
Abstract
We reported earlier that closed head injury (CHI) in mice causes a sharp elevation of brain 2-arachidonoylglycerol (2-AG) levels, and that exogenous 2-AG reduces brain edema, infarct volume and hippocampal death and improved clinical recovery after CHI. The beneficial effect of 2-AG was attenuated by SR141716A, a CB1 cannabinoid receptor antagonist, albeit at relatively high doses. In the present study, we further explored the role of CB1 receptors in mediating 2-AG neuroprotection. CB1 receptor knockout mice (CB1(−/−)) showed minor spontaneous recovery at 24 h after CHI, in contrast to the significant improvement in neurobehavioral function seen in wild-type (WT) mice. Moreover, administration of 2-AG did not improve neurological performance and edema formation in the CB1(−/−) mice. In addition, 2-AG abolished the three- to four-fold increase of nuclear factor κB (NF-κB) transactivation, at 24 h after CHI in the WT mice, while it had no effect on NF-κB in the CB1(−/−) mice, which was as high as in the WT vehicle-treated mice. We thus propose that 2-AG exerts its neuroprotection after CHI, at least in part, via CB1 receptor-mediated mechanisms that involve inhibition of intracellular inflammatory signaling pathways.
Introduction
Traumatic brain injury leads to secondary damage that includes the release of harmful mediators (e.g. glutamate, reactive oxygen species (ROS), inflammatory cytokines). Protective mechanisms are also set in motion, and recently the endocannabinoid system was proposed to be neuroprotective (van der Stelt et al, 2002; Mechoulam, 2002; Mechoulam et al, 2002; Grundy et al, 2001). Cultured rat hippocampal neurons and cerebral cortical neurons are protected from excitotoxicity or ischemia by cannabinoid receptor agonists (Shen and Thayer, 1998; Sinor et al, 2000). In addition, N-methyl-D-aspartate-induced (NMDA) Ca2+ flux is reduced by anandamide, while SR141716A, a cannabinoid receptor (CB1) antagonist, counteracted this activity (Hampson et al, 1998). In vivo models support these observations. Thus, WIN 55212 (a synthetic cannabinoid) reduced ischemic damage in rat brain (Nagayama et al, 1999) and Δ9-tetrahydrocannabinol (THC), the main psychoactive marijuana constituent, reduced neuronal injury in neonatal rats injected with the Na+/K+-ATPase inhibitor ouabain (van der Stelt et al, 2001a). Hansen et al (2002) proposed that N-acylethanolamines, particularly anandamide, are neuroprotective and, indeed, anandamide protected rat brain against ouabain-induced neuronal injury (van der Stelt et al, 2001b). Yet, in contrast, a recent report describes the neurotoxic effects of anandamide in rats, through mechanisms independent of the CB1 receptor and probably mediated, at least in part, via the vanilloid VR1 receptor (Cernak et al, 2004). We have reported that the levels of the endocannabinoid 2-arachidonoylglycerol (2-AG) increased 10-fold within 4 h after closed head injury (CHI) in mice, and that synthetic 2-AG injected after CHI improved outcome (Panikashvili et al, 2001). The neuroprotective effect of 2-AG was attenuated by the CB1 receptor antagonist SR141716A, albeit at relatively high doses. These findings suggest that the neuroprotective effect of 2-AG is apparently cannabinoid receptor mediated.
The mechanisms of neuroprotection or neurotoxicity by cannabinoids are not yet clear. Some cannabinoids, for example, plant derived (THC), synthetic (WIN 55212) or endogenous (2-AG, anandamide), bind to the CB1 receptor; others, such as cannabidiol (CBD), a marijuana constituent (Hampson et al, 1998), or the synthetic dexanabinol (Shohami and Mechoulam, 2000; Knoller et al, 2002) do not. Hence, the effects of cannabinoids could be derived from numerous mechanisms.
Nuclear factor κB (NF-κB), a key regulator of inflammatory response (Perkins, 2000; Karin and Ben-Neriah, 2000) is composed of homo- and heterodimers including p65 and p50. The p65 subunit contains a translocation domain in its C-terminal end. Inactive NF-κB is retained in the cytosol, where its activity is tightly regulated by members of the IκB family. Activation of the IκB kinase by different proinflammatory signals (e.g. endotoxin, tumor necrosis factor-α (TNF-α), IL-1β, oxidative stress; Zingarelli et al, 2003) leads to phosphorylation, ubiquitination, and degradation of IκB (Malek et al, 2001; Tam and Sen, 2001). Nuclear factor κB, thus released, translocates into the nucleus and activates various genes. Studies from our laboratory recently showed robust transactivation of NF-κB at 24 h up to 8 days after CHI. Inhibition of this activation by melatonin was associated with significant improved outcome (Beni et al, 2004).
The present study explores the effect of 2-AG on NF-κB transactivation after CHI in mice. To confirm the role of the CB1 receptor in neuroprotection, the spontaneous and 2-AG-mediated neurobehavioral recovery and NF-κB activation in CB1 receptor knockout (CB1(−/−)) mice were compared with those observed in wild-type (WT) mice.
Materials and methods
Animals
The study was performed according to the guidelines of the Institutional Animal Care Committee. (1) Male C57BL/6J mice (Harlan, Israel) weighing 32 to 35 g were used in this study. The animals were divided into groups, treated with 2-AG or vehicle as described further, and killed at different times after CHI. (2) CB1(−/−) mice were kindly provided by Professor A Zimmer (Bonn, Germany). They were developed as described previously (Zimmer et al, 1999). To transfer the CB1 tm1zim mutation from the MPI2 embryonic stem cell genetic background (129/sv) to a C57BL/6J genetic background, congenic mice were generated by breeding the mutant allele for 10 generations with C57BL/6J mice. Heterozygous mice were bred to yield WT, heterozygous and null mice.
Trauma Model
Mice were subjected to CHI under ether anesthesia, confirmed by loss of pupillary reflex, using a weight-drop device that falls over the left hemisphere, as described elsewhere (Chen et al, 1996) and modified by Yatsiv et al (2002). In brief, after a sagittal scalp incision, mice were immobilized under a cylindrical calibrated weight drop device. A tipped teflon cone was placed 3 mm lateral to the midline and 1 mm caudal to the left coronal suture, and a metal rod (94 g) was dropped on the cone from a height of 11 to 14 cm (adjusted to body weight) to cause CHI. Sham-treated mice were anesthetized with ether, their scalps were incised, but trauma was not induced.
Evaluation of Functional Outcome
At 1 h after CHI, the functional status of the mice was evaluated according to a set of 10 neurobehavioral tasks (neurological severity score (NSS)) that test reflexes, alertness coordination, and motor abilities (Table 1; Beni-Adani et al, 2001). One point was awarded for absence of reflex or failure to perform a particular task. Hence, a score of 10 reflects maximal neurological impairment. Only mice with NSS >4 at 1 h after injury (NSS 1 h) were included in the study. Immediately after evaluation of NSS 1 h, mice were randomly assigned to vehicle or drug treatment (see below), and NSS was evaluated again at 24 h. The extent of spontaneous recovery was calculated as the difference between NSS 1 h and that at 24 h: ΔNSS=NSS (1 h)−NSS (24 h) and compared with that induced by 2-AG treatment.
Neurological severity score for head-injured mice

One point is awarded for failure to perform a task. NSS at 1 h in the range of 8 to 10: severe CHI.
Cerebral Edema
Cerebral edema was evaluated at 24 h after CHI (time for maximal edema, Chen et al, 1996) by determining the tissue water content in the injured brain, as previously described (Chen et al, 1996). The percentage of tissue water was calculated as
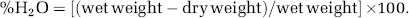
Infarct Volume
At 24 h after CHI brains of WT and CB1(−/−) mice were sliced to 2-mm thick slices using a brain mold. The slices were placed in a 2% solution of 2,3,5-triphenyltetrazolium chloride (TTC) in PBS and photographed using Stereoscope Stemi SV11 (Zeiss, Germany) and digital photocamera Coolpix E990 (Nikon, Japan). Scion Image-Release Beta 4.0.2 program was used to quantify the infarct volume, to examine whether lacking CB1 receptor will affect infarct volume.
Electrophoretic Mobility Shift Assay for Nuclear Factor κB DNA Binding
Mice were decapitated 8 and 24 h after CHI or sham operation. Nuclear extracts were prepared as described previously (Beni et al, 2004). The injured tissue weighing approximately 100 mg was dissected on ice, transferred briefly into ice-cold 0.32 mol/L sucrose and incubated for 5 to 10 mins. Tissues were homogenized in ice-cold 1:4 (w:v) buffer A (0.5 mol/L sucrose; 10 mmol/L HEPES, pH 7.9; 1.5 mmol/L MgCl2; 10% glycerol; 1 mmol/L EDTA) into which 1 mmol/L DDT, 1 mmol/L PMSF and protease inhibitor cocktail (1:25; Roche Diagnostics; Mannheim, Germany) were added before use. After 15-min incubation on ice, centrifugation at 10,000g for 5 mins at +4°C was performed. Supernatants were discarded and pellets were rewashed with 1:1 (v:v) buffer A and centrifuged at 10,000g for 5 min at +4°C. Supernatants were discarded and nuclear pellets were re-suspended in 1:1 (v:v) ice-cold buffer C (20 mmol/L HEPES, pH 7.9; 25% glycerol; 0.42 M NaCl; 0.2 mmol/L EDTA), to which 1 mmol/L DDT, 0.5 mmol/L PMSF were added freshly. After 30-min incubation on ice with frequent vortexing, the samples were centrifuged for 20 mins at 15,000g at +4°C, and the supernatants were stored at −80°C until used as nuclear extracts in the EMSA. The consensus sequence for NF-κB was a double-stranded oligonucleotide (5′-AGT TGA GGG GAC TTT CCC AGG C-3′; 3′-TCA ACT CCC CTG AAA GGG TCC G-5′; Promega, E3291; Madison, WI). Oligonucleotides contained 5′-OH blunt ends that were labeled with [γ-32P] (Perkin-Elmer Life Sciences) using T4 polynucleotide kinase (Promega, M4101) according to the instructions of the manufacturer. The binding reaction mix, containing 10 mmol/L HEPES (pH 7.9), 60 mmol/L KCl, 10% glycerol, 2 μg bovine serum albumin, 0.4 mmol/L DDT, 2 μg poly(dI-dC), 10 μg nuclear protein, and γ-32P-labeled NF-κB (30,000 cpm), was incubated in ice for 1 h. Specificity of the protein-DNA complexes was confirmed by incubation (30 mins) of nuclear extracts with 100-fold excess of unlabeled NF-κB oligonucleotide before the respective γ-32P-labeled probe was added. Supershift assay for NF-κB entailed incubation (30 mins) of nuclear extracts with 3 μl of anti-p65 and anti-p50 monoclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) before addition of the γ-32P-labeled probe. DNA–protein complexes were resolved on a 5% polyacrylamide gel made up in 1 × TGE (50 mmol/L Tris, 400 mmol/L glycine, 2 mmol/L EDTA) at 100 V for 95 mins. The gels were vacuum-dried and exposed to Kodak X-ray films at −80°C. Quantitative data were obtained using Bio-Rad Multi-Analyst (PC Version 1.1). Levels of NF-κB were expressed as the relative optical densities against background within a gel. Each experiment was repeated three to four times, and the data represent the mean of all measurements.
Drugs
2-Arachidonoylglycerol was synthesized in our laboratory according to published procedures (Mechoulam et al, 1995). Emulphor was obtained from Sigma Israel. 2-Arachidonoylglycerol was dissolved in anhydrous ethanol:emulphor: saline (1:1:18) and injected intraperitoneally at 100 μl per 10 g body weight at a dose of 5 mg/kg.
Statistical Analysis
Values represent the mean±s.d. Statistical significance of differences between means was evaluated by the nonparametric Mann–Whitney test for NSS assessment, and by Student's t-test for brain water content, and OD measurement. Probability values (P) smaller than 0.05 were considered to be statistically significant.
Results
Spontaneous Recovery after Closed Head Injury in Wild-Type and CB1 Receptor Deficient Mice
To assess the basal motor activity of CB1(−/−) mice, the NSS was recorded for naive CB1(−/−) and WT mice (n=7 and 9, respectively). Neurological severity score was higher in the CB1(−/−) than in the WT mice (1.79±0.58 versus 0.56±0.88, P=0.008), indicating some basal deficits, as reported earlier (Zimmer et al, 1999). CB1(−/−) and WT mice were then subjected to CHI and their neurobehavioral outcome was evaluated 1 and 24 h later. Owing to the basal difference in NSS, it is hard to claim that similar values of NSS at 1 h indicates similar severity of injury in both groups, rather, similar NSS 1 h may bias towards more severe injury in the WT. Yet, over the next 24 h the WT mice recovered significantly better than the CB1(−/−) mice as depicted by their higher ΔNSS (1.89±0.78 versus 0.71±0.76, respectively, P=0.0164) (Figure 1A). By expressing the recovery as a difference between the NSS at 1 h and that at 24 h, each mouse serves as it own control, and ΔNSS for the individual mice indeed reflects their post CHI recovery, which is independent of the pre-CHI deficits. Thus, it appears that the CB1(−/−) mice are more susceptible to the secondary brain damage after CHI than WT mice, and their spontaneous recovery is slower.
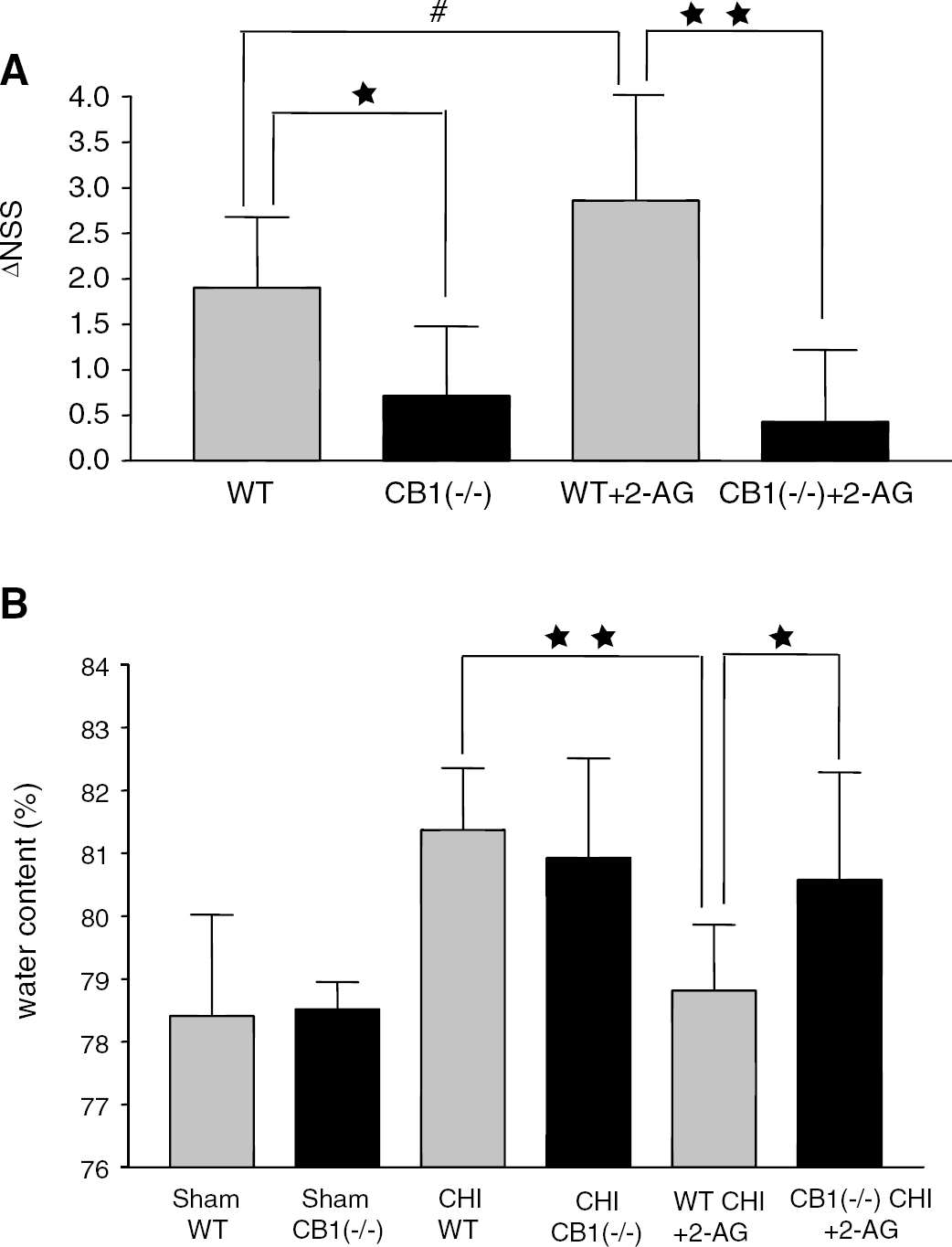
CB1(−/−) mice display smaller spontaneous recovery after CHI as compared with WT mice, and do not respond to 2-AG therapy. (
Infarct Volume and Edema after Closed Head Injury in CB1 Receptor Deficient Mice
Using TTC staining 24 h after CHI infarct volume was measured in WT and CB1(−/−) mice. In spite of the difference in functional recovery, there was no difference in the infarct volumes between the groups 12.3±7.4 versus 15±10.6 respectively, P=0.51. Similarly, basal water content in the cortex of nontraumatized CB1(−/−) mice was similar to that of the WT. After CHI, significant water accumulation was found in both groups (Figure 1B), but edema was not statistically different between these groups (81.37±0.99 versus 80.92±1.59 %).
Effect of Exogenous 2-Arachidonoylglycerol on Neurological Severity Score and Edema in Wild-Type and CB1(−/−) Mice
We next addressed the question whether the protective effects of exogenous 2-AG, which were reported earlier, are mediated via the CB1 receptor. Wild-type and CB1(−/−) mice were treated with 2-AG 1 h after CHI. 2-Arachidonoylglycerol significantly improved the neurobehavioral status of the WT, but not of the CB1(−/−) mice (ΔNSS=2.86±1.46 versus 0.43±0.79, respectively; P=0.0041, Figure 1A). Moreover, treatment with 2-AG effectively reduced edema only in the WT mice (Figure 1B, 81.37±0.99, where water content was down to normal (78.81±1.046), but not in the CB1(−/−), where it remained as high as in the nontreated mice (80.57±1.71, P=0.0308 versus WT treated with 2-AG). It should be noted that basal water content, namely in sham-operated mice, in CB1(−/−) mice was similar to that in the WT (78.52±0.43 versus 78.41±1.61).
Effect of 2-Arachidonoylglycerol on Nuclear Factor κB Translocation
Since inflammation and oxidative stress are major components of the post-CHI responses, we decided to investigate the effect of 2-AG on a key transcription factor of these pathways. The pattern of NF-κB–DNA binding of nuclear extract prepared 24 h after CHI is described in Figure 2A. Three bands were obtained and their specificity was proved by adding excess of unlabeled (cold) oligonucleotide NF-κB and specific antibodies for the p65 and p50 subunits. Supershift assays show that the lower band is mostly composed of p50 and the upper band is p50–p65 heterodimer. Closed head injury in control mice (lanes 5 and 9) induced significant increase in NF-κB–DNA binding as compared with sham (lane 10) (optical density 0.84±0.29 versus 0.27±0.15; Figure 2B). Treatment of WT mice with 2-AG (lanes 1, 3, 6, 8) completely abolished this increase (0.37±0.27; P<0.001). We next investigated NF-κB transactivation after CHI in the CB1(−/−) mice, with and without 2-AG treatment. Figure 3 depicts the results of this study showing that, like in the WT, there is approximately three-fold increase of NF-κB activation 24 h after CHI in CB1(−/−) mice (lanes 2 and 9 versus 6). However, in contrast to WT mice, treatment with 2-AG in CB1(−/−) was not effective (lanes 3, 5, 8 and 10), and did not reduce activity of this transcription factor (optical density 1.37±0.8 versus 1.53±0.62; Figure 3B).
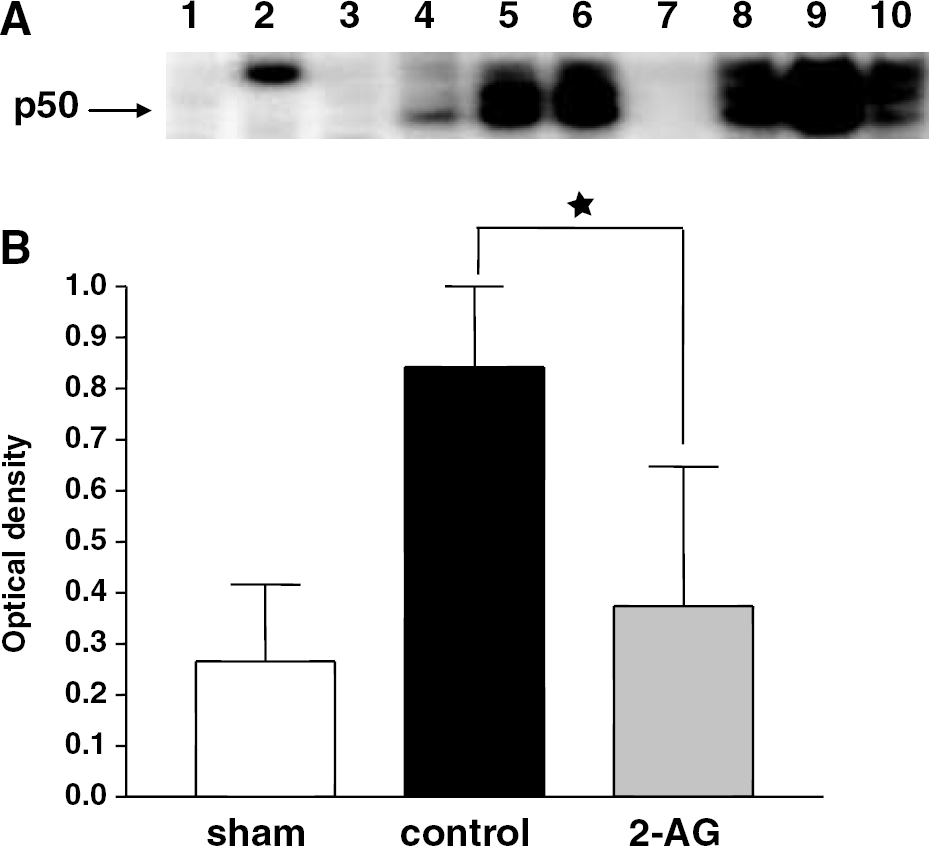
2-Arachidonoylglycerol inhibits NF-κB nuclear translocation 24 h after CHI. (
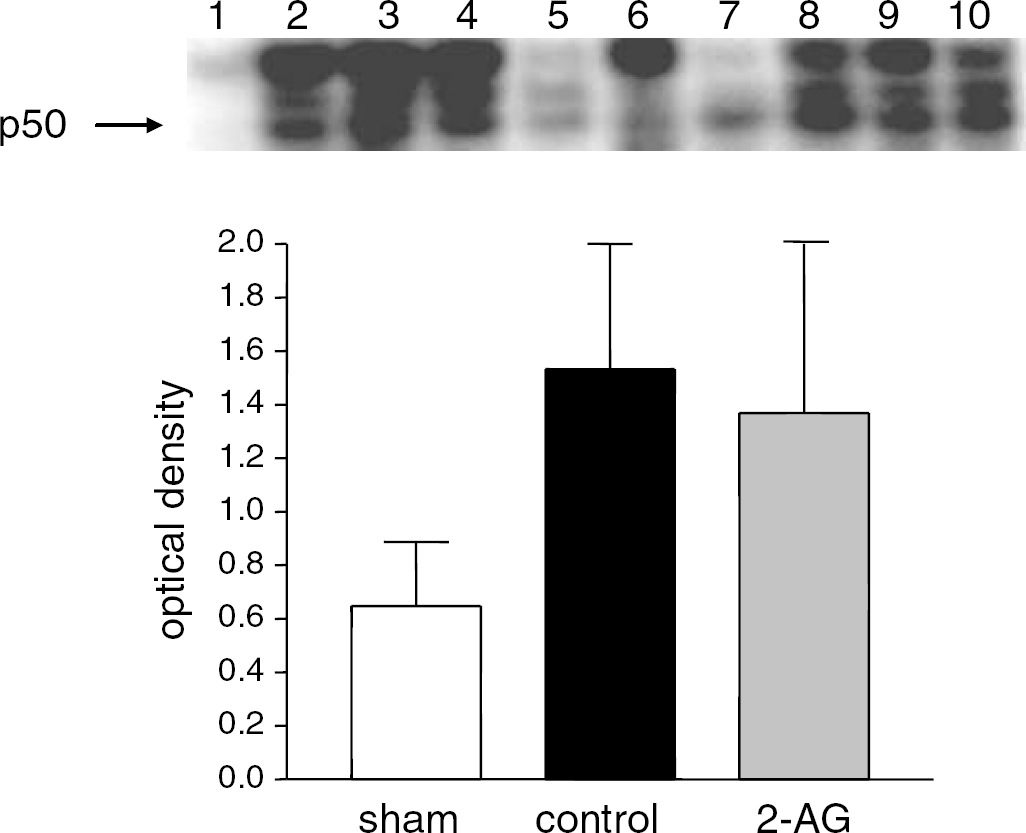
2-Arachidonoylglycerol does not abolish the increase in NF-κB transactivation 24 h after CHI in CB1(−/−) mice. (
Discussion
The present study extends our findings on the beneficial effect of 2-AG after CHI in mice, shows the role of the CB1 receptor in mediating these effects and provides some mechanistic clue to its action. We reported earlier (Panikashvili et al, 2001) that 2-AG treatment decreased edema formation, hippocampal cell death, infarct volume and neurological dysfunction. The CB1 antagonist SR141716A partly inhibited 2-AG protection, albeit at a relatively high dose (20 mg/kg). Since the major brain cannabinoid receptor is CB1, and the specificity of SR141716A as a pure CB1 antagonist is controversial, we decided to use the CB1 knockout mice to study the role of CB1 receptor in neuroprotection. As reported by Zimmer et al (1999), we also found that the naive CB1(−/−) mice display some motor deficits, unrelated to the trauma. As expected, after injury, their spontaneous recovery was extremely slow, as compared with the WT mice, suggesting that the endogenous cannabinoids play a role in the spontaneous recovery after CHI. In another experiment (preliminary data, not shown), NSS was evaluated at 3 and 7 days, and the same pattern was found, namely, ΔNSS of the CB1(−/−) mice remained below 1 during the whole period, while that of the WT slowly, but consistently, increased. These findings agree with those of Parmentier-Batteur et al (2002), who showed increased severity of stroke in CB1(−/−) mice. However, the present findings do not show greater infarct volume or edema in the CB1(−/−), indicating that other endogenous mechanisms are probably involved. To further confirm the role of the CB1 receptor in 2-AG neuroprotection, WT and CB1(−/−) mice were treated with exogenous 2-AG. As expected, no beneficial effects, neither on neurobehavior nor on edema formation, were noted in the CB1(−/−) mice, in contrast to a significant effect on the WT, confirming our earlier reports (Panikashvili et al, 2001).
Proinflammatory cytokines play a crucial role in traumatic brain injury. Tumor necrosis factor-α is released early (within 1 to 4 h) after CHI (Shohami et al, 1997; Stover et al, 2000) and acts on specific receptors. On binding, the cytosolic portions of both TNF receptors recruit multiple intracellular adapter proteins that activate the transcription factor NF-κB, starting with hydrolysis of the inhibitory protein IκB that allows the p65/p50 complex to translocate to the nucleus and to regulate the expression of various (Baeuerle and Henkel, 1994). We have recently reported that inhibition of NF-κB transactivation after CHI (at 1 to 8 days) is associated with better functional outcome (Beni et al, 2004), and therefore decided to investigate the effect of 2-AG on NF-κB activation after CHI. Indeed, treatment with 2-AG completely abolished the robust activation of this transcription factor, which is a key event in the proinflammatory signaling after CHI.
Closed head injury-induced activation of NF-κB in the CB1(−/−) mice was three to four fold higher than in the respective noninjured (sham) CB1(−/−) mice, similar to that observed in the WT. This suggests that the endogenous 2-AG does not affect the inflammatory signaling. However, in response to exogenous 2-AG, WT and CB1(−/−) mice differed dramatically, and the latter did not respond at all to 2-AG treatment. In addition to their antiinflammatory activities, cannabinoids act also as antioxidants. Cannabidiol is a potent antioxidative agent (Hampson et al, 1998) and the nonpsychotropic synthetic cannabinoid, dexanabinol (HU-211), has also antioxidative properties (Shohami and Mechoulam, 2000). 2-Arachidonoylglycerol was also shown to suppress formation of ROS and TNF-α by murine macrophages in vitro after stimulation with lipopolysaccharide (Gallily et al, 2000). The antioxidant properties of 2-AG may well add to its profile as neuroprotectant and inhibitor of NF-κB transactivation. Oxidative stress is one of the major components in the pathophysiology of traumatic brain injury (Lewen et al, 2000), and ample evidence suggests that ROS also regulate signal transduction pathways such as the NF-κB and AP-1 (Vollgraf et al, 1999). Thus, taken together, anti-inflammatory and antioxidant properties of 2-AG may either add or synergize to enhance its activity as a neuroprotective agent.
Cannabinoids produce a variety of neurobehavioral effects, and a major focus of cannabinoid research has been the substantiation of the assumption that the pharmacological actions of cannabinoids are receptor-mediated. Indeed, the CB1(−/−) mice showed spontaneous phenotypes, including hypoactivity, reduced locomotion and rearing, supraspinal hypoalgesia, increased mortality (Zimmer et al, 1999), spontaneous reduction in feeding behavior (Di Marzo et al, 2001), changes in male hormone balance (Paria et al, 2001), and suckling behavior within 1 day of birth Fride et al (2001). For the most part, results observed in mice treated with selective CB1 receptor antagonists mimic the findings observed in the transgenic animals. However, developmental changes may occur to compensate for the lack of CB1 receptors, as has been suggested from studies of neuropeptide expression (Steiner et al, 1999). Our current findings fit into this body of evidence regarding the importance of the endocannabinoid system, acting via the CB1 receptor, in brain function under physiological and pathological conditions. Recently, van der Stelt et al (2002) and Mechoulam (2002) discussed the role of the endocannabinoid system as a general endogenous protection system. The pharmacological picture is further complicated by the fact that there seems to be species differences. While we found 2-AG production to be enhanced in mice after CHI, Panikashvili et al (2001) and Sugiura et al (2000) saw elevation in 2-AG level in picrotoxin-administered rat brain; Hansen et al (2001), in contrast, found anandamide and not 2-AG enhancement in rats after TBI.
Several pharmacological agents have been described to inhibit NF-κB at one or multiple activation steps of the signaling pathway. These agents include proteasome inhibitors, glucocorticoids (such as dexamethasone), nonsteroidal anti-inflammatory drugs and anti-inflammatory cytokines (Zingarelli et al, 2003). Cannabinoids were found to be very effective in different models of inflammation. In the 1970s, Sofia et al (1973a), (1973b), (1974) showed robust anti-inflammatory effects of crude marijuana extract, of the active marijuana constituent THC, as well as of the marijuana nonpsychoactive constituents CBD and cannabinol (CBN) in paw edema inflammation model in rats. Some of these effects were later shown to be CB1 or CB2 receptors mediated (Clayton et al, 2002; Hanus et al, 1999). Cannabinol and 2-AG were shown to inhibit IL-2 expression in activated thymocytes through inhibition of NF-κB (Herring and Kaminski, 1999; Ouyang et al, 1998). Yet, the molecular mechanisms of all these beneficial effects remained unclear. The complexity and controversy in the field of endocannabinoids is further shown in a recent report on the ‘dark side’ of anandamide that describes its toxic effects, both in vitro and in vivo, in rats (Cernak et al, 2004). This study suggests that mechanisms independent of the CB1 receptor, probably vanilloid receptor VR1-mediated, are involved in anandamide's neurotoxicity.
In conclusion, we report for the first time that the endocannabinoid 2-AG exerts neuroprotection after traumatic brain injury, at least in part, by inhibition of NF-κB transactivation through CB1 receptors. We suggest to further study drugs of similar pharmacological profile as novel candidates for treatment of traumatic brain injury.
Footnotes
Acknowledgements
We thank Dr A Zimmer (Bonn, Germany) for providing the CB1(−/−) mice. ES and RM are affiliated with the David R Bloom Center for Pharmacy, of the Hebrew University, and AA is supported by the Israel Ministry of Absorption.