
Abstract
Heat acclimation (HA), a well-established preconditioning model, confers neuroprotection in rodent models of traumatic brain injury (TBI). It increases neuroprotective factors, among them is hypoxia-inducible factor 1α (HIF-1α), which is important in the response to postinjury ischemia. However, little is known about the role of HIF-1α in TBI and its contribution to the establishment of the HA protecting phenotype. Therefore, we aimed to explore HIF-1α role in TBI defense mechanisms as well as in HA-induced neuroprotection. Acriflavine was used to inhibit HIF-1 in injured normothermic (NT) or HA mice. After TBI, we evaluated motor function recovery, lesion volume, edema formation, and body temperature as well as HIF-1 downstream transcription targets, such as glucose transporter 1 (GLUT1), vascular endothelial growth factor, and aquaporin 4. We found that HIF-1 inhibition resulted in deterioration of motor function, increased lesion volume, hypothermia, and reduced edema formation. All these parameters were significantly different in the HA mice. Western blot analysis and enzyme-linked immunosorbent assay showed reduced levels of all HIF-1 downstream targets in HA mice, however, only GLUT1 was downregulated in NT mice. We conclude that HIF-1 is a key mediator in both spontaneous recovery and HA-induced neuroprotection after TBI.
INTRODUCTION
Decades of traumatic brain injury (TBI) research have not led to the development of novel drugs that successfully improve the outcome of injured patients; thus, new research approaches are required. Our research strategy uses the well-established heat acclimation (HA) preconditioning model to enhance endogenous protective mechanisms. Heat acclimation has been shown to confer neuroprotection against TBI in experimental models of closed head injury (CHI). In HA mice, neuroprotection is displayed by enhanced motor recovery, improved cognitive function, and reduced apoptosis postinjury.1–3
Reduced blood flow is one pathophysiological symptom of TBI, leading to secondary ischemia; 4 regenerative mechanisms are then initiated to reduce cellular damage, including the activation of hypoxia-inducible transcription factor 1 (HIF-1). 5 It is a heterodimer composed of constitutively expressed HIF-1β and regulatory HIF-1α, regulated by cellular oxygen concentration. Under hypoxic conditions HIF-1α accumulates, allowing nuclear HIF-1 dimerization and transcription, resulting in a shift to glycolytic ATP production, which counteracts the destructive effects of ischemia. 6 Glucose transporters (GLUTs) are HIF-1 target genes that mediate a favorable brain energy balance. 5 In the brain, glucose transporter 1 (GLUT1) has two glycosylation variants. 55kD GLUT1 delivers glucose from the plasma into the brain and 45kD GLUT1 transports glucose into the astrocyte cell body. 7 Hypoxia-inducible factor 1α also promotes cell survival by activating factors such as VEGF and erythropoietin. 5 Accordingly, mice with neuronal inhibition of HIF-1α showed extensive postischemic injury. 8
Nevertheless, HIF-1α upregulates genes with detrimental cellular consequences, such as apoptosis, 9 blood–brain barrier (BBB) disruption, and edema formation. 10 Edema mediator genes include vascular endothelial growth factor (VEGF) and aquaporin 4 (AQP4).10–12
Enhanced expression of HIF-1α after TBI has been reported by several researchers,13–15 however unlike in brain ischemia, the role of HIF-1α in TBI and its outcome is poorly understood.
Hypoxia-inducible factor 1α is indispensable to the development of the HA phenotype in C. elegans and for cardioprotection in HA rats. 16 After TBI in HA mice, HIF-1α and its downstream prosurvival target genes erythropoietin receptor and p-Akt levels are elevated, suggesting that HIF-1 contributes to neuroprotection conferred by HA.3,17 However, whether HIF-1 is essential to HA-mediated neuroprotection is yet to be established.
Acriflavine, mainly known for its antiseptic properties, inhibits HIF-1 dimerization and prevents HIF-1 transcriptional activity. 18 In this study, we used acriflavine to examine the balance between HIF-1 consensus prosurvival and detrimental pathways and to assess the role of HIF-1 after TBI and in neuroprotection.
MATERIALS AND METHODS
Animals and Maintenance
The study was approved by the Institutional Animal Ethics Committee of the Hebrew University and complied with the guidelines of the National Research Council Guide for the Care and Use of Laboratory Animals (NIH Publication no. 85-23, revised 1996). Male Sabra mice were used. Animals were kept under controlled light conditions of 12 h/12 h light/dark cycle. Food and water were provided ad libitum. The animals were divided into two groups: control normothermic (NT) maintained at an ambient temperature of 24 ± 1°C and heat acclimated (HA) held in a climatic chamber at 34 ± 1°C for 30 days, this period was found to be long enough for the achievement of a stable acclimated state, characterized by a lower basal metabolic rate, lower heart rate, and improved thermotolerance. 19 After HA, and before TBI, mice were transferred to the experimental laboratory at 24 ± 1°C for overnight to adjust them to the acute conditions of the trauma experiment. No difference was found between the weight of NT and HA mice at the end of the acclimation period (NT: 42.0 ± 0.75 g; HA: 40.75 ± 0.65 g, P = 0.18).
Trauma Model
Experimental CHI was induced under isoflurane anesthesia using a modified weight-drop device developed in our laboratory.20,21 Briefly, after anesthesia, a midline longitudinal incision was performed, exposing the skull. A Teflon tipped cone (2 mm diameter) was placed 1 to 2 mm lateral to the midline in the mid-coronal plane. The head was held in place and a 95-g weight was dropped on the cone from a preestablished height, resulting in focal injury to the left hemisphere. After recovery from anesthesia, the mice were returned to their cages and provided with postoperative care and free access to food and water. Sham control mice underwent anesthesia and skin incisions.
Drug Application
In a preliminary experiment, doses of 5, 10, or 15 mg/kg acriflavine were tested in sham-operated mice to determine the optimal acute dose with no side effects. After 10 days of observation neither signs of toxicity, altered body weight nor change in behavior was observed in the treated mice. We chose the highest dose to achieve high HIF-1α inhibition with minimum toxicity. On experiment day, acriflavine 15 mg/kg dissolved in saline or saline alone was injected intraperitoneally into randomly divided HA and NT mice at two time points: immediately after CHI, and 1 hour later, to ensure acriflavine penetration through the BBB into the injured brain.
Neurobehavioral Evaluation
The functional status of the mice was evaluated according to the Neurological Severity Score (NSS) by an observer unaware of the given treatment. This score is a 10-point scale assessing functional neurologic status based on the presence of some reflexes and the ability to perform motor and behavioral tasks such as beam walking, beam balance, and spontaneous locomotion. 22 Animals are awarded one point for failure to perform a task, such that scores range from zero to ten, increasing with the severity of dysfunction. The NSS obtained 1 hour after CHI reflects the initial severity of injury. Therefore, the extent of the recovery (ΔNSS) can be calculated as the difference between the NSS at 1 hour and at any subsequent time point. A positive ΔNSS indicates recovery, zero reflects no change, and a negative ΔNSS indicates neurologic deterioration. The NSS values were measured at 1, 24, and 48 hours after injury. ΔNSS was calculated for 24 hours and for 48 hours time points (n = 9 to 32 mice/group).
Rectal Temperature Measurements
To assess the effect of acriflavine on core body temperature (n = 9 to 32 mice/group), colonic temperatures were measured under basal conditions and 1, 5, 24, and 48 hours after CHI or sham surgery, using a rectal thermistors (Digisense, Thermistor 400 series, Euthech Instruments, Singapore) inserted 3 cm beyond the anal sphincter. 23
Lesion Volume
Mice were subjected to CHI followed by the different treatments (n = 8 to 10 mice/group). Twenty-four hours after CHI, brains were removed and sliced at 2 mm intervals, using a brain mold. Lesion volume was evaluated using 2,3,5-triphenyltetrazolium chloride staining, as described. 2 To avoid inaccuracies because of changes in the ipsilateral hemispheric volume, the lesion volume was calculated as the area of unstained tissue divided by the area of the contralateral hemisphere.
Cerebral Edema
Edema formation was determined 24 hours after CHI, as previously described.
21
Cortices of both ipsilateral and contralateral hemispheres were removed and weighted immediately and then dried for 24 hours in a 95°C oven (n = 10 mice/group). The water content of the tissue was calculated as:

Western Immunoblotting
After 30 days of HA or NT period, mice were subjected to either CHI or sham surgery and were injected with saline or acriflavine as above. Mice were killed 6 or 48 hours later (n = 5 to 6/group). After decapitation, brains were rapidly removed and frontal segments from left, injured, hemispheres were separated and frozen at −70°C until analysis. Sample separation was performed as previously described. Equal protein samples of 25 μg were separated on 10% SDS-polyacrylamide gels. Blots were probed with anti-HIF-1α (1:500; Abcam, Cambridge, UK), anti-GLUT1 (1:200; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), or anti-AQP4 (1:1,000; Abcam). Anti-β-actin (1:1,000; Cell Signaling Technology Inc., Danvers, MA, USA) was used to confirm equal protein loading. Appropriate peroxidase-coupled immunoglobulin G (1:5,000; Jackson Immunoresearch, Soham, Cambridgeshire, UK) was used for secondary incubations. Reactive bands were visualized using the enhanced chemiluminescence system (Biological Industries, Beit Haemek, Israel). Optical density of reactive bands was quantified using Tina software (Raytest, Straubenhardt, Germany) and the protein levels were expressed as the optical density of the examined factor relative to β-actin in the same lane.
Coimmunoprecipitation
The coimmunoprecipitation method was used to determine whether acriflavine inhibits HIF-1 dimerization in the brain after CHI as it does in other organs.18,24 Mice were subjected to CHI, injected with acriflavine or saline as above and killed at 6 hours later. Brains were rapidly removed and frontal segments from injured left hemisphere were homogenated using commercial nuclear extraction kit (Thermo Fisher Scientific Inc., Rockford, IL, USA). Protein concentrations were determined using the Bradford method (Bio-Rad Laboratories, Munich, Germany) and homogenates were kept at −70°C until analysis. For coimmunoprecipitation, 200 μg protein in 200 μl was incubated overnight at 4°C with rotation with anti-HIF-1β (ARNT, 1:50; Cell Signaling Technology Inc.). In all, 30 μl of cleaned Protein Immobilized rProtein (IPA-400HC, Repligen, Waltham, MA, USA) was added and incubated with rotation for 30 minutes at 4°C. After 3 minutes centrifugation at 21,000 g, the pellet was washed five times with homogenization buffer. Finally, sample buffer was added, and the samples were centrifuged at 15,500 g for 5 minutes, and after beads discarding western blot analysis was conducted using either HIF-1α (1:500; Abcam), or anti-ARNT (1:1,000; Cell Signaling Technology Inc.).
Enzyme-Linked Immunosorbent Assay
After 30 days of acclimation or the NT period, mice were subjected to either CHI or sham surgery, injected with saline or acriflavine (n = 5/group) and killed at 48 hours. Brains were removed and the left frontal cortex was frozen in liquid nitrogen. Tissues were homogenized according to manufacturer's recommendations and frozen at −70°C until use. Protein concentrations were determined using the Bradford method (Bio-Rad Laboratories). Levels of VEGF protein were determined using mouse enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Emeryville, CA, USA).
Statistical Analysis
For statistical analyses, we used commercially available computer software (SigmaStat 2.03, Systat Software, San Jose, CA, USA). Treatments were the independent variables and the outcomes of the TBI parameters were the dependent variables. Significance for NSS and temperature experimental series was tested using two-way ANOVA for repeated measures, followed by Tukey post hoc tests. For protein levels, significance was calculated using two-way ANOVA followed by Tukey post hoc tests. Finally, for edema formation or lesion volume one-way ANOVA was used. P values of < 0.05 were considered significant for all comparisons. Data are expressed as mean ± s.e.m.
RESULTS
Hypoxia-Inducible Factor 1 Levels and Dimerization
Co-immunoprecipitation of HIF-1 subunits α and β was used to confirm that acriflavine inhibits HIF-1 dimerization in the brain as shown in other tissues.18,24 Indeed, a reduction in HIF-1α bound to HIF-1β was observed and is shown in Figure 1A.
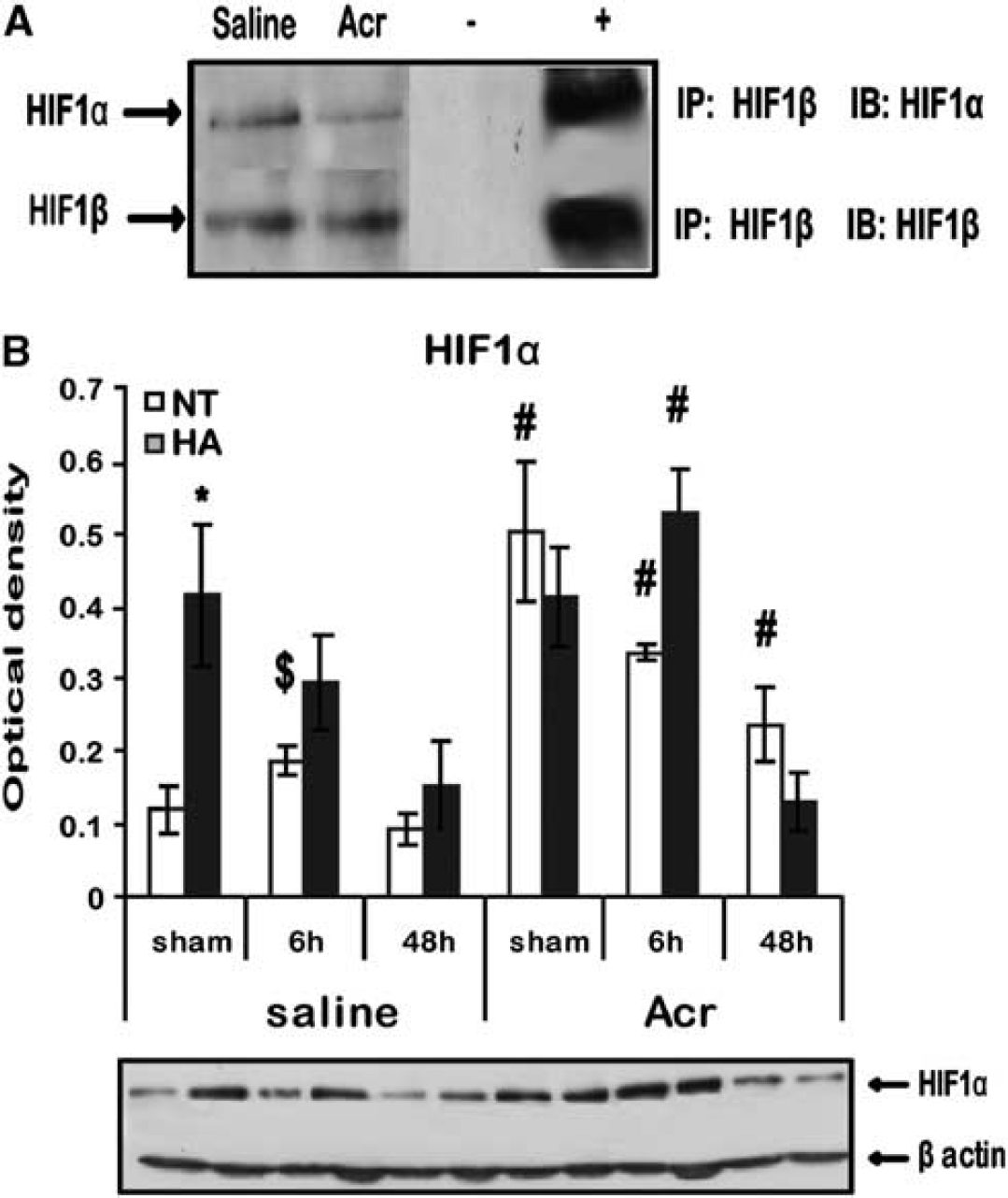
Effects of acriflavine on hypoxia-inducible factor 1 (HIF-1) in the brain. (
We also examined HIF-1α levels after TBI with and without acriflavine (Figure 1B). Basal (sham) HIF-1α levels were higher in saline-treated HA mice than in NT mice (0.41 ± 0.09 versus 0.12 ± 0.03, respectively, P < 0.05). Six hours after TBI, HIF-1α levels were elevated in NT mice (0.19 ± 0.02 versus 0.12 ± 0.03, respectively, P < 0.05). However, after acriflavine treatment higher HIF-1α levels were found at all time points in the NT mice compared with the saline-treated mice (sham 0.50 ± 0.09 versus 0.12 ± 0.03, respectively, P < 0.001; at 6 hours 0.35 ± 0.01 versus 0.19 ± 0.02, respectively, P < 0.001; and at 48 hours 0.24 ± 0.05 versus 0.09 ± 0.06, respectively, P < 0.05). In the acriflavine-treated HA group, HIF-1α levels were only elevated 6 hours after injury (0.236 ± 0.05 versus 0.09 ± 0.06 in saline group, P < 0.05), as seen in Figure 1B.
Recovery of Motor Function
The recovery of each mouse was calculated, at various time intervals, and expressed as ΔNSS (Figure 2). Primary injury severity, estimated by NSS at 1 hour after injury, was similar in HA and NT groups (6.81 ± 0.17 versus 6.64 ± 0.10, respectively, P > 0.05). Spontaneous functional recovery was noted in both saline-treated NT and HA mice, yet it was more robust in the HA group, and reached significance 48 hours after the injury compared with NT mice (ΔNSS = 1.33 ± 0.29 versus 0.43 ± 0.14, respectively, P < 0.001). After treatment with acriflavine deterioration in motor function was observed, compared with saline controls, 24 hours after injury. However, deterioration of the NT mice was less pronounced (ΔNSS = −0.11 ± 0.1 versus 0.21 ± 0.1, respectively, P < 0.05), than that of the HA-treated mice (ΔNSS = −0.64 ± 0.16 versus 0.19 ± 0.06, respectively, P < 0.001). At 48 hours after injury, NT and HA mice treated with acriflavine did not show further recovery whereas saline-treated HA mice recovered significantly (ΔNSS = −0.67 ± 0.19 versus 1.33 ± 0.29 HA acriflavine versus saline, respectively). It is important to note that the condition of the acriflavine-treated HA mice deteriorated after 48 hours; therefore, experiment was stopped and the brains were collected for further analysis.
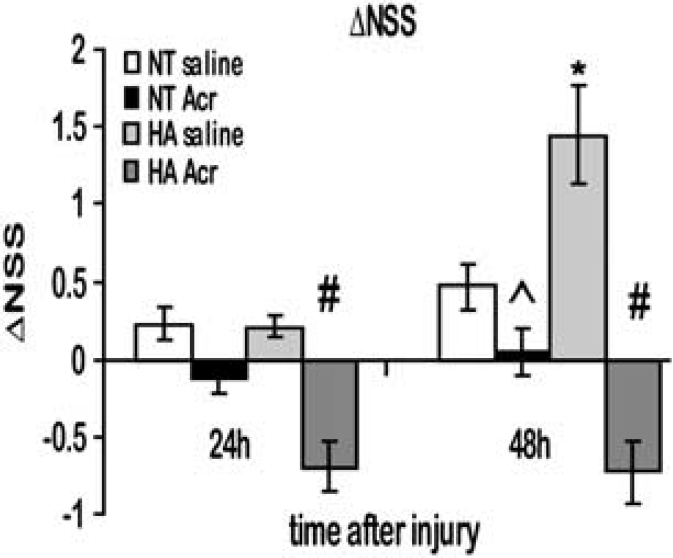
Acriflavine inhibits normothermic (NT) motor recovery after traumatic brain injury (TBI) and causes a deterioration in motor recovery of heat acclimation (HA) mice. Motor function was assessed and expressed using Neurological Severity Score (ΔNSS). The ΔNSS values were significantly higher in HA mice versus NT at 48 hours after injury (*P < 0.05). Acriflavine (Acr) inhibited motor recovery of NT mice (^P < 0.05 versus saline-treated mice), and induced deterioration of motor ability in HA mice at 24 hours and at 48 hours after injury (#P < 0.05 versus saline-treated mice) determined by two-way ANOVA followed by Tukey post hoc tests, n ≥ 9 mice/group.
Lesion Volume
Twenty-four hours after injury, lesion volume was calculated and is shown in Figure 3. As reported 2 saline-treated HA mice showed attenuated lesion volume, compared with NT control mice (0.11 ± 0.01 versus 0.16 ± 0.02, respectively, P < 0.05). However, when treated with acriflavine this effect was abolished and lesion volume of the acriflavine-treated HA mice was larger than that of saline-treated HA mice (0.21 ± 0.02 versus 0.11 ± 0.01, respectively, P < 0.001). After acriflavine treatment, there was no difference between HA or NT mice. Acriflavine did not affect the lesion volume seen in NT mice (0.16 ± 0.02 for saline NT mice versus 0.19 ± 0.02 for acriflavine-treated NT mice).
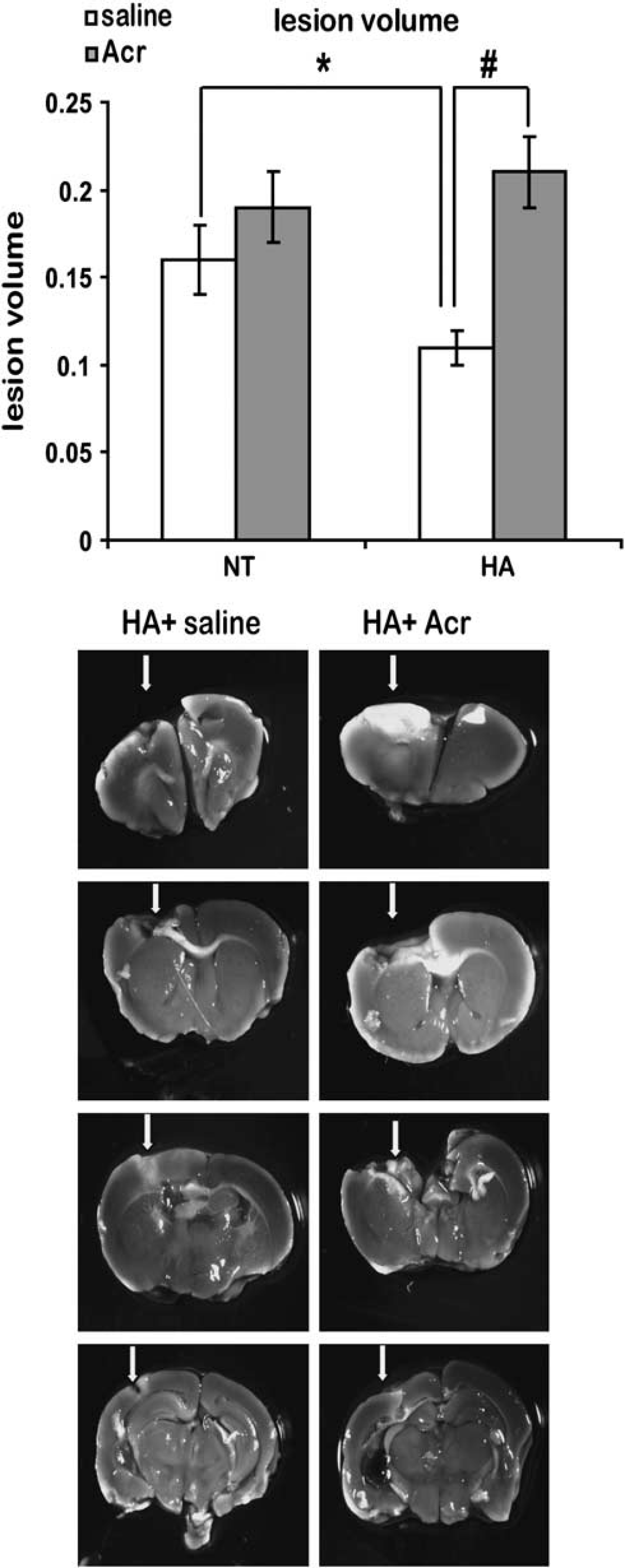
Acriflavine (Acr) increases lesion volume of injured heat acclimation (HA) mice. Lesion volume at 24 hours after injury was measured using 2,3,5-triphenyltetrazolium chloride (TTC) staining as the sum of the percentages of nonstained areas in different brain slices. Arrows point at the nonstained lesion area. The TTC staining quantification revealed significantly reduced lesion volumes in HA mice (*P < 0.05). However, Acr significantly increased lesion volume in HA mice (#P < 0.001) compared with NT mice, determined by one-way ANOVA n = 8 to 12 mice/group.
Core Body Temperature
Core body temperature was the same in HA mice and NT mice (37.75 ± 0.1°C versus 37.44 ± 0.18°C, respectively) at baseline, before CHI. While the sham operation did not affect body temperature for either NT or HA mice, the body temperature of sham mice treated with acriflavine was significantly lower than the temperature measured for saline-treated mice shortly after the surgical procedure, in both NT or HA groups, depicted in Figure 4A. By 24 hours after sham surgical procedure NT body temperature was no longer significantly lower in acriflavine-treated mice. However, body temperature of HA mice treated with acriflavine remained much lower than the HA saline-treated mice (at 24 hours 35.7 ± 0.37°C versus 38.13 ± 0.43°C, respectively, P < 0.01, and at 48 hours 36.06 ± 0.59°C versus 38.33 ± 0.4°C, respectively, P < 0.05). After TBI, there was a dramatic temperature decrease in acriflavine-treated HA mice compared with injured saline-treated HA mice (Figure 4B). The HA acriflavine-treated mice became hypothermic immediately (1 to 5 hours) after the injury, with the lowest temperature of 29.51 ± 1.5°C at 24 hours, remaining reduced up to the last measurement 48 hours after injury (35.31 ± 0.39°C versus 37.0 ± 0.25°C, respectively, P < 0.05). Injured acriflavine-treated HA mice were more hypothermic than their injured saline-treated littermates at all time points, whereas in the injured NT acriflavine-treated mice the hypothermia was transient and only found 1 to 5 hours after injury.
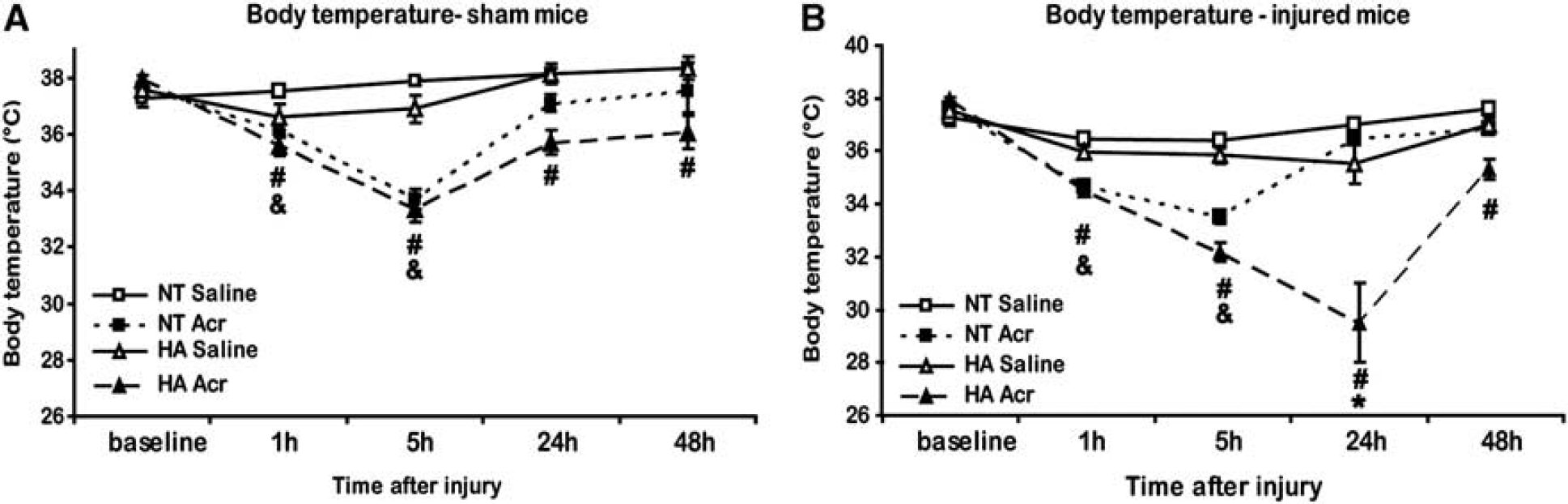
Acriflavine (Acr) induces hypothermia. Baseline core body temperature was measured before closed head injury (CHI), as well as at 1, 5, 24, and 48 hours after Acr administration and sham operation (
Edema Formation
Twenty-four hours after injury, brain water content was evaluated to estimate edema formation (Figure 5). Both in NT and HA mice acriflavine treatment led to a significant attenuation of water accumulation compared with the saline-treated groups (81.79 ± 0.28% versus 82.6 ± 0.28%, respectively, in NT mice, P < 0.01 and 81.38 ± 0.28% versus 82.76 ± 0.37%, respectively, in HA mice, P < 0.001).
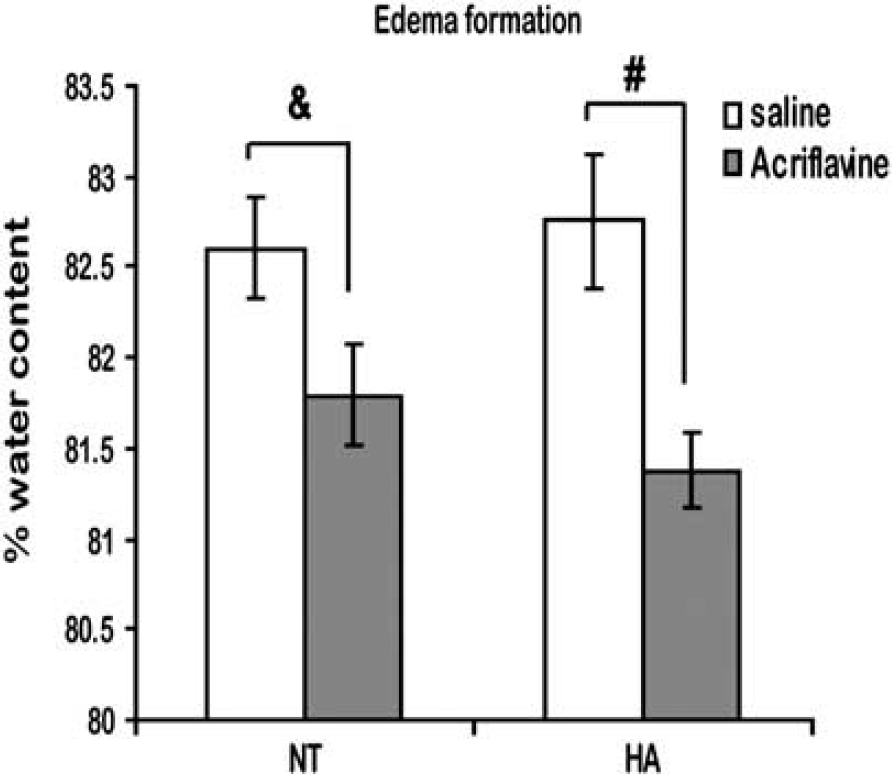
Acriflavine (Acr) reduces postinjury brain edema. Edema formation was determined 24 hours after closed head injury (CHI). Acriflavine reduced edema formation both in normothermic (NT) and in heat acclimation (HA) mice. (&P < 0.01, #P < 0.001) determined by one-way ANOVA. n = 10 mice/group.
Hypoxia-Inducible Factor 1 Downstream Targets Glucose Transporter 1, Vascular Growth Factor, and Aquaporin 4
To explore the cellular mechanisms of acriflavine action, we performed western blot analysis for GLUT1 and VEGF, two of the downstream targets of HIF-1. Basal levels of 55kD GLUT1 were higher in HA mice than in NT mice (0.31 ± 0.06 versus 0.139 ± 0.02, respectively, P < 0.05). Injury induced similar temporal changes in 55kD GLUT1 levels in NT and HA mice, with significantly higher levels (Figure 6A) found 6 hours after injury in both NT (0.36 ± 0.08, P < 0.05 versus sham) and HA mice (0.54 ± 0.06, P < 0.05 versus sham). At 48 hours, GLUT1 levels reverted to baseline and no significant difference from sham was noted in either group. Acriflavine treatment led to a significant increase in 55kD GLUT1 levels in NT mice (0.59 ± 0.06, P < 0.001 versus saline) but not in sham HA mice. A robust, threefold increase in 55kD GLUT1 levels was found 6 hours after injury in HA acriflavine-treated mice (sham 0.20 ± 0.04 versus 0.69 ± 0.11, P < 0.01), in contrast, in the acriflavine-treated NT these levels decreased at 6 and 48 hours (sham 0.59 ± 0.06 versus 0.40 ± 0.015, P < 0.05 and versus 0.13 ± 0.02, P < 0.001, respectively). In both NT and HA mice, acriflavine treatment resulted in 55kD GLUT1 reduction after 48 hours compared with saline-treated mice (in NT 0.13 ± 0.02 versus 0.26 ± 0.06, P < 0.05, respectively, and in HA 0.17 ± 0.02 versus 0.27 ± 0.03, respectively, P < 0.05). However, GLUT1 45kD levels were unchanged in NT mice after either TBI or acriflavine treatment (Figure 6B). However, in HA mice, TBI increased 45kD GLUT1 levels at 6 and 48 hours (0.36 ± 0.08 in sham versus 0.86 ± 0.15 and 0.85 ± 0.2 at 6 and 48 hours, P < 0.05). This increase in 45kD GLUT1 was completely abolished by acriflavine treatment.
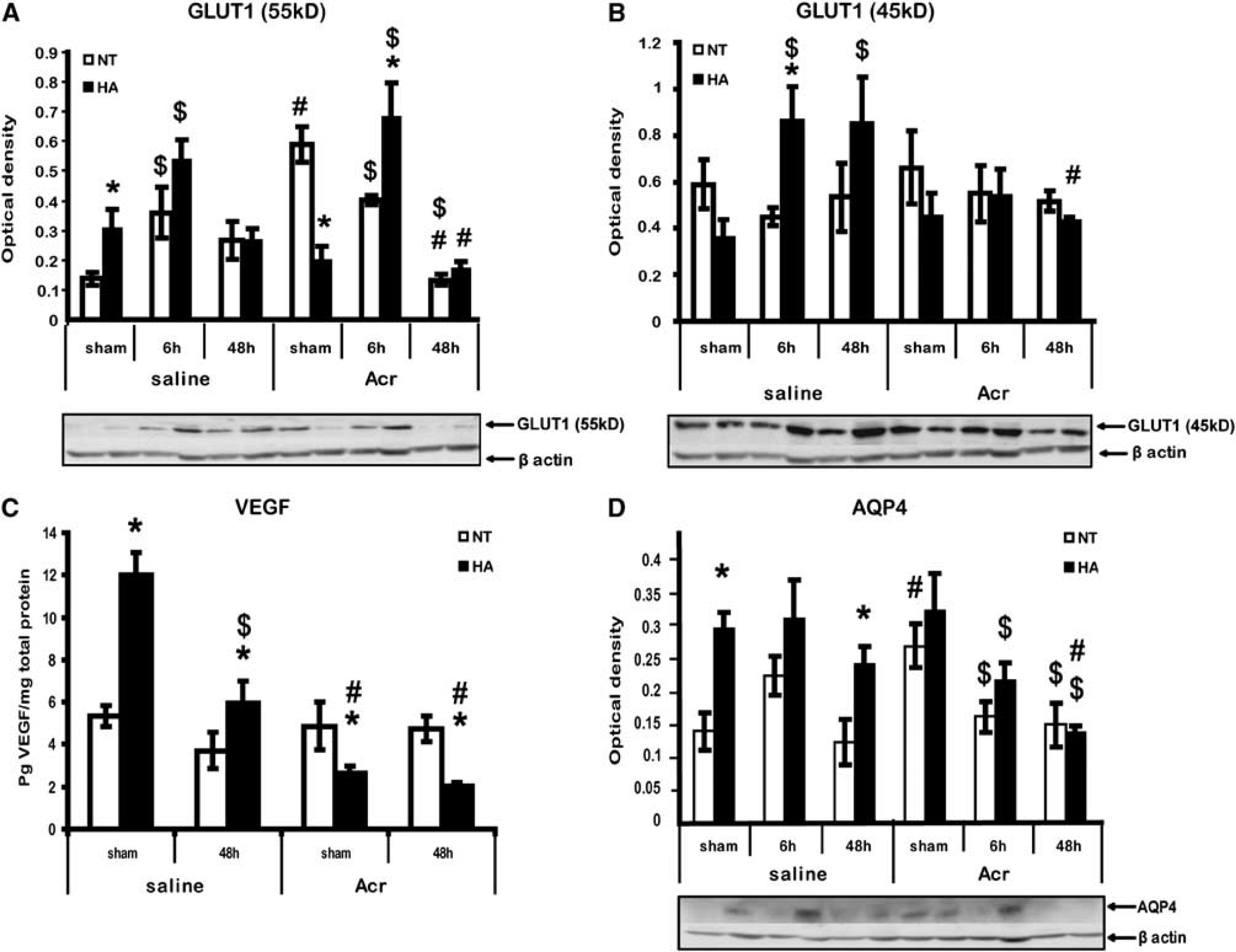
Acriflavine (Acr) alters protein levels of hypoxia-inducible factor 1 (HIF-1) downstream transcription targets. Extracts from injured tissue or sham controls were separated on SDS–PAGE gels and analyzed using western blotting. Optical density normalized to β-actin is shown. Quantified results are presented for 55kD GLUT1 (
Levels of VEGF were detected by enzyme-linked immunosorbent assay (Figure 6C). In NT mice, neither TBI nor acriflavine changed VEGF protein levels, however, in HA saline-treated sham mice higher basal levels of VEGF were found (12.03 ± 1 versus 5.3 ± 0.5 in NT sham mice, P < 0.001) and declined 48 hours after TBI (from 12.03 ± 1 to 5.98 ± 1, P < 0.001). Interestingly, acriflavine dramatically reduced basal levels of VEGF (from 12.03 ± 1 to 2.64 ± 0.3 in sham HA and acriflavine HA mice, P < 0.0001). Traumatic brain injury did not further reduced VEGF levels in HA acriflavine-treated mice.
Aquaporin 4 is regulated by VEGF and is involved in water balance and edema formation. Its levels were assessed and found to be affected by HA (Figure 6D). In saline-treated mice, higher basal levels of AQP4 were measured in HA mice compared with NT mice (0.30 ± 0.02 versus 0.14 ± 0.03, respectively, P < 0.01). These levels remained high at 6 and 48 hours after TBI. Acriflavine treatment significantly increased AQP4 levels in NT sham mice (0.14 ± 0.03 versus 0.27 ± 0.03 after acriflavine treatment, P < 0.05). In HA mice, AQP4 levels were reduced by acriflavine at 6 and 48 hours after injury compared with saline treatment (0.24 ± 0.03 versus 0.14 ± 0.02 after acriflavine treatment, P < 0.01).
DISCUSSION
Hypoxia-inducible factor 1 has a dual role in cellular responses after ischemic and traumatic brain insult. The impact of HIF-1 on angiogenesis, glycolysis, and erythropoietin signaling suggests a role in enhancing cellular survival. 5 However, HIF-1 also mediates detrimental effects of TBI such as disruption of the BBB, brain edema, and apoptosis.9,25,26 Thus, pharmacological inhibition of HIF-1 activation after TBI will shed light on the significance of HIF-1 after TBI and its importance in the HA preconditioning model. The present findings (1) provide evidence that the balance between HIF-1 pro/anti survival effects favors spontaneous recovery from TBI and (2) demonstrate unequivocally that HIF-1 is crucial for the establishment of HA-mediated neuroprotection.
Acriflavine Inhibits Traumatic Brain Injury Spontaneous Recovery
In the present study, a high nonlethal dose of acriflavine (that was determined by preliminary experiments), inhibited the spontaneous recovery of injured control mice, thus indicating that HIF-1 mediates endogenous healing process after CHI. Consistent with previous findings HIF-1α levels were upregulated shortly after CHI. 15 However, Ding et al 25 found that pharmacological inhibition of HIF-1 attenuated synaptic loss 4 hours after the impact, suggesting an early detrimental role of HIF-1 after TBI. Here, an inhibitory effect between 24 and 48 hours after insult was observed. We argue that the beneficial impact of HIF-1-mediated molecular signaling is a delayed effect. The destructive effects of TBI can be divided into primary effects, triggered by the mechanical impact, and secondary cellular processes, and HIF-1 upregulation may have different implications at different time points after injury. Interestingly, inhibition of HIF-1 in NT sham mice increased cellular levels of HIF-1, probably as a compensatory mechanism. However in HA mice, higher basal levels of HIF-1 prevented this phenomenon. 16
Hypoxia-Inducible Factor 1α Inhibition Is Deleterious to Adjustment of Body Temperature
Inhibition of HIF-1 resulted in an acute deterioration of motor ability of HA mice. Importantly, treated sham mice were not affected by acriflavine. This dramatic deterioration implies that injured HA mice are extremely susceptible to HIF-1 inhibition, highlighting the importance of this factor in HA-mediated neuroprotection. Deterioration of motor function in HA mice was accompanied by increased lesion volume, supporting previous data indicating that the HIF-1-regulatory Akt pathway is essential for HA-induced neuroprotection. 17 An interesting finding of this study is the hypothermia induced by acriflavine. Transient hypothermia was also observed in sham mice. Injured acriflavine-treated mice developed an early-onset hypothermia, whereas in the HA mice, hypothermia was extremely dramatic and sustained. The link between temperature and HIF-1 has been investigated before. However, most studies focused on the effects of temperature on HIF-1α levels rather than the other way around. Tanaka et al 27 suggested that postischemic hypothermia reduces HIF-1α levels, and others provided evidence that hyperthermia upregulates HIF-1α.28,29 Here, we showed that inhibition of HIF-1α augments post CHI hypothermia and that this effect was more pronounced in the HA group. Previously, we have found greater post CHI hypothermia in HA mice than in controls. 23 Our current data link HIF-1 to the developed hypothermia. Further studies are required to understand the underlying mechanisms of this phenomenon. A reasonable explanation for this response, however, may be the energy depletion associated with HIF-1 inhibition that attenuates glycolysis and ATP production under stressful conditions; however, examination of these pathways was beyond the scope of the current study. Glycolysis is the main route of ATP production in the brain and is enhanced after TBI, 30 as is glucose transport. 31 Since both are regulated by HIF-1, acriflavine may cause an imbalance between brain energy demand and heat production, resulting in rapid significant hypothermia. Since acriflavine was injected intraperitoneally we cannot exclude mechanisms mediated by peripheral thermoregulatory organs such as skeletal muscle and adipose tissue.
Zhang et al 32 suggested that HIF-1 mediates biogenesis and thermoregulation in brown adipose tissue. Given these findings together with HIF-1α upregulation in HA mice, 3 we suggest that HA mice are more susceptible to HIF-1 inhibition. Unlike other flavones, there is no data whether acriflavine causes vasodilatation which in turn can induce hypothermia as some anesthetics do. To address this question, we used an isolated rat aorta preparation and found no direct effect on vascular tone (data not shown). Mild-to-moderate hypothermia reduces tissue oxygen demand, 33 and thereby induces neuroprotection, thus it is used to treat TBI victims. 34 In the present study, the severe hypothermia in HA mice was accompanied with regression of recovery, and this may be because the hypothermia was secondary to alternations in cellular pathways, or to the severity of the hypothermia.
Hypoxia-Inducible Factor 1α Affects Vascular Growth Factor, Aquaporin 4, and Glucose Transporter 1 Involvement in Edema Formation and Bioenergetics
Edema formation after CHI peaks at 24 hours after TBI sustains for up to 2 weeks. 35 We followed the accepted protocol of edema evaluation 24 hours after injury, and evaluated ΔNSS up to 48 hours after injury. The HIF-1 downstream markers were sampled at 6 and 48 hours and the effects of acriflavine were only significant at 48 hours.
Hypoxia-inducible factor 1 regulates the transcription of VEGF, a key mediator of vascular permeability, 11 which may disrupt the BBB allowing the extravasation of plasma water and solutes into the brain parenchyma resulting in vasogenic edema. 12 Vascular endothelial growth factor induces BBB permeability mainly via activation of matrix metalloproteinases 9 and proteolytic degradation of BBB components.10,36
Expression of AQP4 is regulated by HIF-1 and VEGF 37 and is the most abundant water channel in the brain. Aquaporin 4 is mostly expressed by astrocytes near capillaries and in epedymal cells that line brain cavities, i.e., sites with fluid interfaces and water movement. It controls water transport in and out of the brain, which makes it a leading candidate for edema mediation. 14 However, the role of AQP4 in edema formation after TBI is controversial, as it mediates both types of edema involved in TBI pathology. However, AQP4 induces cytotoxic edema formation, but on the other, it protects from vasogenic edema. 38 Nevertheless, in stroke models using AQP4-deficient mice as well as in TBI models using AQP4 inhibition, AQP4 inactivation reduced edema formation without alternation in BBB function or morphology.10,39
In agreement with other studies that showed the harmful effects of HIF-1 post TBI, leading to BBB disruption, 40 the current study reinforces this hypothesis because the edema was reduced in all treated mice. Notably, edema attenuation was more significant in the HA mice. However, despite having less edema, the HA mice had pronounced behavioral damage and hypothermia. In this group only, significant decreases in both VEGF and AQP4 were detected, suggesting greater susceptibility of preconditioned mice to HIF 1α inhibition. Edema attenuation in control mice was accompanied by a slight decrease in VEGF and AQP4, implying involvement of additional pathways such as hypothermia per se 33 or reduced levels of other HIF-1 targets such as GLUT1. The 55kD GLUT1, expressed exclusively in the endothelial cells of the BBB, is responsible for brain glucose uptake from serum 7 and may also have a role. Here, we show that all injured mice that were treated with acriflavine had lower levels of 55kD GLUT1 48 hours after TBI. Since GLUT1 also transports water molecules 41 it has a potential role in edema formation. Moreover, basally induced 55kD GLUT1 seen in HA mice may imply a favorable basal energy balance enabling the brain to better cope with subsequent injury.
Interestingly, astrocyte-specific 45kD GLUT1 is rapidly upregulated after TBI in HA mice only, and acriflavine treatment abolished this effect. 45kD GLUT1 transports glucose into astrocytes where it is used to produce ATP. A suggested important role for glucose uptake by astrocytes is a glucose-derived lactate shuttle to neurons, supplying neuronal energy. 42 Hence, it is tempting to speculate that upregulation of 45kD GLUT1 in the injured HA mice may attenuate neuronal energy depletion contributing to HA-mediated neuroprotection. Thus, acriflavine causes poor outcomes in these mice by abolishing this protective effect. The 45kD GLUT1 was unchanged in NT mice, correlating with other TBI studies. 31
Summary and Conclusions
In this study, we used a unique experimental model in which, HIF 1α is basally and constitutively up regulated, we aimed to explore the balance between HIF-1 prosurvival and detrimental pathways and to assess its role after TBI and in neuroprotection. We approached this question by testing a wide range of HIF-1 targets and dissected the results obtained using, as a tool, available published material rather than taking longitudinal approach and study one parameter only. Hence, our study opens new future questions.
Taken together, for now, we report for the first time that HIF-1 inactivation inhibits spontaneous recovery after TBI, independent of the severity of edema, and accompanied by significant hypothermia. These findings suggest that HIF-1 prosurvival pathways are predominant. New insights into the mechanisms of HA-mediated crosstolerance were discussed. The effects of HIF-1 on the regulation of body temperature were revealed for the first time, and were especially noticeable in our model of thermal stress.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The authors thank the Hoffman Leadership and Responsibility Program of the Hebrew University and the Professor Jashovam Shani Fund for generous support to GU.