
Abstract
Peroxisome proliferator-activated receptor-γ (PPARγ) is a transcription factor that regulates the expression of various gene products that are essential in lipid and glucose metabolism, as well as that of the peroxisome-enriched antioxidant enzyme, catalase. Activation of PPARγ is linked to anti-inflammatory activities and is beneficial for cardiovascular diseases. However, little is known about its role in intracerebral hemorrhage (ICH). 15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) acts as a physiologic agonist for PPARγ. In this study, we found that injection of 15d-PGJ2 into the locus of striatal hematoma increased PPARγ-deoxyribonucleic acid (DNA) binding activity and the expression of catalase messenger ribonucleic acid (mRNA) and protein in the perihemorrhagic area. Additionally, 15d-PGJ2 significantly reduced nuclear factor-κB (NF-κB) activation and prevented neutrophil infiltration measured by myeloperoxidase (MPO) immunoassay, and also reduced cell apoptosis measured by terminal deoxynucleotide transferase dUTP nick-end labeling (TUNEL). In addition, 15d-PGJ2 reduced behavioral dysfunction produced by the ICH. Altogether, our findings indicate that injection of 15d-PGJ2 at the onset of ICH is associated with activation of PPARγ and elevation of catalase expression, suppression of NF-κB activity, and restricted neutrophil infiltration. All these events predicted reduced behavioral deficit and neuronal damage.
Introduction
Spontaneous intracerebral hemorrhage (ICH) accounts for 10% to 20% of all strokes in the United States, and ⅔ of all hemorrhagic strokes (Broderick et al, 1993, 1999). No medical or surgical therapy to date has been approved to significantly reduce morbidity or mortality after ICH (Auer et al, 1989; Batjer et al, 1990; Juvela et al, 1989; Morgenstern et al, 1998; Poungvarin et al, 1987; Tellez and Bauer 1973; Yu et al, 1992).
The rapid accumulation of blood within the brain parenchyma leads to increase of local pressure and disruption of the normal anatomy. The clotting cascade and thrombin production account for two of the earlier events after ICH (Gong et al, 2001; Xi et al, 1998; Xue and Del Bigio, 2001). Activation of proinflammatory transcription factor nuclear factor-κB (NF-κB) (Hickenbottom et al, 1999; Wagner et al, 2004), and infiltration of hematogenous inflammatory cells (Aronowski and Hall, 2005; Del Bigio et al, 1996; Mayne et al, 2001; Peeling et al, 2001; Xue and Del Bigio 2000a, b), characterizes the inflammatory response.
The peroxisome proliferator-activated receptors (PPARs), including α, β, and γ, are members of the nuclear hormone receptor superfamily of ligand-activated nuclear transcription factors (Kliewer et al, 1994; Mangelsdorf et al, 1995). The initial study of PPARγ was focused on its role in adipogenesis (Auwerx, 1992; Berger and Moller, 2002). More recently, this nuclear receptor emerged from a role limited to metabolism (diabetes and obesity) to a player in general transcriptional control of numerous cellular processes, with implications in inflammation (Bishop-Bailey, 2000; Blanquart et al, 2003; Bocher et al, 2001; Delerive et al, 2001) and atherosclerosis (Ailhaud, 1999; Duez et al, 2001; Duval et al, 2002; Francis et al, 2003).
15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) is a nonenzymatic break-down product of prostaglandin D2, and it is postulated to act as an endogenous ligand for PPARγ (Kliewer et al, 1995; Straus and Glass, 2001). More recently, analysis of 552 patients with an acute stroke admitted within 24 h after symptom onset showed that increased plasma 15d-PGJ2 concentration is associated with good early and late neurologic outcome and smaller infarct volume (Blanco et al, 2005), suggesting beneficial role for 15d-PGJ2.
Catalase is ubiquitous to all cell types including glia and neurons (Girnun et al, 2002; Moreno et al, 1995), and localized predominantly in peroxisomes, the membrane-bound organelles that house various oxidation reactions in which toxic peroxides are generated as side products (Chance et al, 1979), and serves to protect cells from the toxic effects of hydrogen peroxide (H2O2) by catalyzing its decomposition. The functional PPAR-responsive element (PPRE) was located at the catalase gene promoter, indicating that catalase expression is directly regulated by PPARγ (Girnun et al, 2002).
In this study, we characterized the effects of 15d-PGJ2 on PPARγ and NF-κB activity, catalase expression, neuronal death, inflammation, and behavioral dysfunction after experimental ICH in rats.
Materials and methods
Double Blood Injection Model and Brain Tissue Specimen Preparation
Intracerebral hemorrhage was induced using the double blood injection model, as described in our previous work (Felberg et al, 2002; Hickenbottom et al, 1999). Briefly, male Sprague-Dawley rats (weight, 250 to 350 g) under chloral hydrate anesthesia (0.45 g/kg; intraperitoneally (i.p.)) were immobilized in a stereotaxic frame. A 1-mm-diameter burr hole was produced in the skull (0.7 mm anterior and 3 mm lateral to bregma), and a 22-G stainless-steel cannula was inserted for blood injection into the caudate putamen (5 mm deep to bregma). Hemorrhage was induced first by an injection of 15 μL of autologous blood (over 3 mins), 7 mins after the first injection, another 30 μL blood injection followed (over 5 mins). The blood was drawn from the femoral artery within 60 secs before injection.
For biochemical studies, animals were fatally anesthetized with chloral hydrate (1.0 g/kg, i.p.) and perfused with ice-cold phosphate-buffered saline (PBS). The brains were excised, immediately placed in ice-cold PBS, and subdissected. The ipsilateral striata were snap-frozen in −80°C 2-methylbutane, and stored in a −80°C freezer for further RNA isolation or protein extraction.
For immunohistochemistry, the brains were quickly removed, snap-frozen in −80°C 2-methylbutane, and stored in a −80°C freezer before cryosectioning.
Behavioral Measurements
All behavioral tests were conducted in a quiet and low light room by an experimenter masked with respect to the treatment groups. The footfault and forelimb placing tests were performed according to previously published methods (Bland et al, 2001). The postural reflex and circling tests were performed as described previously (Bederson et al, 1986; Bland et al, 2001). The neurologic deficit score (NDS) was determined 30 mins before and 72 h after ICH. We used pretesting to determine the behavioral baseline and to exclude the abnormally behaving rats. Only rats with < 20% of footfault asymmetry, normal forelimb placing, and rectal temperature of 37.7 ± 0.5°C were included and subjected to ICH (Zhao et al, 2005). The total NDS (0 to 16) was calculated by combining the score on the following four tests.
Postural flexing test: The degree of abnormal posture was estimated by suspending rats by their tails 20 cm above a tabletop. Normal rats extended both forelimbs toward the table surface. Rats displaying this behavior were recorded a score 0. Rats with only flexing of the contralateral limb toward the body were recorded as 2. Rats that additionally rotated the contralateral forelimb toward the tail were graded as 4.
Circling or sidewalk: Rats that circled or sidewalked toward the paretic side were tested on 10 trials. A score of 2 or 4 was given to each rat according to the severity of their deficits.
Forelimb placing (wisker): Animals were held by their torsos with forelimbs hanging freely. Contralateral and ipsilateral forelimb placing responses were induced by gently brushing the respective vibrissae on the edge of a tabletop for 10 trials. Percent successful placing responses were determined. A score of 0 to 4 was given to each rat according to the severity of the deficit by calculating the percent nonplacing × 0.04.
Footfault: Animals were placed on an elevated grid, with openings of 2.3 cm2. As the animals traversed the grid, a footfault was scored each time the contralateral forepaw slipped through an opening in the grid. The total number of steps was also counted. The percent footfault was calculated as the number of footfaults/total steps × 100. A score of 0 to 4 was given to each rat according to the severity of the deficit by calculating the percent footfaults × 0.04.
Administration of 15-Deoxy-Δ12,14-Prostaglandin J2
Ten micrograms of 15d-PGJ2 (Cayman, Ann Arbor, MI, USA) in 4 μl of saline was injected over 5 mins into the same area as the blood injection, 1 mins before blood injection, and repeated once again 30 mins later. The rats were killed at 1 h, 3 h, 24 h, and 3days after the ICH. Saline (vehicle) infusion was used as control.
Nuclear Extraction
The nuclear extraction was performed as described previously by Hickenbottom et al (1999). Briefly, the tissue was homogenized with a Potter homogenizer in ice-cold hypotonic lysis buffer ((10 mmol/L HEPES, pH 7.9, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, 1 mmol/L DTT) containing a mixture of proteinase inhibitors (0.5 mmol/L PMSF, 2 μg/mL leupeptin, 2 μg/mL aprotinin, 0.5 mg/mL benzamidine, 5 mmol/L sodium fluoride, 2 mmol/L sodium pyrophosphate, 1 mmol/L sodium orthovanadate)) and the lysates were incubated on ice for 30 mins. The cell lysates were vortexed and microcentrifuged at 10,000 g for 1 min at 4°C. The supernatants were kept and stored at −80°C as the cytosolic fraction. The pellets were washed once with the same lysis buffer and resuspended in ice-cold hypertonic nuclear extract (NE) buffer (20 mmol/L HEPES (pH 7.9), 420 mmol/L NaCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT containing the mixture of proteinase inhibitors). After incubation on ice for 30 mins and centrifugation at 14,000 g for 5 mins, the NE was collected and the aliquots stored at −80°C. Protein concentrations were determined using the Bradford method.
Electrophoresis Mobility Shift Assay (EMSA) for Peroxisome Proliferator-Activated Receptor-γ and Nuclear Factor-κB
Ten microgram of NE protein was incubated with 32P-labeled double-stranded oligonucleotides (2 pmol for PPARγ and 40 fmol for NF-κB) containing the PPRE (sequences in rat acyl CoA oxidase promoter) 5′-AGCTGG GACCAGGACAAAGGTCACGTT-3′ or NF-κB-responsive element sequences (from the HIV-LTR) 5′-TTGTTACAAGG GACTTTCCGCTGGGGACTTTCCAGGAGGCGTGGG-3′, as described previously (Hickenbottom et al, 1999; Tugwood et al, 1992). Binding reactions were prepared in a final volume of 20 μL (2 μg poly-(dI-dC), 25 mmol/L HEPES (pH 7.9), 0.5 mmol/L EDTA, 0.5 mmol/L DTT, 1% NP-40, 5% glycerol, and 50 mmol/L NaCl) and incubated for 30 mins at 37°C in a water bath. To confirm specificity of the binding a supershift assay using 2 μg of rabbit anti-PPARγ Ig (Santa Cruz) added to the reaction was performed (data not included). Also, the competition assays using 100-fold radioinert competitor oligonucleotides that were included in the reaction mixture confirmed the binding specificity (data not included). Nucleoprotein–oligonucleotide complexes were resolved electrophoretically on a 7% nondenaturing polyacrylamide gel in 0.5 × TBE buffer. The gel was dried and autoradiographed. Optical density was determined using Kodak Analysis (EDAS) 290 system.
Western Blot
For protein electrophoresis and immunoblot, we followed previously published methods (Hickenbottom et al, 1999). Briefly, equal amounts of protein were prepared in 4 × loading buffer containing 0.5 mol/L Tris (pH 6.8), 0.4 mol/L DTT, 16% SDS, 0.02% bromophenol blue, and 40% glycerol, and heated at 95°C for 10 mins. Samples were resolved in a vertical electrophoresis chamber (Bio-Rad) using a 4% stacking gel over a 9% acrylamide resolving gel for 1 h at 200 V. For immunoblotting, separated proteins were laterally transferred to nitrocellulose membranes (0.2 μm) using a transfer buffer consisting of 0.192 mol/L glycine and 0.025 mol/L Tris (pH 8.3) and 10% methanol. Coomassie blue and Panceau red (Sigma, St Louis, MO, USA) were used to stain gels and nitrocellulose membranes (respectively) to confirm that equal amounts of protein were loaded in each lane. Blots were blocked overnight in 3% nonfat milk in TBST (20 mmol/L Tris, 0.15 mol/L NaCl, and 0.005% Tween-20) at 4°C.
For immunoblotting, the membrane was incubated with the rabbit anti-catalase antibody (Cat. #: AB1877-100, Abcam, Cambridge, MA, USA; 1:1000) in 1% BSA-TBST. Goat anti-rabbit-HRP (1:5000) in 3% milk–TBST was used to visualize catalase with ECL (Pierce, Rockford, IL, USA). Semiquantification of immunoreactive bands on X-ray film was achieved by analyses of optical density using the computer-assisted Kodak Analysis (EDAS) 290 system.
RNA Isolation
Total RNA was prepared using Trizol Reagents (Gibco, Gaithersburg, MD, USA). Briefly, the ipsilateral striata were homogenized in the Trizol reagents (Invitrogen) and extracted with chloroform, precipitated with isopropanol, and washed with 70% ethanol. The RNA quality was checked by the ratio of absorbance at A260/280.
Real-Time Reverse Transcriptase–Polymerase Chain Reaction (RT–PCR)
Complementary deoxyribonucleic acid (cDNA) was prepared by RT with an oligo-dT primer (Promega, Madison, WI, USA). A reaction containing 0.5 μg of total RNA and 10 U of ImProm-II Reverse Transcriptase (Promega, Madison, WI, USA) in a total volume of 20 μl was incubated at 42°C for 60 mins. One-fourth of the cDNA from the RT reaction was amplified by TaqMan 7500 probe-based real-time PCR systems (Applied Biosystems, Foster City, CA, USA). The amount of the PCR product was measured as fluorescent signal intensity after standardizing to rat β-actin internal control. A set of rat catalase primers and probe (Rn00560930, Applied Biosystems) was used. Rat β-actin (Rn00667869, Applied Biosystems) was used as an internal control. Quantities of catalase products in test samples were normalized to the rat β-actin gene and values were normalized to vehicle-treated control. Relative expression levels were calculated by the 2(–Delta Delta C(T)) method (Livak and Schmittgen, 2001).
Immunohistochemistry
Coronal cryosections (10 μm thick) were cut using a Leica model CM1800 cryostat. Sections were collected on glass microscope slides, dried on a 55°C slide warmer, and fixed with 95% methanol + 5% acetic acid at −15°C for 10 mins before immunostaining. After blocking in 1% normal goat serum, the cryosections were incubated with rabbit anti-myeloperoxidase (MPO) (Cat. #: A0398, Dako, Carpinteria, CA, USA; 1:500), mouse NeuN (Cat. #: MAB377, Chemicon, Temecula, CA, USA, 1:1000) or rabbit anti-catalase (Cat. #: AB1877-100, Abcam, Cambridge, MA, USA) antibody in 3% milk in PBS. Antigen was visualized using secondary antibodies conjugated to Alexa-Fluor 488 (Molecular Probes, Carlsbad, CA, USA; 1:200). The sections were counterstained with Hoechst 33258 and coverslipped in fluorescence mounting media (Vector) to prevent fading. For double staining, a goat anti-rabbit secondary antibody conjugated to Alexa-Fluor 546 (1:200) was employed. To show catalase expression changes, we also employed immunohistochemistry using the DAB detection system, to avoid interference associated with strong autofluorescence around the hematoma area. The cryosections were prepared exactly as those used for immunofluorescence, except that 3% H2O2 treatment was introduced to quench endogenous peroxidase. A biotinylated horse anti-rabbit Ig (secondary antibody) was detected using an ABC Elite kit (Vector) and 3.3′-diamino-benzidine (Vector). Separate set of cryosections, representing area of the brain at 0.2 mm posterior to the blood injection site, were employed for hematoma size determination, according to morphometrical analysis (Hickenbottom et al, 1999) of the cresyl violet-stained section.
Terminal Deoxynucleotide Transferase dUTP Nick-End Labeling (TUNEL) Stain and Neuron Double Labeling
Coronal cryosections (10 μm thick) were first immunoassayed as described above. Then, the slides were labeled with an in situ cell death detection kit (ApopTag kit 7165, Intergen, Purchase, NY, USA) following the manufacturer's instructions. Briefly, the immunostained sections were equilibrated and labeled using terminal deoxynucleotidyl transferase (TdT) and a mixture of digoxigeninlabeled nucleotides, followed by incubation with anti-digoxigenin Rhodamine-labeled antibody. Finally, the slides were counterstained with Hoechst 33258 and coverslipped with fluorescence mounting medium (vector). Negative controls were performed by omitting the TdT enzyme from the incubation buffers.
Capturing Images and Counting Positive-Stained Cells
To avoid problem with the sampling methods during cell counting, we took advantage of a motorized stage system (Zeiss Axioskop 2 microscope equipped with a motorized stage and MS-2000 (ASI) control system) and program (MetaMorph software 6.2) that allowed image stitching. This computer-controlled image acquisition system permitted automatic, unbiased, cell counting. Two consecutive 10 μmol/L thick sections through the ICH core (1 mm anterior to the bregma) were used for analysis. Four randomly selected rats were analyzed for each treatment group. Sixty-four images were captured under × 20 objective for a total area of 9.6 mm2 (each image equal to 448 × 335 μm) for each analyzed section. The fluorescence-labeled cells were visualized using the following set of filters: Ex/Em of 490/520 for Alexa 488, Ex/Em of 550/575 nm for Alexa 546, and Ex/Em of 365/480 nm for Hoechst 33258. Epifluorescence was captured using a CoolSnap ES (Photometrics) CCD camera driven by the MetaMorph software 6.2.
For each section, the total number of MPO-positive, NeuN-positive, TUNEL-positive, as well as NeuN/TUNEL-double-positive cells, and catalase-positive cells were determined.
Statistical Analysis
The data were analyzed using a t-test for direct comparison between two groups. ANOVA supplemented by the Newman–Keuls' multiple range test was used for comparison of more than two groups. A P-value < 0.05 was regarded as significant.
Results
15-Deoxy-Δ12,14-Prostaglandin J2 Improved Sensory-Motor Recovery after Intracerebral Hemorrhage
The total NDS in rats treated with saline or 15d-PGJ2 is illustrated in Figure 1. Whisker placing, footfault and circling deficit in animals treated with 15d-PGJ2 were 40.5%, 68.7%, and 68% of saline control (data not shown), respectively. Overall there was a significant reduction in the total NDS by 53.0%, as compared with the saline control group.
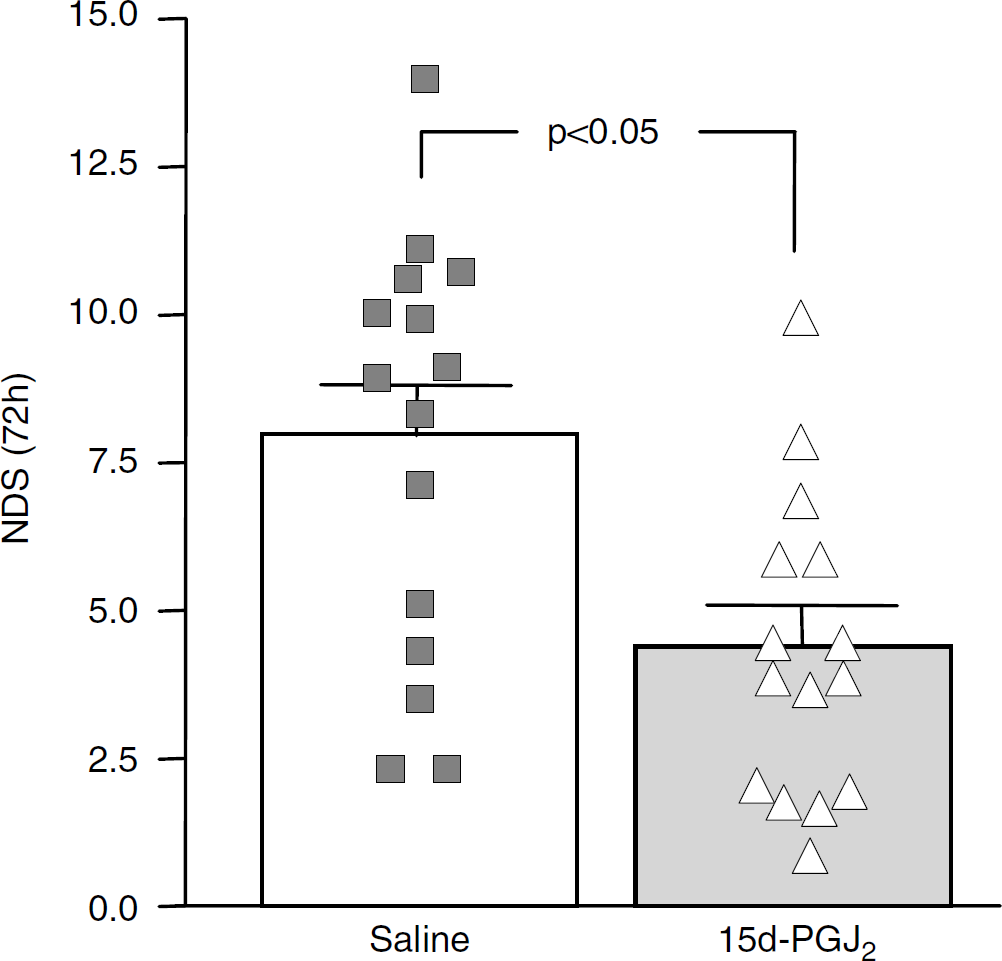
Neurologic deficit score (NDS) in animals 3 days after intracerebral hemorrhage (ICH). Rats received intrastriatal 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) (n = 15; open triangles) or saline (n = 15; closed squares) immediately before blood injection to produce ICH. The data are expressed as mean ± standard deviation (s.d.) *P < 0.05. Each symbol represents NDS of an individual rat.
15-Deoxy-Δ12,14-Prostaglandin J2 Increases Peroxisome Proliferator-Activated Receptor-γ-Deoxyribonucleic Acid Binding Activity and Inhibits Nuclear Factor-κB Deoxyribonucleic Acid Binding Activity
Intrastriatal injection of 15d-PGJ2 (as compared with saline) resulted in increased PPAR-DNA binding activity, as assessed by EMSA. This increase was evident as early as 1 h after ICH (Figure 2A). Interestingly, in contrast to PPARγ activation, the NF-κB DNA binding activity was reduced at 1 h after ICH (Figure 2B). Quantitative analysis of band density showed a 265% increase (P < 0.01) in PPAR–DNA binding in response to 15d-PGJ2 treatment that coincided with 208% decrease in NF-κB–DNA binding activity in terms of the intensity of the top band on the EMSA gel (Figure 2B).
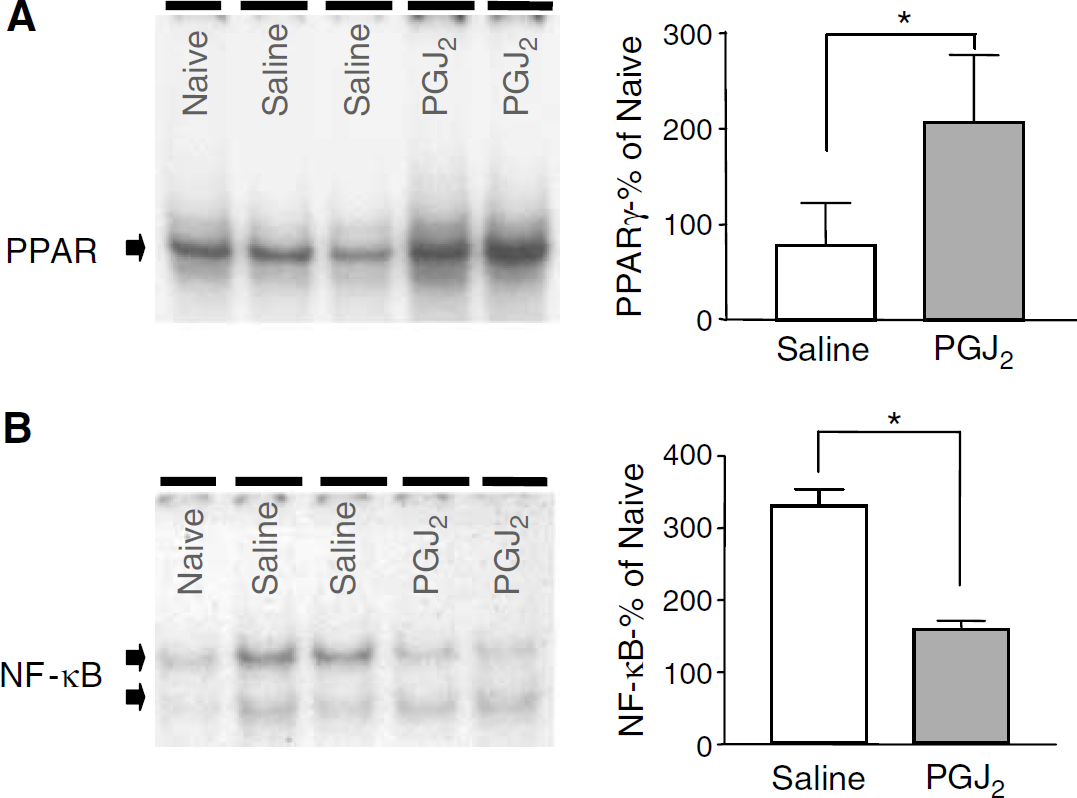
Electrophoretic mobility shift assay (EMSA), assessing peroxisome proliterator-activated receptor-γ (PPARγ)–deoxyribonucleic acid (DNA) and nuclear factor-κB (NF-κB)–DNA binding in nuclear extracts from striatum harvested 1 h after induction of hermorrhage in animals treated with saline or 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2). Representative gels show increased PPAR binding to the DNA probe representing PPAR-responsive element (
15-Deoxy-Δ12,14-Prostaglandin J2 Increases Catalase Expression
Catalase messenger ribonucleic acid (mRNA) expression (induction) after 15d-PGJ2 was 1.6-, 2.1-, and 1.7-fold higher than saline control at 1, 3, and 24 h after ICH, with a P < 0.05 at 3 and 24 h (see Figure 3A). Although we observed a strong trend for increased catalase mRNA expression with 15d-PGJ2 treatment in sham groups (without blood injection) at 3 h, this difference was not statistically significant.
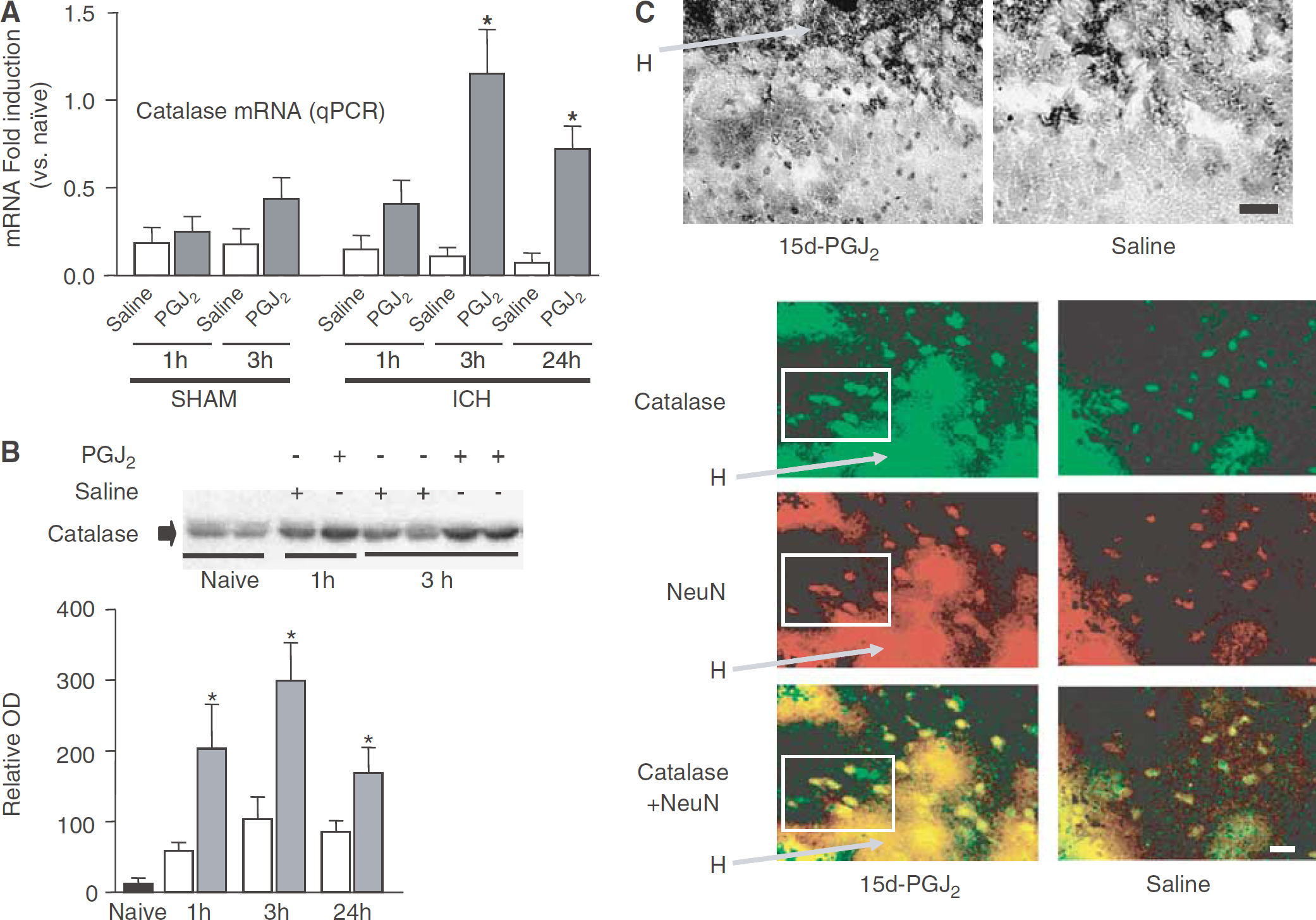
Analyses of catalase expression in the ipsilateral striatum of rats subjected to intracerebral hemorrhage (ICH) with or without treatment with 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2). (
In agreement with the mRNA data, catalase protein in brain tissue homogenates was also significantly upregulated in animals treated with 15d-PGJ2, as compared with saline control at 1, 3, and 24 h after ICH (Figure 3B).
Most catalase-immunopositive cells were detected in the parenchyma at the border zone of the hematoma (Figure 3C). The total number of catalase-immunopositive cells was increased, as well as the signal intensity (as determined using DAB and fluorescence staining intensity assessment) was stronger in the 15d-PGJ2-treated animals (Figure 3C). Double immunolabeling for NeuN (neuronal marker; red epifluorescence) and catalase (green epifluorescence) showed that among cells showing robust catalase immunopositivity, the majority were neurons (Figure 3C; bottom panel).
15-Deoxy-Δ12,14-Prostaglandin J2 Prevents Neutrophil Infiltration
Intracerebral hemorrhage produced robust infiltration of MPO-positive cells into the affected striatum. The MPO-positive cells were present primarily at the border zone between hematoma and parenchyma, resulting in a tight encapsulation of the hematoma.
Treatment with 15d-PGJ2 produced 57.2% reduction (P < 0.05) of the total number of MPO-immunopositive cells in the affected striatum (Figure 4A). This reduced inflammation was unlikely because of reduced size of hematoma in 15d-PGJ2-treated rats, since brains' sections generated from animals treated with saline or 15d-PGJ2 had similar hematoma size (1.93 ± 1.8 versus 1.9± 1.3 mm2, respectively).
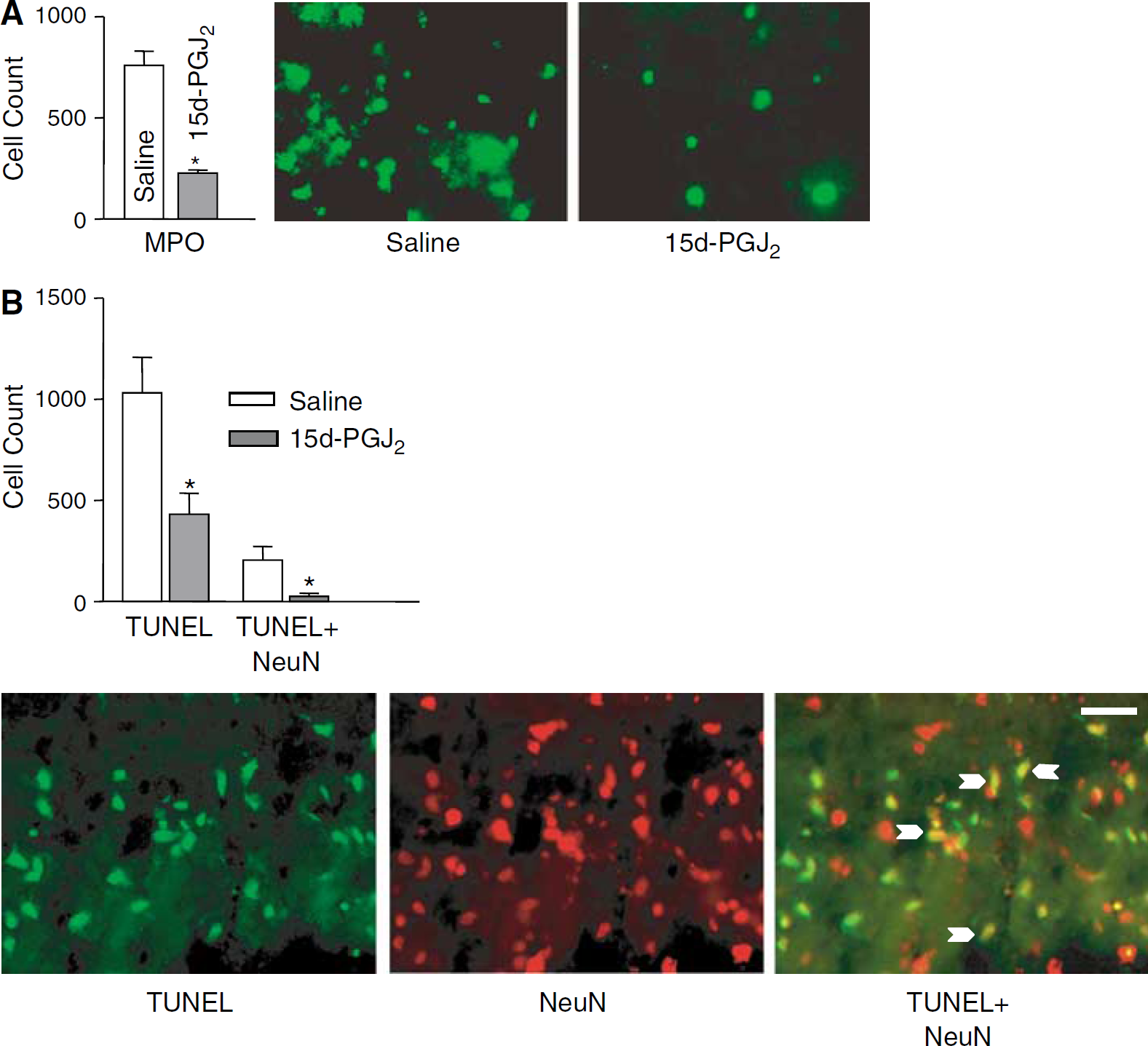
(
15-Deoxy-Δ12,14-Prostaglandin J2 Reduces Number of Terminal Deoxynucleotide Transferase dUTP Nick-End Labeling-Positive Cells after Intracerebral Hemorrhage
We observed a robust increase in the number of cells with fragmented DNA (TUNEL-positive cells) in the parenchyma at the border zone of the hematoma. We also showed a substantial (58.2%) reduction of TUNEL-positive cells in the affected striatum of rats treated with 15d-PGJ2, as compared with saline control (Figure 4B).
However, since dying cells could encompass any of the brain cell types, including infiltrating neutrophils, we determined the number of TUNEL-positive neurons by counting cells that were positive for both NeuN and TUNEL. In saline-treated rats, 19.9% of the TUNEL-positive cells were neurons. The number of TUNEL-positive neurons was significantly reduced in rats treated with 15d-PGJ2, and amounted to only 6.0% of the total TUNEL-positive cells (Figure 4B).
Discussion
In this report, we have shown that intrastriatal injection of 15d-PGJ2 at the onset of ICH coincided with the activation of PPARγ and elevation of catalase expression, suppression of NF-κB activity, and restricted neutrophil infiltration. All these events predicted reduced behavior deficits and neuronal damage.
Peroxisome proliferator-activated receptor-γ has recently emerged as an important physiologic and pathologic regulator of the cerebrovascular unit. Peroxisome proliferator-activated receptor-γ agonists from the thiazolidinedione class of drugs reduce damage produced by focal ischemia (Aronowski, 2002; Shimazu et al, 2005; Sundararajan et al, 2005). It has been postulated that the beneficial effect of PPARy agonists in stroke is because of their anti-inflammatory effect (Sundararajan and Landreth, 2004).
Unlike synthetic thiazolidinediones, 15d-PGJ2 act as an endogenous ligand for PPARγ (Kliewer et al, 1995; Negishi and Katoh, 2002; Ricote et al, 1998; Rohn et al, 2001; Straus and Glass, 2001). 15-Deoxy-Δ12,14-prostaglandin J2 is a downstream product of prostaglandin D2, a prostaglandin uniquely enriched in brain along with its synthesizing enzyme (prostaglandin-D synthase), suggesting a high likelihood for 15d-PGJ2 to play a physiologic and pathologic role in brain function (Mase et al, 1999).
The catalase gene contains the functional PPRE within its promoter region (Girnun et al, 2002). Since, 15d-PGJ2 activates PPARγ in hemorrhagic brain, it was important to determine whether 15d-PGJ2 can stimulate catalase expression. In agreement with the role of 15d-PGJ2 in activating PPARγ, and PPARγ playing role in catalase gene expression, animals treated with 15d-PGJ2 showed a significant increase in catalase mRNA and protein in the affected striatum. It is important to note that the 15d-PGJ2-triggered activation of PPARγ occurred at a time frame (1 to 3 h) corresponding to increased catalase expression, arguing for a functional role of PPARγ in transactivation of the catalase gene. The implication of these findings is that increased expression of catalase could contribute to the neuroprotective effect of 15d-PGJ2. The inducibility of the catalase gene could play an important role in combating the oxidative stress induced by ICH, as the constitutive level of catalase in the brain is low (Ho et al, 2004) and may not be sufficient to neutralize oxidative stress (Aronowski and Hall, 2005; Nakamura et al, 2005; Wagner et al, 2002). In agreement with this notion, virus-mediated overexpression of the catalase gene in neurons (hippocampal or cortical primary neuronal cultures) increased neuronal resistance to damage produced by excitotoxin or oxygen-glucose deprivation (Wang et al, 2003a). In support of this in vitro study, overexpression of catalase in rat striatum (achieved through virus-mediated gene transfer) decreased the vulnerability of catalase-overexpressing cells to damage produced by middle cerebral artery occlusion (Gu et al, 2004). In light of these studies, it is likely that increased catalase expression in response to 15d-PGJ2, similar to a virus-mediated catalase gene transfer, could be cytoprotective.
The release of cytokines and chemokines, adhesion molecule expression, activation of microglia, transmigration of hematogenous cells (including neutrophils) across the blood-brain barrier to brain parenchyma, and activation of numerous proinflammatory enzymes have been shown in numerous studies evaluating ICH (Aronowski and Hall, 2005; Gong et al, 2000; Del Bigio et al, 1996; Mayne et al, 2001; Peeling et al, 2001; Xue and Del Bigio, 2000a, b). Although inflammation is triggered primarily to remove the blood and other debris left by the hemorrhage, the by-products of this response are cytotoxic and lead to further brain tissue damage. In agreement with this concept, pharmacologic approaches aimed at reducing inflammation through inhibiting tumor necrosis factor alpha (TNFα), interleukin (IL)-1β, or preventing microglia activation have shown efficacy in reducing damage produced by ICH (Aronowski and Hall, 2005; Masada et al, 2003; Mayne et al, 2001; Power et al, 2003; Wang et al, 2003b). Most of the proinflammatory responses are amplified and regulated by the transcription factor, NF-κB (Barnes and Karin, 1997). Peroxisome proliferator-activated receptor-γ agonists (including 15d-PGJ2) have been shown to inhibit NF-κB, either directly by interacting with its subunits, competing for common transcription coactivators or through upregulation of its inhibitory protein, inhibitor κBα (Cernuda-Morollon et al, 2002; Delerive et al, 2000; Dowell et al, 1997; Gelman et al, 1999; Heneka et al, 2003; Li et al, 2000; Nolte et al, 1998). 15-Deoxy-Δ12,14-prostaglandin J2 also inhibits NF-κB through inactivating the inhibitor kappaB kinase (IKK), the enzyme responsible for liberation of NF-κB (Rossi et al, 2000). It is therefore likely that the inhibition of NF-κB by 15d-PGJ2, as shown in the present study, may provide an explanation for the anti-inflammatory effect of 15d-PGJ2. It is also likely that this anti-inflammatory effect could be achieved, at least in part, through ameliorating activation of microglia. Microglia contribute to ICH-related damage, as the microglia inhibition by tetracycline derivatives (Power et al, 2003) or microglia inhibitory factor (Wang et al, 2003b) reduced brain damage after ICH. Since, 15d-PGJ2 was shown to reduce microglia activation (Petrova et al, 1999; Storer et al, 2005), it is likely that this aspect of 15d-PGJ2 activity may also play an important role in protecting brain from damage produced by ICH. Finally, we hypothesize that such anti-inflammatory effect of 15d-PGJ2 could play an important role in inhibiting neutrophil infiltration, microglia activation, and consequently contribute to improved neurologic outcome after ICH.
In summary, we have tested the effect of 15d-PGJ2 on outcome from ICH. We showed that 15d-PGJ2 can ameliorate behavioral dysfunction and neuronal damage produced by ICH, and that the underlying mechanism of 15d-PGJ2 protection may involve activation of PPARγ, expression of antioxidant catalase, reduction of NF-κB, and reduction of inflammation. This study was designed as a proof of concept. Therefore, the administration protocol for 15d-PGJ2 called for the intraparenchymal administration of the drug to avoid the peripheral effect of the prostaglandin. Although less clinically relevant, intrahematoma injection of neuroprotective substances could be considered during surgery for blood clot evacuation. We believe that our study provides strong justification for additional work characterizing 15d-PGJ2 such as optimization of concentration (picomolar to micromolar range of doses ought to be evaluated), time window of opportunity for effective treatment, and route of administration and bioavailability.