show HIF-1α protein bands and corresponding β-actin bands representative for each experimental group. Lower panels include levels of HIF-1α protein expression measured by densitometry using samples obtained from five animals in each experimental group. HIF-1α expressions increased significantly in the hippocampus, brain stem, and cerebellum (
In parallel to the immmunohistochemical approach for all brain regions, VEGF protein content was verified in the brain stem, which showed the most elevated water content indicating enhanced edema formation. Western blot analysis with anti-VEGF antibody showed approximately 38 kDa protein band, representing VEGF homodimeric native form (Ferrara et al, 2003), which was significantly increased (2.23-fold increase, P<0.001 versus sham) in the brain stem at 24 h after SAH. Barely detectable VEGF expression was found in sham brains. HBO significantly reduced the expression of VEGF in the brain stem (ANOVA, P<0.01 versus SAH); however, VEGF protein level remained slightly and insignificantly elevated (by 19.64%) above control values (Figure 8A).
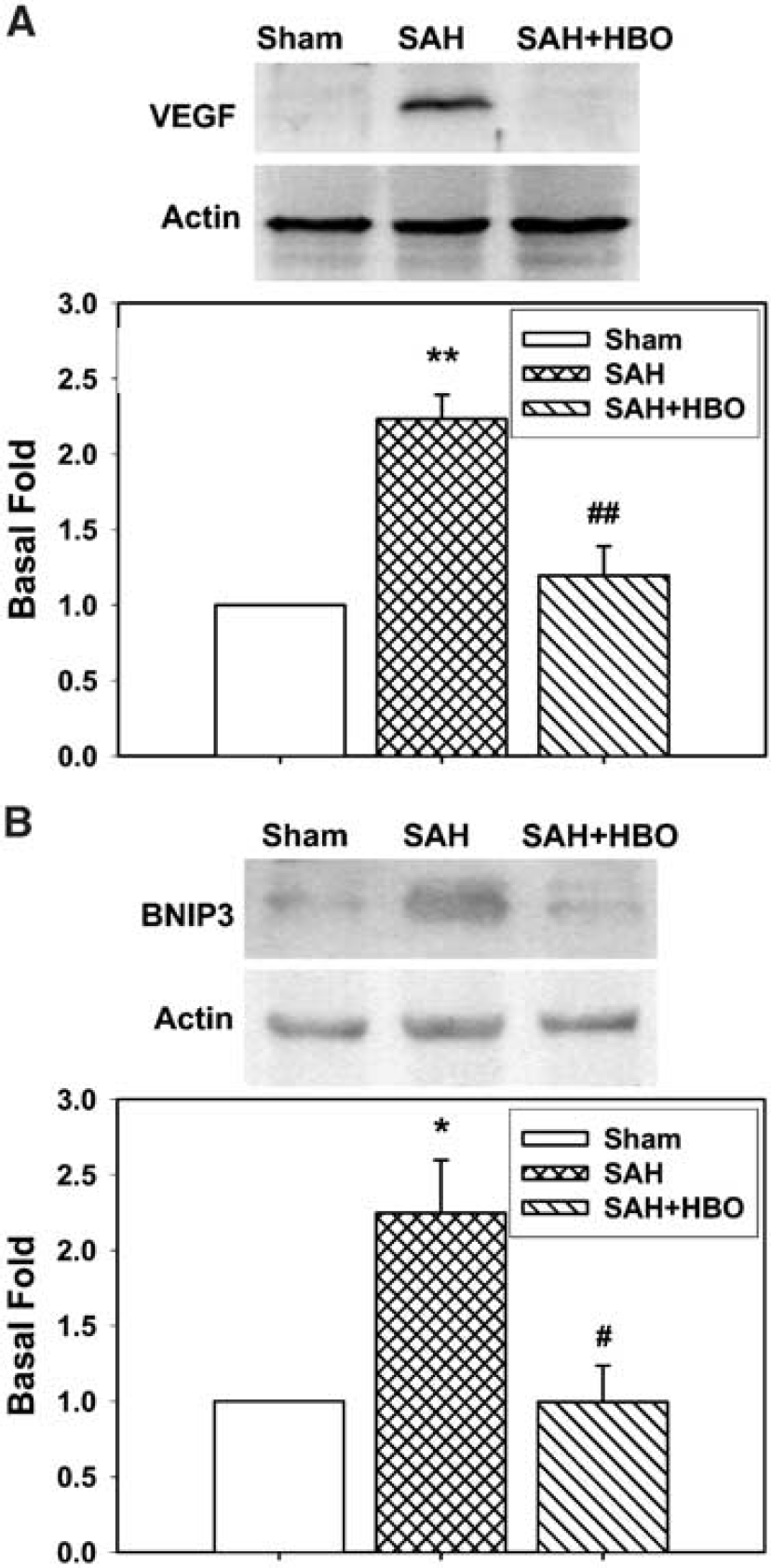
(
Western blot analysis of hippocampal samples showed that BNIP3 protein migrated in the gel as approximately 60 kDa dimer; it showed weak expression in the sham tissues. In the hippocampus, a significant increase in BNIP3 protein expression was noted (by 124.67%) in the no treatment SAH group (Figure 8B). BNIP3 level was normalized after HBO treatment (99.65% of control).
Discussion
This study demonstrated high mortality and neurologic functional disability in the first 24 h after SAH, accompanied by disruption of BBB, brain swelling, brain edema, reduction of CBF and elevation of ICP. In addition, molecular changes of HIF-1α and its target genes, VEGF and BNIP3 are noted, which related to apoptotic cell death and enhanced BBB permeability. HBO reduced the expression of HIF-1α, VEGF, BNIP3, corrected physiologic responses including BBB, brain swelling, CBF, ICP, and CPP, and decreased mortality and neurologic deficits. Some of the actions of HBO seem to relate to the prevention of apoptosis by reducing BNIP3 and to attenuation of disruption of BBB by suppression of VEGF. We speculate that HIF-1α and target genes, activated in pathophysiologic conditions attributable to SAH, mediated early brain injury and in turn aggravated SAH pathophysiology.
Early Brain Injury—Intracranial Pressure, Cerebral Blood Flow, CPP
One of the factors for early brain injury after SAH is acute cerebral ischemia. Measurements of cortical microflow provided evidence that acute cerebral ischemia occurred after SAH induction in the present study. Although ischemic threshold resulting in complete energy failure is considered to be below 20% of regional CBF in the norm, decline to 26% of baseline, as observed in this study, is capable of producing marked acidosis and disturbed protein synthesis and contributing to edema formation. This range of CBF decrease may contribute to cytotoxic edema formation because of loss of sodium and potassium ionic gradients across the cell membrane (Hossmann, 1994). Recent studies provided evidence that even CBF decrease to 40% of baseline triggers profound metabolic disarrangements in cerebral tissues progressing in time (Hossmann, 2003). Initial CBF recovery at 1 h, approximately 80% of baseline, was followed by developing hypoperfusion sustained at 24 h, similar to the results reported previously by others (Sun et al, 2003). In addition, it is likely that brief cerebral contralateral ischemia is accompanied by longer lasting and more profound ischemia in the ipsilateral hemisphere. Jarus-Dziedzic et al (2003) measured CBF bilaterally in the area supplied by middle cerebral artery in perforation SAH model and found more profound decrease in CBF ipsilaterally as compared with CBF values on the contralateral side.
The relationship between raised ICP and CBF impairment is well documented. Cerebral ischemia occurs at cerebral perfusion pressure below 40 mm Hg. However, an increasing number of observations suggest that higher CPP values, between 70 and 90 mm Hg, are required to avoid cerebral ischemia (Lang and Chesnut, 1994; Artru, 1997; Aoki et al, 2002). In our study, CPP (calculated as difference between mean arterial blood pressure and ICP) decreased rapidly after SAH induction to 35.4±5.65, below the ischemic threshold. After initial recovery at 5 mins, CPP decreased again, contributing to hypoperfusion sustained at 24 h. It has been proven that hypoperfusion of long duration may contribute to neuronal damage (Ni et al, 1994; Farkas et al, 2002). HBO attenuated cerebral ischemia by improving CBF and CPP, and decreasing ICP in SAH rats.
Early Brain Injury—Brain Swelling, Brain Edema and Blood–Brain Barrier
At 24 h after SAH, brains showed marked swelling resulting from increase in brain water content and CBV and reflected by increased brain wet weight and brain volume. Marked increase in brain water content associated with enhanced BBB permeability, occurring as early as 24 h, emphasizes the severity of early brain injury. One of the possible reasons for brain swelling is the retention of blood in brain tissues after severe cerebral ischemic injury because of dysfunction of autoregulation. Indeed, CBV, which represents the blood content in brain tissues, also showed marked increase, exceeding 80% after SAH in our study. Such high increases were previously reported from clinical investigations, but not in animal models. Grubb et al (1977) measured rCBV, rCBF, and rCMRO2 in SAH patients and found depressed CBF and a significant (P<0.001) increase in CBV (to 58% above normal) in severely neurologically impaired patients with vasospasm.
We measured brain hemoglobin levels to calculate CBV. In theory, acute ischemia after SAH may result in hemorrhagic transformation (Hamann et al, 1999) and affect the results. However, we evaluated the presence of hemorrhagic transformation in brain sections stained with hematoxylin and eosin and found neither petechial nor confluent petechial haemorrhage in the brain parenchyma at 24 h after SAH (data not shown). Appearance of hemorrhagic transformation depends on duration of artery occlusion and usually occurs after longer ischemic insults than observed in our study. In experimental ischemia induced by middle cerebral artery occlusion in rats, the probability of hemorrhagic transformation increased at longer time of occlusion. Only one out of the nine animals reperfused after 1 h of middle cerebral artery occlusion developed hemorrhagic transformation. One and a half hour middle cerebral artery occlusion fitted 25% probability of hemorrhagic transformation (Fagan et al, 2003). It seems likely that the microvascular injury after SAH may not develop as rapidly as in ischemic stroke. Additionally, cerebral infarction, where hemorrhagic transformation usually occurs, did not develop within 24 h in the model of SAH.
In addition, an increase of BBB permeability observed mainly ipsilaterally might raise question whether acute ischemia after SAH results in BBB disruption. Even though greater BBB permeability on the ipsilateral side may be accountable for a greater degree of CBF reduction ipsilaterally, as evidenced by others in the perforation SAH model (Jarus-Dziedzic et al, 2003), blood components including thrombin formation may contribute to BBB disruption after SAH. The role of blood components in BBB disruption after SAH has been examined previously (Sasaki et al, 1986).
HBO reduced brain wet weight, brain volume, and CBV. Some discrepancy appeared as HBO abolished BBB rupture, whereas brain water content was not significantly changed, leading to the supposition that HBO does not reduce edema formation after SAH. Several explanations for this finding may be proposed. First, comparable water content but improved BBB maintenance after HBO may point to a cytoreactive edema present mostly in swollen astrocytes, since we did not observe swollen neuronal perikarya. This phenomenon is an active reaction of astrocytes for neuronal protection, scavenging potassium, and for maintenance of neuronal energetic metabolism (Ito et al, 2003). Possibly, the phase of cytoreactive edema was present in the no treatment group only in the early time interval followed by cytotoxic edema and superimposed vasogenic edema as indicated by widespread IgG staining. The question arises whether a single HBO treatment permanently prevents neuronal injury or produces a delay in SAH-related sequelae. Second, the effect of HBO on BBB integrity may be incomplete and efficient for decrease in permeability for protein molecules (such as IgG) but not substantial in case of water and small solutes. This interpretation raises a question about the possibly differentiated effect of HBO on the expression of tight junctions forming proteins and aquaporins, key mediators of water movement in the brain (Papadopoulos et al, 2002). Third, although it seems that HBO did not reduce brain water content, this negative finding should be interpreted with caution. HBO significantly reduced mortality from 47.62% to 20%, thus keeping alive animals presenting advanced pathologic conditions. In conclusion, the overall effect of HBO on BBB maintenance remains to be clarified and requires further multidirectional studies involving different exposure times and parameters related to hyperbaricity.
Early Brain Injury—Morphology
Because classical histologic techniques are not sensitive enough to detect early neuronal changes after SAH, other indicators of injured cells, including heat shock proteins, immediate early genes, nitrotyrosine, and free radical end products were proposed (Matz et al, 1996; Harada et al, 1997; Sayama et al, 1999). However, the induction of these factors was also linked to blood components including hemoglobin and thrombin, and the inflammatory mediators. Other biochemical findings, including highly elevated brain glutamate, more specifically suggest that cerebral ischemia plays a significant role in early brain injury after SAH (Sehba et al, 1999).
One striking observation emerging from our study is the early appearance of histologically documented brain injury associated with remarkable induction of HIF-1α and protein expression of target genes BNIP3 and VEGF. Pyramidal neurons, in the sector CA1 of the hippocampus and in the cerebral cortex showed cytoplasmic condensation and nuclear pyknosis, markers of evolving necrosis. Neuronal loss was relatively less accentuated, being in line with the reported delayed time course of cell death in the above-mentioned brain regions after global ischemic stroke. Postischemic neuronal loss reached 50% of cells in CA1 after 7 days (Kawai et al, 1992). The spatial pattern of neuronal changes observed in our study resembled global ischemia-induced alterations. Sector CA1 of the hippocampus was severely affected as were numerous foci in cerebral cortex. In human autopsy SAH studies, cortical necrotic foci were revealed after SAH (Dreier et al, 2002). Abundance of injured cells without features of apoptosis in acute SAH stage may lead to the supposition that necrotic cell changes dominate. On the contrary, we may speculate that many affected neurons (not showing evident eosinophilia) do not represent irreversible cell damage, thus making them still amenable to HBO treatment (Csordas et al, 2003). HBO alleviated morphologic features of cell damage as revealed by histologic examination. It prevented appearance of dark neurons with cytoplasm shrinkage and nuclear pyknosis in the CA1 hippocampal sector and in the cerebral cortex. The beneficial effect of HBO in the cerebral cortex was mediated at least partially by abolition of apoptotic cascades, because TUNEL positive cells in brain samples after HBO were virtually absent.
Widespread distribution of HIF-1α-positive staining seems related to the occurrence of apoptotic cell death. In small cortical foci, HIF-1α colocalized with TUNEL in nuclei indicating apoptotic cells. It may suggest that HIF-1α triggers apoptotic cascades in cortical neurons possibly via caspase-8 and p53-related mechanisms as reported previously (Halterman and Federoff, 1999). Immunochemistry revealed that strong HIF-1α staining was present in damaged CA1 cells; however, in other sectors many cells remained morphologically unchanged despite positive HIF-1α staining. This may indicate different time courses of HIF-1α-mediated cell death. In global ischemia, neuronal damage in CA3 sector or dentate gyrus occurred later than in CA1 (Hossmann et al, 2001).
Mortality and Neurologic Deficits after Early Brain Injury
The severity of pathophysiologic disturbances noted in our study was reflected by profound neurologic impairment and high mortality rate. A score below 6 and mortality over 47% is rarely reported in ischemic stroke, either focal ischemia induced by middle cerebral artery occlusion or global ischemia after cardiac arrest. In previous studies originating from our lab, 48 h mortality in the rat perforation model was 56.7% (Gules et al, 2002). Veelken et al (1995) reported that no animal in which perforation was not accompanied by clamping common carotid artery (to reduce bleeding) survived the 3 h observation period. The lower mortality rate noted in our study is possibly related to the diameter of the filament used (4–0 versus 3–0 used by others), reducing the amount of discharged blood. Prunell et al (2002) reported that in animals injected with 250 μL of blood prechiasmatically, the mortality rate was 50%, whereas 300 μL injection resulted in 100% mortality rate.
Our study revealed that severity of early brain injury is a key factor contributing to overall mortality related to SAH. In experimental studies showing amelioration of vasospasm, reduction of mortality was not obtained (Kamii et al, 1999). In addition, HBO substantially improved the neurologic scores of SAH rats. If the applied scale yielded several grades as in the clinical situation, animals treated with HBO would represent milder grade than not treated ones. A relatively small number of animals presented very profound acute ischemia of duration longer than 5 mins. In these rats, mortality was 80% in the no treatment group versus 33% in the HBO group. Therefore, lower scores from these survivors could influence and underestimate HBO results. In this study, for the first time, a significant effect of HBO on mortality rate after SAH was reported (P=0.036). Moreover, none of the HBO-treated animals died between 1 and 3 h, while being oxygenated, unlike in the no treatment group. This provides an additional benefit of HBO therapy to keep severely affected individuals alive.
HIF-1α and BNIP3 in Early Brain Injury
The observation of expression of HIF-1α in apoptotic cells suggests its role in apoptosis because of one of its target genes: BNIP3. Indeed, expression of BNIP3, a proapoptotic protein, was found in the vulnerable sector CA1 of the hippocampus in this study. However, the presence of BNIP3 in brain structures may offer another mechanism for early cell death after SAH. It was shown that BNIP3 transfected into cultured cells resulted in necrosis rather than apoptosis (Sowter et al, 2001). BNIP3-related pattern of cell death does not fit classical criteria for necrosis or apoptosis alone, because evident cell membrane damage may be accompanied by positive TUNEL stain; however, the latter being much less intense than in ‘regular’ apoptosis, for example after tBID transfection. Interestingly, although BNIP3 opens mitochondrial permeability transition pore, it does not trigger cytochrome c release into the cytoplasm and initiation of classical apoptotic cascade (Vande et al, 2000). Ischemic cell death of CA1 neurons was found 2 days after global cerebral ischemia as was nuclear BNIP3 staining in degenerating neurons (Schmidt-Kastner et al, 2004). In our study, only a moderate increase in BNIP3 staining as compared with the background was seen in the CA3 sector and in the cerebral cortex. Because increase in HIF-1α protein content in the cortex did not reach statistical significance (Figure 8E), we did not assay BNIP3 in this brain region. The mechanism for salvage of CA1 neurons may be linked to the suppression of BNIP3 expression, revealing the novel mechanism of HBO neuroprotection provided in our study.
Hypoxia-Inducible Factor-1α and VEGF in Early Brain Injury
In contrast to selective BNIP3 expression, VEGF staining and protein content followed changes related to HIF-1α. This close relation was already reported by others in animal models of other neurologic diseases (Forsythe et al, 1996; Mu et al, 2003; Pichiule et al, 2003). Interestingly, an increase in VEFG expression, including the hippocampus, as shown by immunohistochemistry correlated with enhancement of IgG staining, suggesting that VEGF may contribute to BBB disruption after SAH.
Although both HIF-1α and VEGF immunoreactivities were increased in the cerebral cortex, the increase in HIF-1α protein content was insignificant. This may because of a limited injury of irregular pattern in cerebral cortex as observed in animals and in patients (Matz et al, 1996; Dreier et al, 2002), since the tissue for Western blot was collected from the same region of frontal cerebral cortex. The severely affected brain regions included brain stem that significant brain edema occurred with higher level of HIF-1α; a parallel increase in VEGF protein content was also observed after SAH. In response to HBO, both factors showed decreased expression, as assessed by Western blot. Therefore, even though our results allowed the assumption that HIF-1 mediates VEGF upregulation in our experimental settings, other factors including IGF-1, PDGF, TGF-α, and TGF-β upregulate VEGF through MAPK pathways and IL-1 and IL-6 may upregulate VEGF expression upon local hypoxia (Vinores et al, 1998; Proescholdt et al, 1999; Zhang et al, 2000). Most of these factors are related to the permeability of BBB, consistent with our observation that VEGF expression positively correlated with increased IgG stain. We have explored the relation between VEGF and brain edema after SAH previously (Kusaka et al, 2004). Control of VEGF expression by HIF-1 is well established in other types of strokes; however, comprehensive mechanism of VEGF activation after SAH remains elusive.
Josko (2003) found increase in VEGF expression 48 h after intracisternal injection of hemolysed blood. VEGF increases BBB permeability by a mechanism involving an increase in endothelial cell calcium influx, synthesis/release of nitric oxide, increased synthesis of platelet-activating factor, or an increased release of products via activation of the cyclooxygenase pathway (Mayhan, 1999). It was shown that VEGF antagonism reduces edema and infarct size in the mouse model of cortical ischemia (Ferrara et al, 2003). One of our previous studies showed that inhibition of Src tyrosine kinase resulted in a remarkable drop of VEGF expression and reduction of brain edema (Kusaka et al, 2004). HBO reduced VEGF expression providing a molecular basis for controlling vascular permeability after SAH.
Mechanism of HBO in Subarachnoid Hemorrhage—Hypoxia-Inducible Factor-1α
Although there are several papers collecting data on the beneficial effect of HBO on infarct size and functional outcome, applied both before and after the ischemic insult (Prass et al, 2000; Schabitz et al, 2004), relatively small number of studies investigated molecular mechanisms of HBO-mediated neuroprotection. They included inhibition of apoptosis, inflammation, or ischemia-triggered release of dopamine (Yin et al, 2002; Yang et al, 2002; Calvert et al, 2003). Presumably, most authors consider that functional and structural improvement after HBO merely follows improved oxygen delivery. This hypothesis has been questioned recently: HBO inhibited neuronal death and improved neurologic outcome after canine cardiac arrest; however, neither oxygen delivery nor metabolic rate for oxygen was different in animals treated with HBO as compared with not treated ones (Rosenthal et al, 2003). Authors have concluded that the mechanism of HBO neuroprotection is not because of stimulation of oxidative cerebral energy metabolism. It leads to the thesis that the benefit of HBO treatment may be attributable to a variety of unknown molecular mechanisms.
Hypoxia-inducible factor-1α has been proposed to control adaptive response in hypoxic tissues, thus providing a neuroprotective mechanism (Bergeron et al, 2000). Functional HIF-1 complex is formed by regulatory subunit-α (HIF-1α) and a constitutively expressed β-subunit (Semenza, 2001). An increase in DNA binding by HIF-1α results in enhanced transcription of gene for erythropoietin, a potent neuroprotectant (Prass et al, 2003). However, HIF-1α DNA binding was reduced in response to oxygen glucose deprivation, if preceded by hypoxic preconditioning (Ruscher et al, 1998). Such an observation may indicate that hypoxic stress occurring 48 h after preconditioning was attenuated but may also suggest that HIF-1α and its target genes participate in pathogenesis of oxygen and glucose deprivation-mediated damages, because preconditioning provided neuroprotection associated with reduced HIF-1α-binding capacity. In several other papers, HIF-1α is strongly included in the preconditioning paradigm. It is believed to stimulate angiogenesis and glycolysis, increases in brain levels of glycolytic and tricarboxylic-acid intermediates or improved ATP production (Dirnagl et al, 2003). On the contrary, a number of papers address mechanisms of neuronal death mediated by HIF-1 activation (Guo et al, 2001; Moritz et al, 2002; Lee et al, 2004). In this study, for the first time, we showed increase in the HIF-1α protein in hippocampus, brain stem, cerebellum, and focally in the cerebral cortex at 24 h after SAH. This study provides background for further investigations of HIF-related molecular pathways of cellular injury after SAH and strongly supports the hypothesis that hypoxic mechanisms of brain injury are activated in this type of stroke. However, because of the distinct and complex pathophysiology of SAH, patterns of such activation may occur substantially differently from those observed in ischemic insults.
Hypoxia-inducible factor-1α expression sustained throughout the brain at 24 h after SAH indicates that tissue hypoxia may present at this time period, especially in hypoperfused tissue. Indeed, brain tissue hypoxia (prolonged for several days after the induction of ischemia) was observed in the global ischemic stroke model (Chavez and LaManna, 2002). The other explanation is persistent MAPK activation stimulated by blood components. These two factors may act simultaneously because it was reported previously that MAPK stabilizes HIF-1 under hypoxia (Berra et al, 2000). Remarkable diminution of HIF-1α protein levels in response to HBO may speak in favor of hypoxic stimulation. However, the existence of combined stimulation of HIF-1α is supported by the fact that incomplete recovery of basal HIF-1α protein level was noted after HBO in the brain stem and cerebellum. Also, this prolonged HIF-1α expression seems to be related to more potent HIF-1α stimulation by SAH as compared with some other hypoxic insults. Six hours of hypoxia in adult rats produced only minimal changes in HIF-1α expression, probably because of a rapid ubiquitin-mediated HIF-1α degradation once hypoxia is reversed (Tang et al, 2002). The time course of HIF-1α expression after SAH and the corresponding effect of HBO still need to be determined. The mechanism of HIF-1α reduction possibly involves increased oxygen supply to hypoxic tissues in the hyperbaric environment. Exposure to HBO is associated with several fold increased saturation values in blood and peripheral tissues (Veltkamp et al, 2000). Depending on this explanation, however, the link between HBO and diminished HIF-1α protein expression should be maintained much longer because treatment ended 3 h after SAH and diminished protein content was measured at 24 h after SAH.
HBO Application in Subarachnoid Hemorrhage
In this study, we showed that HBO had a complex impact on SAH-related pathophysiology. It improved CBF, reduced ICP, and increased CPP. The underlying mechanism comprised reduced CBV concomitant with decreased wet brain weight and brain volume. It is known that CBV has a significant impact on ICP (Grande et al, 2002). Reduction in ICP might contribute to the improvement of cortical microflow observed at 24 h after SAH (Oertel et al, 2002). It is conceivable that improvement of CBF was accompanied with relief of tissue hypoxia because of improved CPP. Theoretically, HIF-1α-related pathways could be attenuated by the action of HBO on ICP and CBF through an independent mechanism; however, the observed sequence of phenomena do not confirm this hypothesis but speak in favor of a causative role of HIF-targeted genes for early brain injury after SAH. Shortly after hyperbaric oxygenation, the values of ICP, CPP, and CBF did not differ significantly between the HBO and no treatment group. Delayed improvement of SAH-related pathophysiologic alterations seems to indicate that targeting HIF-1α-dependent mechanisms of neuronal injury results in general improvement in brain function. It seems that HIF-1α-dependent molecular changes, triggered by SAH-induced ischemia and ICP elevation, provided mechanisms for subsequent aggravation of SAH pathophysiology. Therefore targeting HIF-1α may be beneficial for brain tissues affected by SAH, resulting in the reduction of pathophysiologic alterations and improvement of functional outcomes.
It seems that HBO may have two main applications in SAH patients. First, it may provide supportive therapy allowing for better control of ICP and yielding CBF improvement. There are clinical reports on improved CBF in patients with traumatic brain injury after HBO treatment (Rockswold et al, 2001). Other indications may emerge from this study. HBO treatment is worth considering in patients with severe neurologic deficits, poor clinical grade and disqualified from operation. In some of these patients, clinical attempts are made to reduce ICP and qualify the patients for surgery. It seems that HBO may provide an additional efficacious tool for such efforts. Several considerations should be mentioned here. First, the effect of HBO treatment initiated at a later time point is not known. In ischemic model of stroke, HBO treatment started after 12 h proved to be detrimental (Badr et al, 2001). Second, it is not known whether continuation of HBO therapy beyond the acute phase would be beneficial.
In the present study, all outcome measures are at 24 h. We used 24 h after SAH as the end time point because nearly 50% mortality occurred within 24 h. It is certain that this time period does not cover the overall early brain injury timing and additional studies are required to examine at least the first 3 days after SAH. Because ICP and brain water content tend to recovery and back to normal range at 3 days after SAH, other physiologic and neurobehavioral markers may be studied. In addition, the mechanisms responsible for early brain injury could be different between the first 24 h and later at 72 h. Thus, the investigation of mechanisms of SAH-induced brain injury beyond 24 h requires distinct measures and different study design.
In addition, the endovascular perforation model of SAH as used in the present study is designed to investigate early brain injury and most closely mimics acute pathophysiologic sequelae of SAH in humans, including increased ICP, reduced CPP and CBF (Grote and Hassler, 1988; Bederson et al, 1995; Veelken et al, 1995). However, this model does not provide a prolonged vasospasm as reported by us previously (Gules et al, 2002), even though there is number of studies showing vasospasm in the perforation model of SAH in rats and mice (Sayama et al, 1999; Kamii et al, 1999; Saito et al, 2001). HBO has been used as an adjunct treatment of SAH to counter cerebral ischemia, particularly for cerebral vasospasm (Isakov et al, 1985; Kohshi et al, 1993). However, the mechanisms of HBO therapy in cerebral vasospasm that represents the most significant sequalae of SAH contributing to mortality and morbidity in this disease require further studies.
In conclusion, HBO reduces early brain injury after SAH, probably by inhibition of HIF-1α and its target genes, which prevents ischemic cell change, and leads to the decrease of apoptosis and preservation of BBB function. This mechanism may underlie HBO neuroprotection resulting in reduced mortality and improved neurologic function after SAH.