
Abstract
Immunophilin ligands, such as cyclosporin A and FK506, have neuroprotective effects in experimental stroke models, although the precise mechanism is unclear. Cyclophilin C-associated protein (CyCAP) is a natural cellular ligand for the immunophilin, cyclophilin C, and has a protective effect against endotoxins by downmodulating the proinflammatory response. Expressions of CyCAP and cyclophilin C mRNA in a rat middle cerebral artery (MCA) occlusion ischemia model were investigated by Northern blotting and in situ hybridization. Both CyCAP and cyclophilin C mRNAs were ubiquitously distributed in the neurons of the normal brain. Expression increased in neurons of the periinfarct zone up to 7 days after MCA occlusion. The neuronal distribution was confirmed by counterimmunostaining of NeuN. Both mRNAs were predominantly expressed in microglia of the ischemic core at 7 days, confirmed by immunostaining with the microglial marker, ED1. The quantification of CyCAP and cyclophilin C mRNAs at 7 days by Northern blot analysis showed the 8.5-fold increase (P<0.005, n=6) and 6.8-fold increase (P<0.005, n=6), respectively, in ischemic core compared with control. The coincidence of CyCAP and cyclophilin C expression in neurons and microglia suggests distinct roles in each cellular population. In particular, the early increase in penumbral neurons might be related to protection in periinfarct neurons.
Introduction
Cerebral ischemia is a powerful stimulus for the de novo expression and upregulation of numerous gene systems (Barone and Feuerstein, 1999; Koistinaho and Hokfelt, 1997). Such changes in gene regulation may provide important insights into the mechanism of cellular damage and potential recovery. In particular, cells in the penumbra display a complex response to ischemia, including selectively increased mRNA levels of genes associated with stress, apoptosis, transcription, and inflammation. More than 8000 genes associated with transient global ischemia in rats were analyzed by the DNA microarray technique (Lockhart and Winzeler, 2000) to show that expression levels of 246 transcripts were increased and 213 were decreased after ischemia (Kawahara et al, 2004).
Ischemia in the central nervous system (CNS) is followed by a robust and prolonged inflammatory response (Barone and Feuerstein, 1999; Barone and Parsons, 2000) that is a major determinant of the destiny of the cells in the periinfarct zone (Emsley and Tyrrell, 2002; Kreutzberg, 1996; Streit et al, 1999). Inflammation is mediated by both molecular components, including cytokines, and cellular components, mainly microglia, many of which have pro- and/or antiinflammatory properties. The immunosuppressant drugs cyclosporin A, and FK506 have neuroprotective effects in animal models of stroke (McCarter et al, 2001; Sharkey and Butcher, 1994; Shiga et al, 1992) by binding with immunophilins, protein receptors for cyclosporin A and FK506. The cellular role of immunophilins remains somewhat unclear, but immunophilins including cyclophilins and FK506-binding proteins, which bind to cyclosporin A, and FK506, respectively, are likely to be important mediators of neuroprotection (Guo et al, 2001).
Cyclophilin C is a cyclophilin that was cloned and characterized as a new binding protein for cyclosporin A (Friedman and Weissman, 1991). Cyclophilin C-associated protein (CyCAP) was identified on the basis of its ability to bind cyclophilin C (Friedman et al, 1993; Friedman and Weissman, 1991). CyCAP was also identified independently as a cell surface-associated antigen on mouse macrophages and was named MAMA (Chicheportiche and Vassalli, 1994). Cyclophilin C-associated protein is a member of the scavenger receptor cysteine-rich domain superfamily including a variety of cell-surface molecules and secreted glycoproteins. These proteins are expressed by B cells, T cells, and macrophages, which are implicated in host defense and immune regulation. Cyclophilin C-associated protein is regarded as a candidate for the natural cellular ligand for cyclophilin C because the interaction is inhibited by cyclosporin A (Friedman and Weissman, 1991). Moreover, CyCAP has a protective effect in the response to endotoxins by downmodulating the proinflammatory response in vivo (Trahey and Weissman, 1999). Therefore, CyCAP is regarded as an endogenous immunophilin ligand with a potentially potent neuroprotective antiinflammatory effect during cerebral infarction like immunosuppressant drug cyclosporin A and FK506. However, little is known about how CyCAP and cyclophilin C respond to ischemia. The sites of CyCAP expression in brain have also not yet been precisely determined.
In the present study, CyCAP mRNA expression was studied with in situ hybridization and Northern blotting in the rat ischemia model to quantify and map spatial distribution at 2 hours to 7 days after middle cerebral artery (MCA) occlusion, classify cell types expressing CyCAP, and identify CyCAP and cyclophilin C colocalization in individual cells. These observations should provide important information for understanding the mechanism of action and the effect in brain protection of immunophilins, which show particular promise as a novel class of neuroprotective agents for use in stroke treatment.
Materials and methods
Induction of Focal Cerebral Ischemia
The research project was approved by the Guidelines for the Care and Use of Laboratory Animals of Gunma University Graduate School of Medicine. Male Sprague–Dawley rats (300 to 330 g) were anesthetized with halothane in nitrous oxide–oxygen (70:30), then intubated and artificially ventilated. A femoral artery was cannulated for recording arterial pressure and blood gases. Rats were maintained normotensive (MABP∼80 mm Hg), normocapnic (PaCO2<44 mm Hg), adequately oxygenated (PaO2>100 mm Hg), and normothermic during anesthesia.
Focal cerebral ischemia was induced using a modification of the permanent MCA occlusion method (Tamura et al, 1981). Briefly, a 2-cm skin incision was made, then the left temporal muscle was incised and stripped subperiosteally from the lateral and ventral aspects of the temporal bone to enter the infratemporal fossa from the foramen opticum rostrally to the foramen ovale caudally. A small subtemporal craniectomy was made, centered 3 mm rostral to the foramen ovale, and a linear incision made in the dura with a 25-gauge needle. Cerebral ischemia was then induced by electrocoagulation of the MCA from a point proximal to the origin of the lenticulostriate artery to the point crossing the inferior cerebral vein. The MCA was then transected at the olfactory tract to ensure completeness of occlusion. The time of transection was taken as the exact time of MCA occlusion. The craniectomy wound was then sutured and the animal was allowed to recover from the anesthesia. A subcutaneous injection of 1 ml of physiologic saline was administered to prevent postanesthetic dehydration.
Tissue Processing and Histological Identification of Ischemic Damage
Rats were decapitated at 2 hours, 24 hours, 3 days, and 7 days after ischemia. The brains were rapidly removed, embedded in Tissue-Tek® OCT compound (Sakura, Torrance, CA, USA), frozen, and stored at −80°C until use. Sections of 20 μm thickness were cut on a cryostat, thawed, and mounted onto silan-coated slides, and dried immediately with a hair dryer. These sections were stained with hematoxylin and eosin, and examined by light microscopy.
Cyclophilin C-Associated Protein and Cyclophilin C cDNAs
Normal adult rats and ischemic rats were decapitated under deep anesthesia. The brains were quickly removed and frozen in liquid nitrogen. Total RNA was extracted from these samples using Isogen reagent (NipponGene Inc., Toyama, Japan), following the manufacturer's protocols. The total RNA was subjected to reverse transcription using a Ready-To-Go T-Primed First-Strand Kit (Amersham Biosciences, Buckinghamshire, UK) according to the manufacturer's protocols. For CyCAP, we used the sense primer: 5′-TTC AAC AGC TTC CGC GTG GTC A-3′ and antisense primer: 5′-AGA CAG TGG AAG TTA AGG GA-3′ (GenBank accession number C07012, homolog to rat peptidylprolyl isomerase that is 85.6% homologous to mouse CyCAP (Friedman et al, 1993), 39 to 218, 180 bp), and for cyclophilin C, sense primer: 5′-CGG TGG AAA ACT TCG TGG CTC T-3′ and antisense primer: 5′-CCT CAA CCA CAA GGG TGT T-3′ (GenBank accession number NM008908 (Friedman and Weissman, 1991), 248 to 676, 428 bp). The reverse transcription solution (1 μL) was used for polymerase chain reaction (PCR). Polymerase chain reaction was performed in a final volume of 100 μL containing 250 μmol/L each of dATP, dCTP, dGTP, and dTTP, 1 μmol/L of each primer, 1.25 U AmpliTaq Gold polymerase (Applied Biosystems, Foster City, CA, USA), 1 × PCR Gold Buffer, and 3 mmol/L MgCl2. The mixture was incubated in a thermal controller (GeneAmp PCR System 9700; Applied Biosystems) for 45 cycles using the following profiles: CyCAP, (1) 95°C for 9 mins, (2) repeat cycles at 94°C for 30 secs, 58°C for 30 secs, and 72°C for 30 secs, and (3) 72°C for 7 mins; cyclophilin C, (1) 94°C for 9 mins, (2) repeat cycles at 94°C for 30 secs and 64°C for 1 min, and (3) 64°C for 10 mins. The PCR product was gel-purified and subcloned into the SrfI site of pPCR-script Amp SK(+) plasmid (Stratagene, La Jolla, CA, USA). We confirmed the insert to correspond with the correct position of each cDNA by sequence determination. These products were used as probes to detect CyCAP and cyclophilin C mRNA in Northern blot analysis and in situ hybridization.
Northern Blot Analysis and Quantification of mRNAs
The cRNA probes for CyCAP and cyclophilin C were synthesized with [α-32P]UTP (Perkin-Elmer Life Science Inc., Boston, MA, USA) and T3 RNA polymerase.
The brains were rapidly isolated and immediately immersed in ice-cold saline. The brains were then cut into six coronal sections using a brain slicer (Muromachi Kikai Co. Ltd., Tokyo, Japan). Coronal slices (2-mm thick) were cut at the level of the septal nuclei and globus pallidus (anterior coordinates of 8.7 to 6.7 mm (Paxinous and Watson, 1986)). Adjacent slices were treated with 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma, Saint Louis, MO, USA) solution for 20 mins to confirm the areas of ischemic damage and intact brain. Based on the results of TTC staining, the anatomic regions such as the ipsilateral remote cortex, ischemic core cortex, and contralateral cortex were dissected to sample the tissue for Northern blotting. Control rat brains were dissected in the same way without TTC staining. Total RNA was isolated from these dissected regions by the method of Chomczynski and Sacchi (1987).
RNA (20 μg per lane) was electrophoresed on a 1.4% agarose gel containing 0.66 mol/L formaldehyde and transferred overnight in 20 × SSC (1 × SSC=150 mmol/L sodium chloride and 15 mmol/L trisodium citrate) to a nylon membrane (Biodyne; Pall BioSupport Corp., East Hills, NY, USA). RNA was crosslinked to the nylon membrane with a UV crosslinker (Stratagene). The membranes were prehybridized with the hybridization buffer (50% formamide, 0.2% SDS, 5% dextran sulfate, 50 mmol/L HEPES, 5 × SSC, 5 × Denhart's solution, and 100 μg/mL denatured salmon sperm DNA) at 68°C for 2 hours. Subsequently, the membrane was hybridized at 68°C overnight with the hybridization buffer containing a CyCAP cRNA probe. The membrane was washed twice in 2 × SSC–0.1% SDS at 25°C for 15 mins and twice in 0.1 × SSC–0.1% SDS at 68°C for 1 hour. Autoradiography was performed by sandwiching the filter for 6 hours with an X-ray film (XAR-2; Eastman Kodak Co., Rochester, NY, USA) at −70°C. After the detection of CyCAP mRNA, the probe was stripped off, and the blots were rehybridized with cyclophilin C mRNA probe. Hybridization and washing were performed as described above, and autoradiography of the membrane performed for 52 hours. mRNA levels were quantified by densitometry using NIH Image version 1.63. The optical density of the CyCAP (2.4 kb) and cyclophilin C (1.4 kb) bands was corrected for 28S density for comparison of the same blot.
In Situ Hybridization
Digoxigenin (DIG)-labeled antisense cRNA for CyCAP and cyclophilin C was synthesized by the T3 RNA polymerase (Stratagene) reaction with EcoRI-digested plasmid as template, using a DIG RNA labeling kit (Roche Diagnostics, Tokyo, Japan) according to the supplier's instructions. Sense cRNA was synthesized in a similar manner using T7 RNA polymerase and NotI-digested plasmid. In situ hybridization was performed as previously described (Imai et al, 1997) with modification. Sections were digested with 1 μg/mL proteinase K for 15 mins at 37°C, then immersed in 0.1 mol/L triethanolamine containing 0.25% acetic anhydride at room temperature for 10 mins, and subsequently dehydrated in 70%, 95%, and 100% ethanol. Hybridization was performed in a solution containing 50% formamide, 10% dextran sulfate, 600 mmol/L NaCl, 1 × Denhart's solution, 10 mmol/L Tris–HCl (pH 8.0), 200 μg/mL tRNA, 1 mmol/L EDTA, 0.2 mg/mL sodium heparin, 0.2% SDS, and 1 μg/mL of DIG-labeled RNA probe at 55°C for 16 hours. After rinsing in 5 × SSC and washing in 2 × SSC containing 50% formamide, the slides were incubated in 50 μg/mL RNase A at 37°C for 30 mins to exclude the nonspecific probe reactions. Sequential washing was performed twice in 2 × SSC at 55°C for 15 mins, and twice in 1 × and 0.1 × SSC at room temperature for 10 mins each. After washing, sections were blocked with 10% sheep serum, and then incubated overnight at 4°C with 1:1000 dilution of alkaline phosphatase-conjugated anti-DIG antibody (Roche). Specimens were treated with 4-nitroblue-tetrazolium chloride and 5-bromo-4-chloro-3-indolyl-phosphate (Roche) to develop color for an appropriate time.
Combined In Situ Hybridization/Immunohistochemistry by Double- Immunofluorescence Labeling
For double-labeling with specific marker proteins for each cell type, hybridized and washed slides were incubated with fluorescein-conjugated anti-DIG antibody (Roche; 1:10 dilution) after blocking. After overnight incubation at 4°C, cell type-specific monoclonal antibody against NeuN (Chemicon International, Temecula, CA, USA; 1:100 dilution) for neurons, GFAP (Sigma; 1:100 dilution) for astrocytes, and ED1 (Serotec Ltd., Oxford, UK; 1:100 dilution) for microglia/macrophages was applied overnight at 4°C. Tests of the specificity of this ED1 antibody showed this antibody recognizes the membrane of cytoplasmic granules, like phagolysosomes in tissue macrophages (Dijkstra et al, 1985; Damoiseaux et al, 1994). Although ED1 is not detected in resting microglia, the activated microglia in the border zone of ischemic core are strongly stained by this antibody (Ito et al, 2001). These monoclonal antibodies were visualized using TexasRed-conjugated anti-mouse IgG horse antibody (Vector Laboratories, Burlingame, CA, USA; 1:100 dilution). Images were analyzed with a fluorescent microscope with a CCD camera (BH2-RFL-T fluorescence system; Olympus, Tokyo, Japan), and figures were generated using Adobe Photoshop® software (Adobe Systems, San Jose, CA, USA).
Statistical Analysis
One-way ANOVA followed by post hoc Fisher-protected t tests using StatView 5.0 (SAS Institute Inc., Cary, NC, USA) were used to compare the temporal expression of mRNAs. A value of P<0.05 was considered significant.
Results
Histopathology
Ischemic damage was identified consistently in the ipsilateral MCA territory including the frontal cortex, dorsal parietal cortex, and the lateral part of the caudate-putamen. The anatomic areas investigated are depicted in Figure 1. The infarct area was divided into three types of lesions according to Garcia et al (1995), with modification: (1) the area of irreversible ischemic damage where neuronal perikarya showed the morphologic characteristic features of ischemic damage (Brierly and Graham, 1984), that is, microvacuolation, shrinkage, triangulation of the nucleus and cytoplasm, and increased eosinophilia of cytoplasm (dark gray area in Figure 1); (2) the periinfarct area where only a limited number of neurons showed such morphologic signs (light gray area in Figure 1); and (3) the necrotic area where all tissue components had undergone coagulation necrosis, and edema and resorption were also observed (black area in Figure 1). Marked expansion of the necrotic area was observed from 2 to 24 hours after ischemic insult. At 2 hours after ischemia, there was only a small necrotic area in the ventral cortex and a relatively extensive periinfarct area in the adjacent ipsilateral cortex. Most of the periinfarct area had undergone irreversible damage at 24 hours after ischemia, and showed necrotic features at 3 days. Microglia/macrophages and some neutrophils had migrated to the necrotic area to form clusters of cells in the subacute phase (3 to 7 days after ischemia).
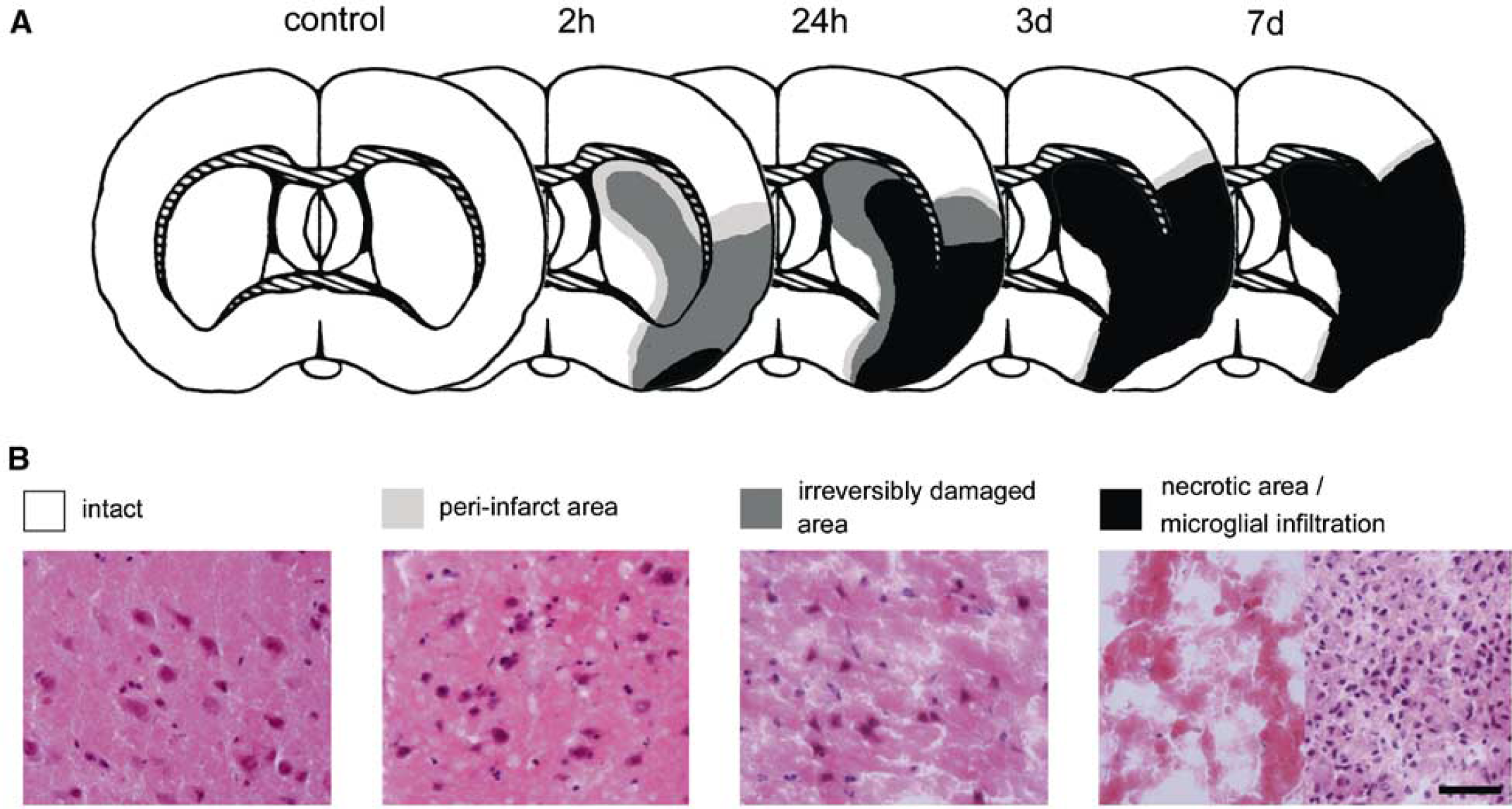
Progression of the ischemic lesion. (
Northern Blot Analysis
Northern blot analysis revealed CyCAP mRNA as a single 2.4-kb band (Figure 2A). Cyclophilin C-associated protein mRNA was detected in both the normal cortex and ipsilateral cortex with focal cerebral ischemia. Quantitative analysis revealed that the difference in induction of the mRNAs depends on the ischemic damage and time course after ischemia. Cyclophilin C-associated protein mRNAs started to increase in the periinfarct zone at 2 hours after MCA occlusion and gradually increased by 24 hours (Figure 2B). Cyclophilin C-associated protein mRNAs showed a 4.1-fold increase compared with the control (P<0.005) at 7 days after MCA occlusion. The pattern of increase of CyCAP mRNAs in the ischemic core seemed broadly identical to that in the periinfarct zone, but the increase rate at 7 days after ischemia was larger than in the periinfarct zone, showing a 8.5-fold increase compared with the control (P<0.005). No significant changes were noted in the contralateral side (Figure 2B).
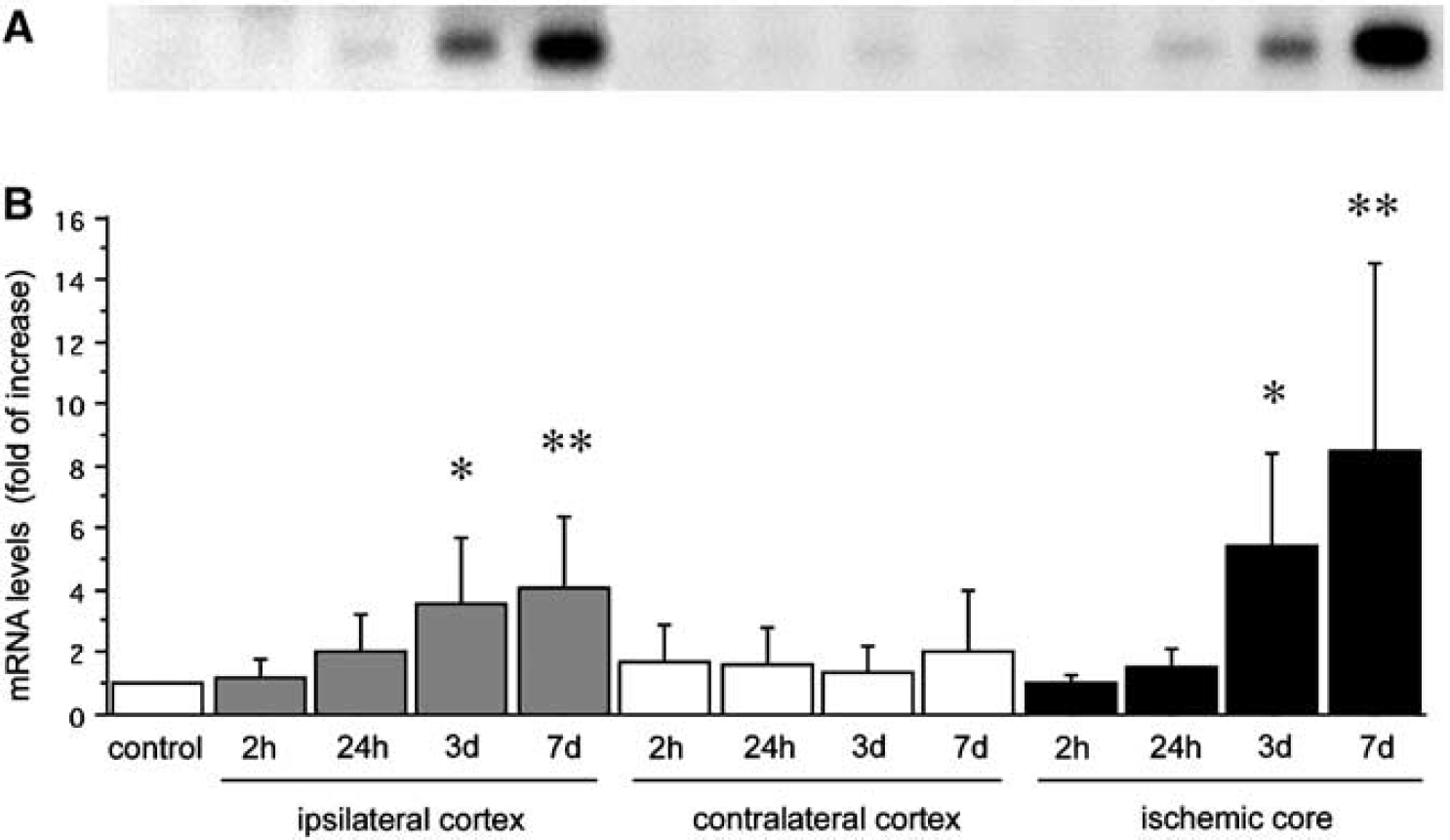
Time course of CyCAP mRNA expression after focal cerebral ischemia. (
In Situ Hybridization and Immunohistochemistry
Cyclophilin C-associated protein mRNA distribution in control rats:
Cyclophilin C-associated protein mRNA exhibited ubiquitous distribution throughout the brain, with primarily neuronal localization (Table 1, Figure 3). Strong CyCAP expression was found in regions where large neurons are abundant, such as the pyramidal cell layer of the hippocampus (Figures 3A to 3D), mitral cell layer of the olfactory bulb (Figures 3F and 3G), thalamus (Figure 3H), several nuclei of the brain stem and hypothalamus (Figure 3I), Purkinje cells of the cerebellum (Figures 3J and 3K), and neocortex (Figure 4, control). White matter structures, such as the corpus callosum and internal capsule, exhibited little expression of CyCAP (Figure 3A). Sense probe for CyCAP used as the control produced essentially no hybridization signals (Figure 3E).
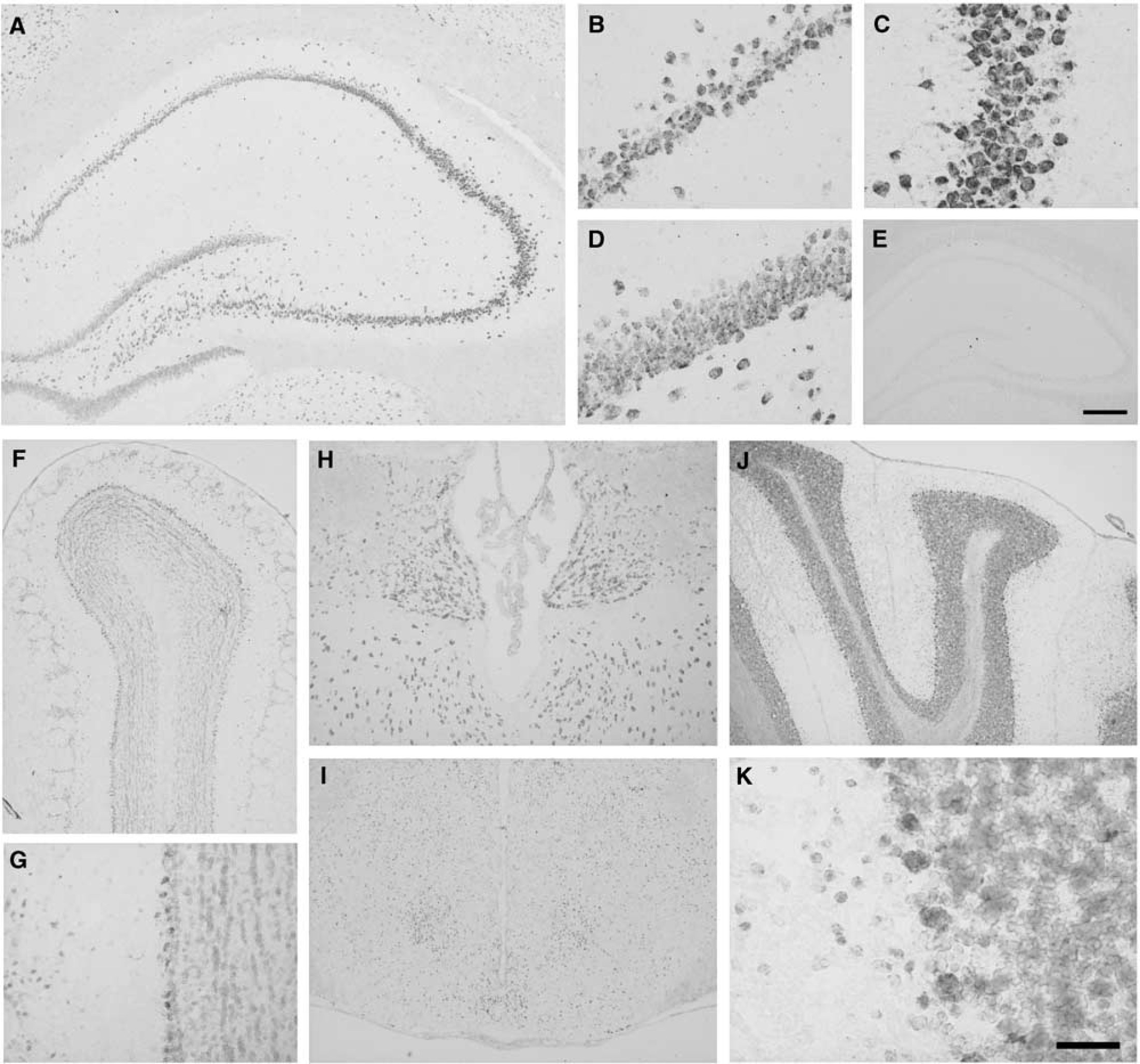
Cyclophilin C-associated protein mRNA expression in a control rat. (

In situ hybridization of CyCAP mRNA expression in periinfarct cortex in rats 2 hours, 24 hours, 3 days, and 7 days after focal cerebral ischemia and control rats. The inset (upper left) indicates regions encompassing the periinfarct MCA cortex (box). Other insets indicate magnified regions of layer V of the cortex. Ischemia resulted in marked increase of CyCAP mRNA level in all cell layers of the cortex. Note the accumulation of highly CyCAP-expressed cells in the transitional zone of the periinfarct area and necrotic area at day 7 (arrowheads). Sense probe detected no signal at 2 hours after MCA occlusion. Bar=60 μm for insets, and 300 μm for original photomicrographs.
Regional expression of CyCAP and Cyclophilin C in normal rat brain
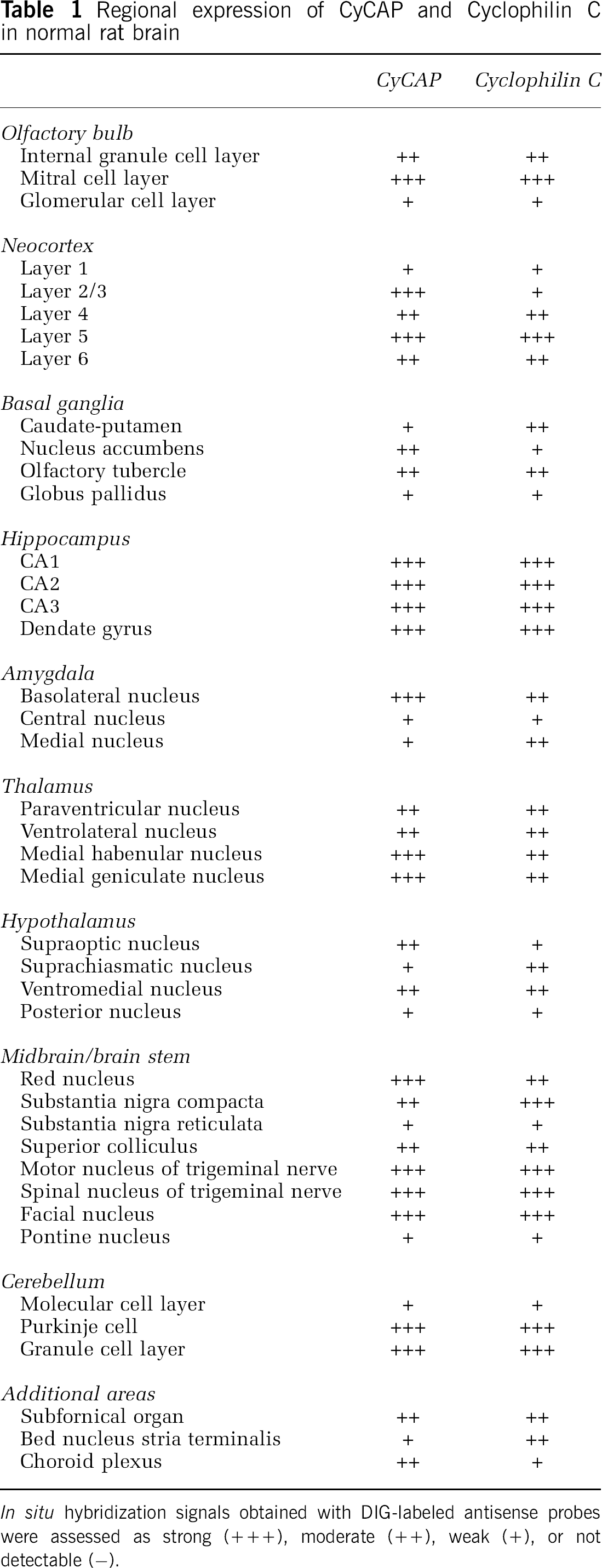
In situ hybridization signals obtained with DIG-labeled antisense probes were assessed as strong (+++), moderate (++), weak (+), or not detectable (−).
Cyclophilin C-associated protein mRNA expression in focal cerebral ischemia:
After ischemia, CyCAP mRNA started to increase in the periinfarct cortex at 2 hours after MCA occlusion (Figure 4). Cyclophilin C-associated protein mRNA-expressing cells were distributed predominantly in the ipsilateral cortex, with most cells displaying neuronal morphology, particularly in neuronal layer V, but some cells displaying small and round morphology. This increased expression pattern in the periinfarct zone was maintained until 7 days after MCA occlusion, with increased staining over time. These findings are consistent with the result of Northern blotting quantification of CyCAP. However, CyCAP expression in the ischemic core was reduced markedly at 2 hours after MCA occlusion and completely lost at 24 hours. Cyclophilin C-associated protein-expressing cells started to emerge around the periinfarct zone in the subacute phase. Cyclophilin C-associated protein mRNA-expressing cells had migrated in the ischemic core to construct clusters of cells with high cellular density at 7 days after MCA occlusion. Many of these cells showed mitotic figures, suggesting active proliferation.
Characterization of Cyclophilin C-associated protein mRNA-Expressing Cell Types
Most CyCAP mRNA-expressing cells in the periinfarct cortex in the acute phase expressed NeuN and were thus identified as neurons (Figure 5). Some CyCAP mRNA-expressing cells were relatively small and likely to be microglia/macrophages. Cyclophilin C-associated protein mRNA-expressing cells in the periinfarct zone at 3 days after MCA occlusion showed colocalization of NeuN and ED1, suggesting that microglia/macrophages as well as neurons express CyCAP mRNA. Some of the spindle-shaped reactive astrocytes in the periinfarct cortex expressed CyCAP moderately at 7 days after MCA occlusion (Figures 6D to 6F). In this phase, most CyCAP mRNA-expressing cells in the ischemic core showed ED1 immunoreactivity and were identified as microglia/macrophages (Figures 6G to 6I). There was no evidence of neutrophils in the ischemic area (data not shown).
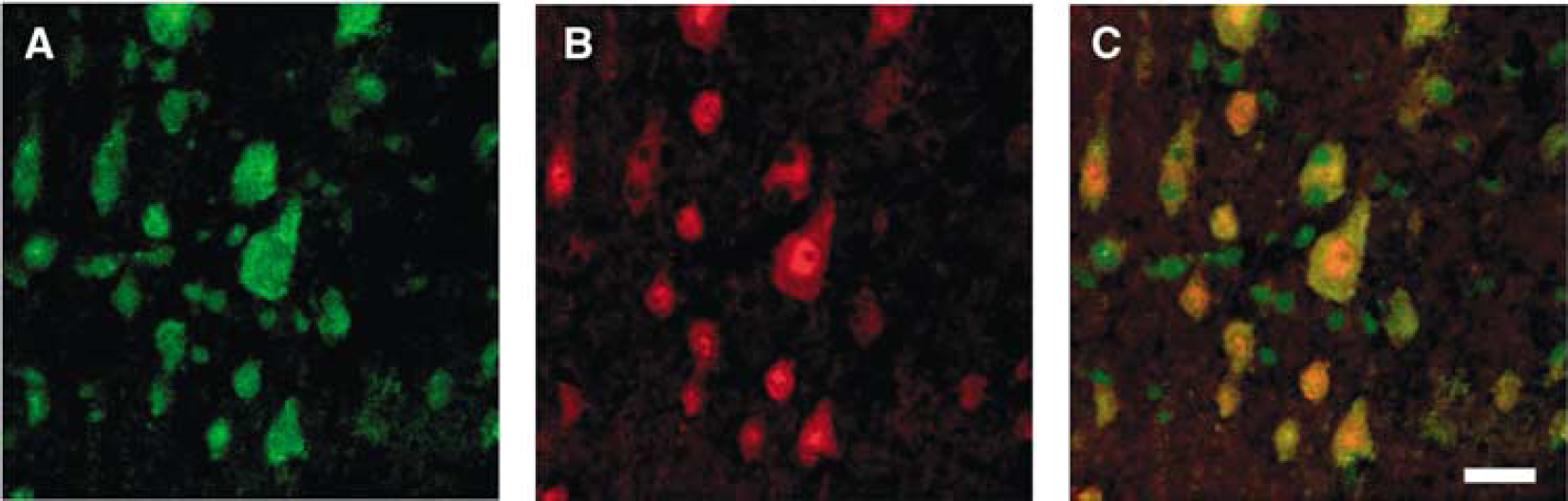
Double-labeling analysis of CyCAP mRNA and NeuN protein in the periinfarct cortex 3 days after MCA occlusion. (
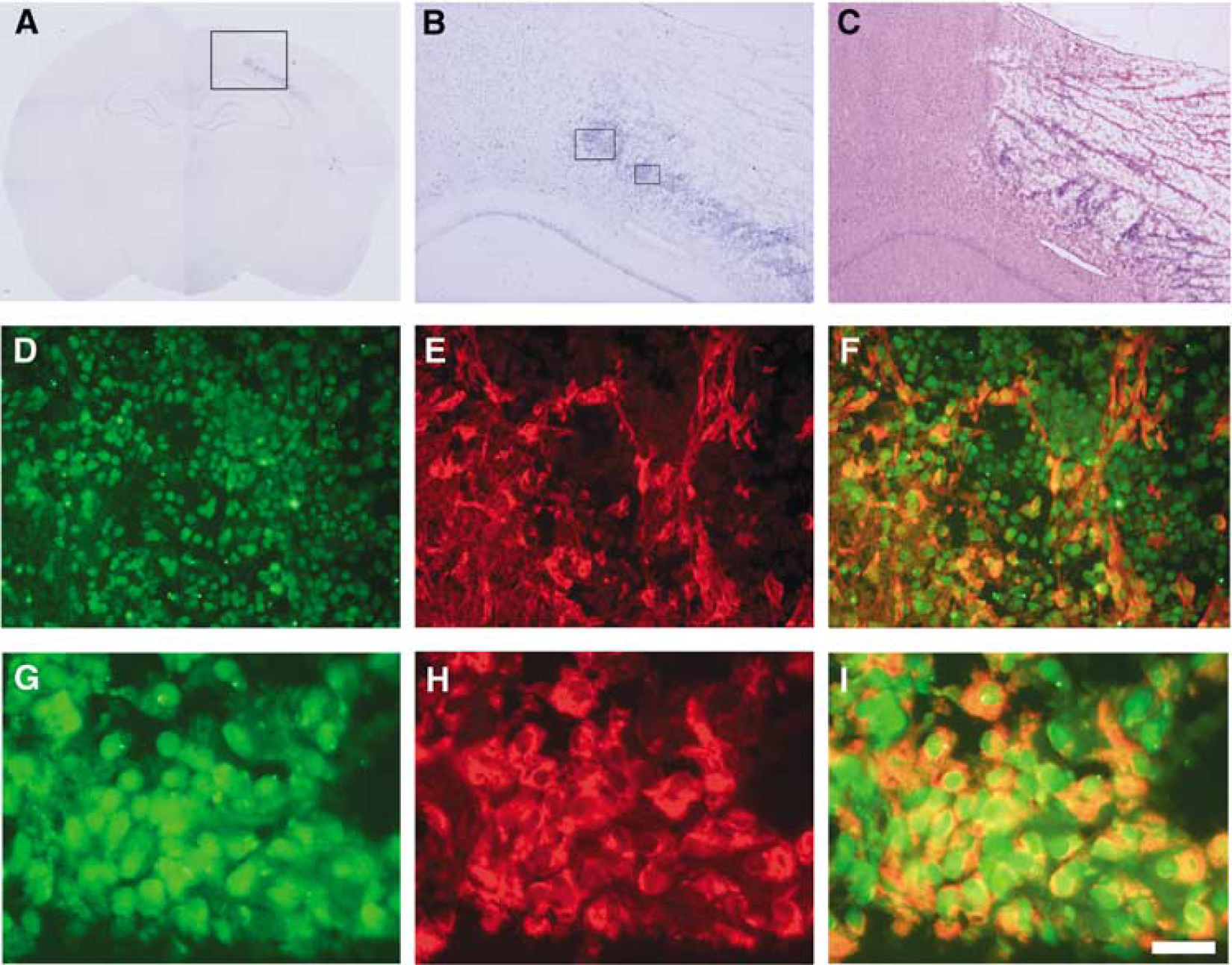
Induction of CyCAP mRNA in the periinfarct cortex 7 days after MCA occlusion. (
Expression of Cyclophilin C mRNA
Northern blot analysis revealed cyclophilin C mRNA as a single 1.4-kb band with increased expression in the ipsilateral cortex and ischemic core similar to CyCAP mRNA (Figure 7A). Quantitative analysis revealed that cyclophilin C mRNAs started to increase in the ipsilateral cortex and ischemic core at 24 hours after MCA occlusion and gradually increased. At 7 days after MCA occlusion, the increased rates of cyclophilin C mRNAs in the ipsilateral cortex and ischemic core were 5- and 6.8-fold increase, respectively (compared with control; P<0.005). No significant changes were noted in the contralateral side (Figure 7B).
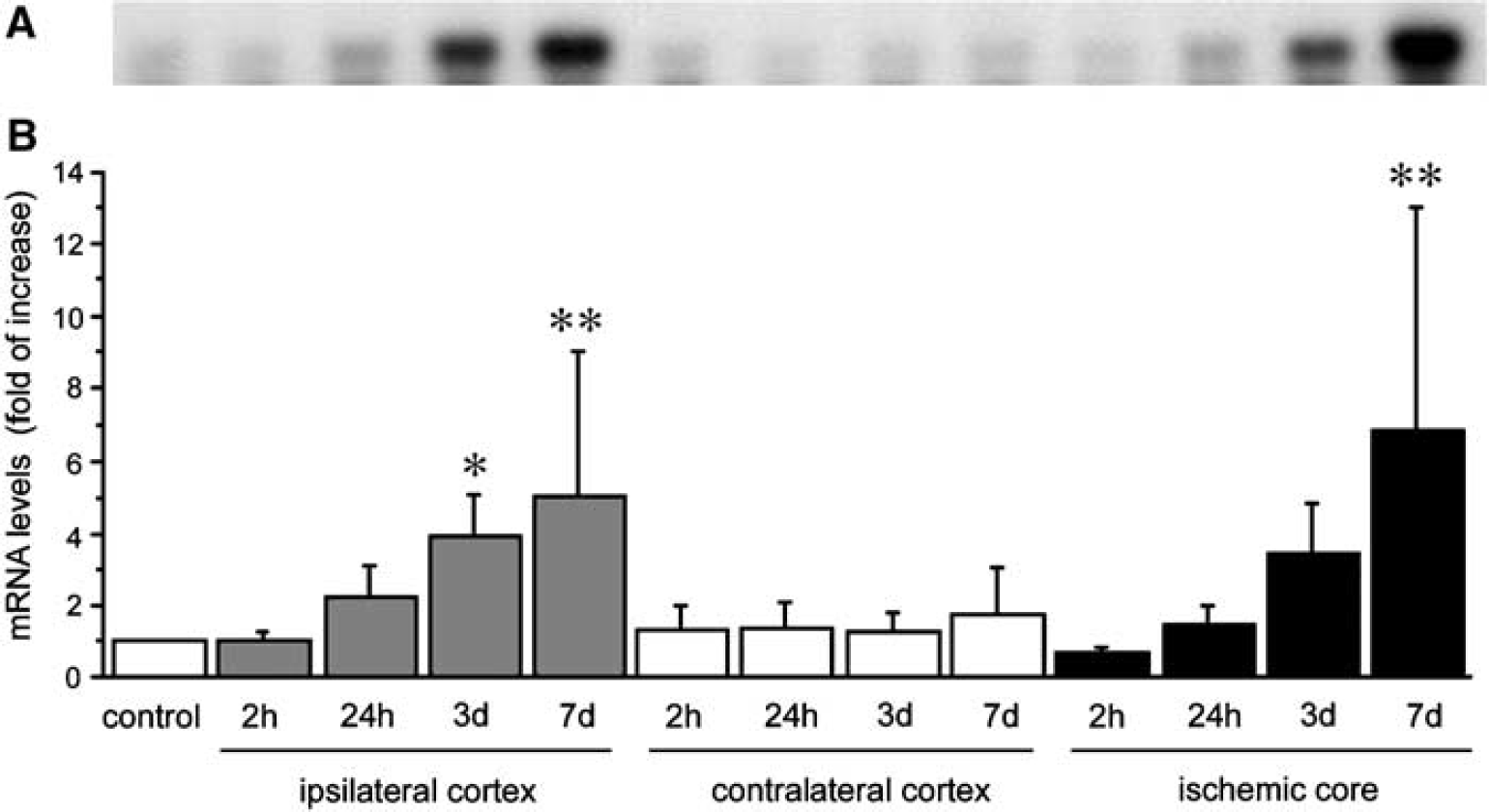
Time course of cyclophilin C mRNA expression after focal cerebral ischemia. (
In situ hybridization revealed that the expression of cyclophilin C mRNA in focal cerebral ischemia was broadly similar to that of CyCAP in terms of spatial and temporal profiles. The expression of cyclophilin C mRNA in the acute phase was predominantly in the neurons in the periinfarct area. The dominancy of expression of cyclophilin C mRNA was shifted from the neurons in the periinfarct area in the acute phase to the microglia of the ischemic core in the subacute phase (Figure 8).
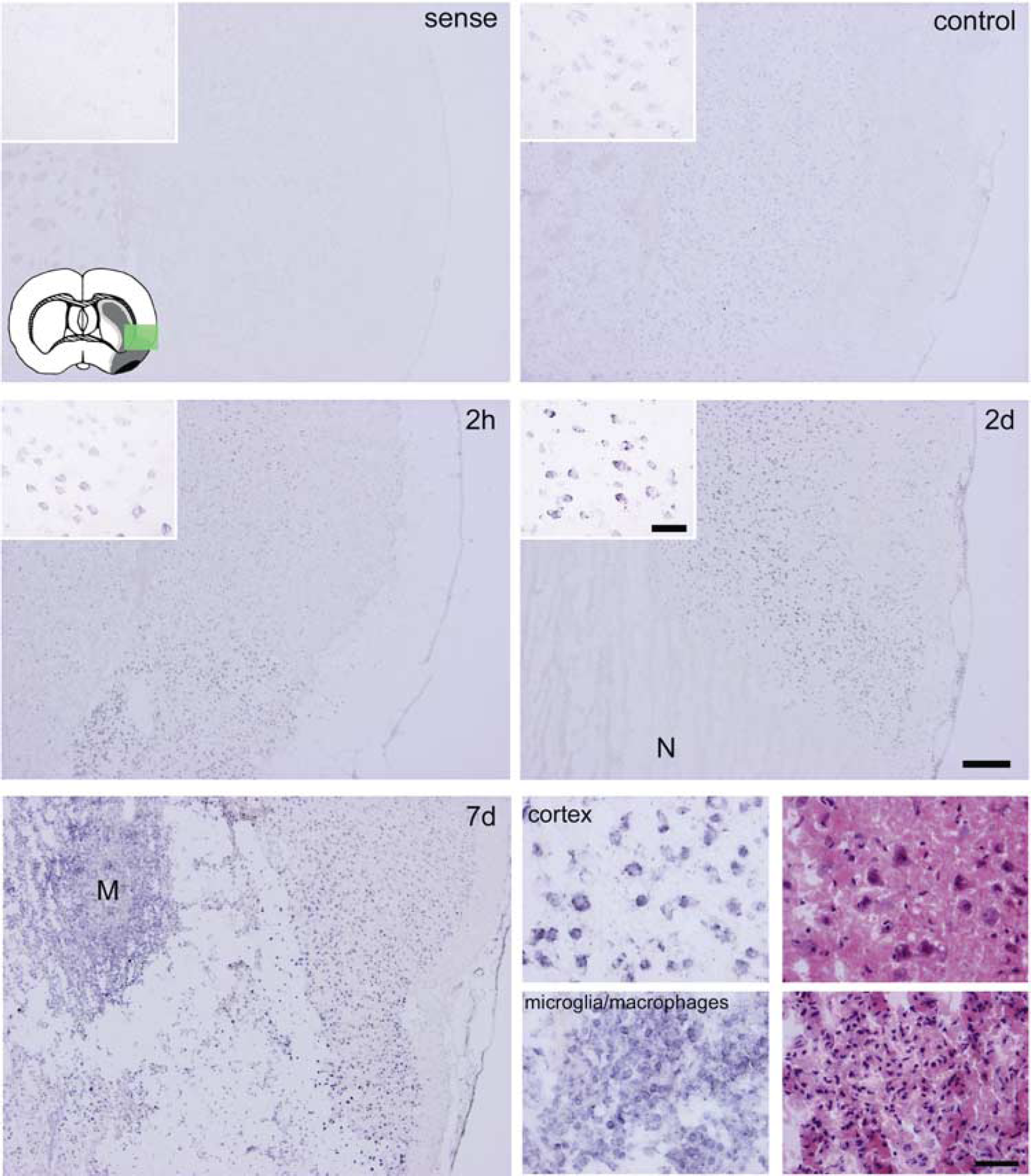
In situ hybridization of cyclophilin C showing enhanced expression in the periinfarct cortex. Inset (upper left) indicates regions encompassing the periinfarct MCA cortex (green square). Other insets indicate magnified regions of layer V of the cortex. Ischemia resulted in marked increase of CyCAP mRNA level in all cell layers of the periinfarct cortex. No signals were detected in necrotic areas (N). Cyclophilin C was strongly expressed in microglia/macrophages, which had migrated to the necrotic area to form clusters at 7 days after ischemia (M). Four photomicrographs (lower right) show magnifications of these clusters of microglia/macrophages and cortex, with hematoxylin and eosin staining of adjacent sections. Sense probe detected no signals. Bar=50 μm for insets and lower right, and 300 μm for original photomicrographs.
Schemes of the present results including anatomic areas of infarction and distribution of CyCAP mRNA expression are summarized in Figure 9.
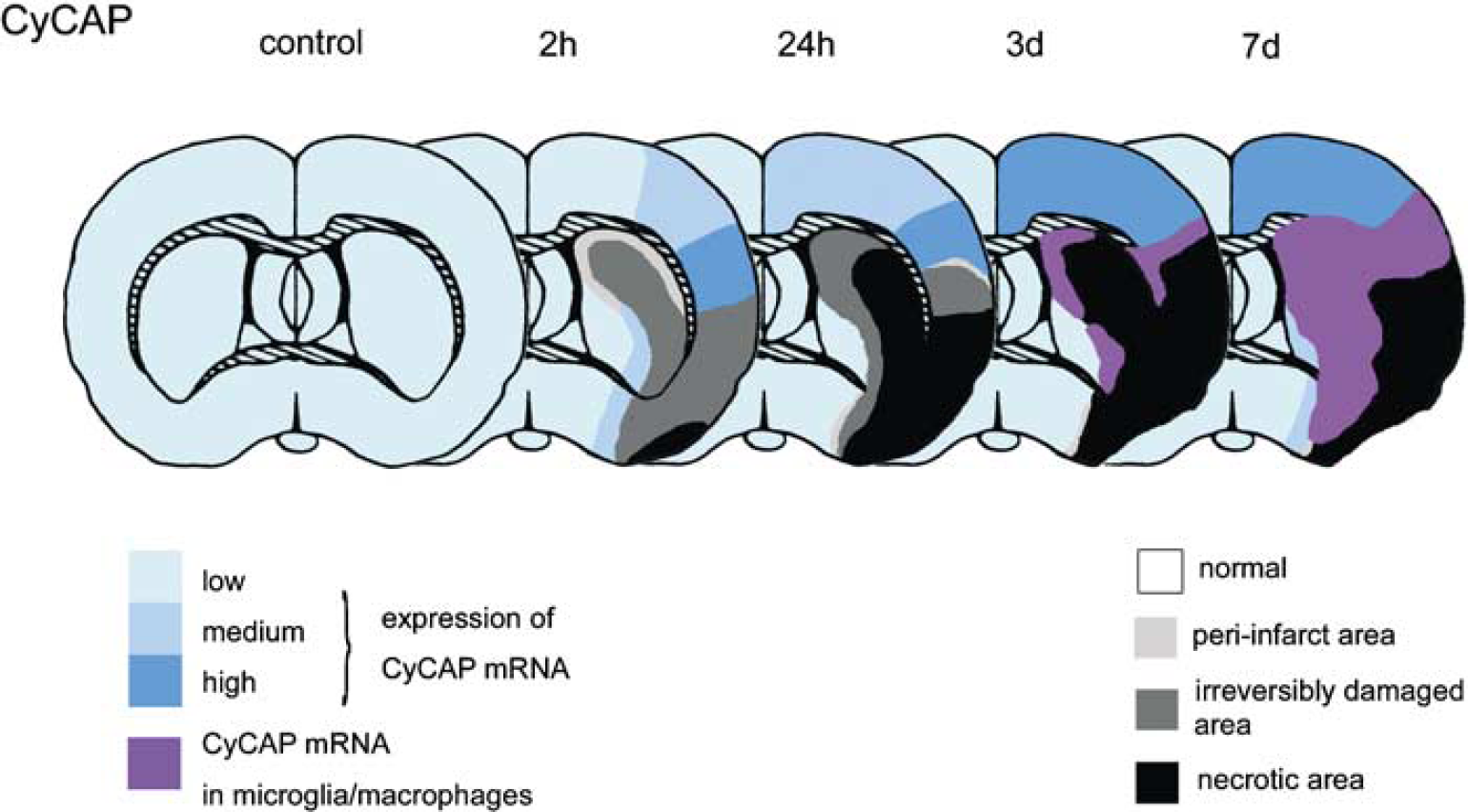
Schematic diagrams summarizing temporal and spatial expression of CyCAP. Cyclophilin C-associated protein mRNA is significantly upregulated in the periinfarct zone compared with the intact region and is enriched in the discrete neuronal population in the postischemic brain from the acute to subacute phases (blue). The distribution of mRNA expression is changed in microglia/macrophages in the core with dramatically increased expression of mRNAs (pink) rather than predominant neurons in the periinfarct area at 7 days after MCA occlusion.
Discussion
The present study examined the regulation of CyCAP and cyclophilin C mRNA expression in a well-characterized rat model of permanent focal cerebral ischemia by in situ hybridization and Northern blotting. The major observations were that both CyCAP and cyclophilin C mRNA were significantly upregulated in neurons of the periinfarct zone from the acute phase to the subacute phase, the distributions of mRNA expression were both changed in the microglia/macrophages in the ischemic core with dramatic increases in the periinfarct area at 7 days after MCA occlusion, and the expression patterns of both CyCAP and cyclophilin C mRNAs were synchronized in both spatial and temporal profiles.
Cyclophilins in the CNS show a heterogeneous distribution, mainly in the neurons (Dawson et al, 1994). Cyclophilin localizations largely correspond to those of calcineurin, which suggest related functions (Dawson et al, 1994; Friedman et al, 1993). In the present study, CyCAP transcription was distributed throughout the intact brain predominantly in neurons, although the signal intensities were somewhat variable (Table 1). Interestingly, CyCAP as well as cyclophilin C mRNAs were induced at 24 hours after MCA occlusion and increased until 7 days in the periinfarct area. This region is perfused by collateral channels from the anterior and posterior cerebral arteries and is called the penumbra.
Focal ischemia caused by MCA occlusion results in necrosis at the infarct core and activation of complex signal pathways for cell death and cell survival in the penumbra (Barone and Feuerstein, 1999; Sharp et al, 2000; Soriano et al, 2000). Calcineurin, a calcium-calmodulin-dependent protein phosphatase, is pivotal in this delicate balance. Activation or upregulation of calcineurin leads to apoptotic cell death in many cell systems since calcium-calmodulin-dependent protein phosphatase is stimulated by calcium (Ankarcrona et al, 1996; Asai et al, 1999). In this scheme, calcium induces cell death by activating the calcium-calmodulin-dependent phosphatase calcineurin, which dephosphorylates Bad, leading to its activation and translocation to mitochondrial membranes (Wang et al, 1999). The immunosuppressants cyclosporin A and FK506 indirectly inhibit calcineurin via their effects on peptidyl-cis/trans isomerase (‘rotamase’) activity (Steiner et al, 1997a). Specific increased expression of CyCAP mRNA accompanied by upregulation of cyclophilin C mRNA in the potentially salvageable area at the boundary of the infarct, combined with the inhibition of oxidative stress generation, suggests a beneficial influence on cell survival. These findings imply that the immune system has an intrinsic capacity to provide a natural mechanism of organ protection during harmful environmental conditions.
Cyclophilin C-associated protein and cyclophilin C mRNA expression synchronously extends from the penumbra to the ischemic core from the acute phase to the subacute phase. mRNA expression was predominantly found in activated microglia/macrophages in the ischemic core in the subacute phase. Microglia/macrophages are likely to cluster in the ischemic core at 7 days after ischemia (Figure 6). The microglia/macrophage response to injury might be either beneficial by scavenging necrotic debris or detrimental by facilitating cell death in neurons that would otherwise recover (Danton and Dietrich, 2003). Microglia become activated within 2 to 24 hours after ischemia, and proliferate and migrate toward the site of damage (Kreutzberg, 1996). The primary purpose as phagocytic cells is to remove debris after irreversible damage, to promote tissue repair by secreting growth factors, and to facilitate the return to tissue homeostasis (Schwartz, 2003; Streit et al, 1999). Cyclophilin C-associated protein was identified originally as a cell surface-associated antigen, membrane glycoprotein on mouse macrophages, which might be related to macrophage adhesion and migration to the site of injury (Chicheportiche and Vassalli, 1994). This action of CyCAP might be related to beneficial macrophage activation, proliferation, and migration to the site of injury in this phase.
Microglia account for 5% to 20% of the total glial cell population (Barron, 1995), and also contribute to the inflammatory response to ischemia. Activated microglia and macrophages are the principal CNS sources of cytotoxic substances such as glutamate, nitric oxide, reactive oxygen intermediates, matrix metalloproteinases and their inhibitors, hydrolases, cathepsins, plasminogen activators, and cytokines such as interleukin (IL)-1β, tumor necrosis factor-alpha (TNF-α), and tumor growth factor-β (Gregersen et al, 2000; Lehrmann et al, 1998; Rothwell, 1999). The release of cytotoxins might be the main cause of microglia- and neutrophil-mediated damage during and after ischemia. A gene-targeted study provided interesting results with regard to the function of CyCAP, showing more sensitivity to the lethal effects of endotoxin (Trahey and Weissman, 1999). Cyclophilin C-associated protein-deficient mice overproduced proinflammatory cytokines such as IL-12, interferon-γ, and TNF-α in response to endotoxin. Furthermore, macrophages stimulated in vitro with endotoxin in serum deficient in CyCAP secreted more TNF-α. These studies suggest that CyCAP specifically downregulates the endotoxin signal and proinflammatory response in vivo.
Moreover, the immunophilin ligands, GPI-1046 (Steiner et al, 1997a, 1997b) and L685818 (Becker et al, 1993; Steiner et al, 1997a) that specifically bind to immunophilins showed that these ligands exert neurotrophic and neuroregenerative effects in neural injury models (Guo et al, 2001). Interestingly, such effectiveness was recognized with both early and late administration (7 to 28 days after injury) (Steiner et al, 1997b; Zhang et al, 2001). The present study observed a striking colocalization of CyCAP and cyclophilin C mRNAs in the activated macrophages of the ischemic core. The increased induction of these mRNAs in the subacute phase might have potent neuroregenerative effects rather than neuroprotective effects like the neurotrophic and neuroregenerative effects of the neuroimmunophilin ligands, GPI-1046 and L685818, in the chronic phase.
The observed changes in CyCAP and cyclophilin C mRNA expression in response to permanent focal cerebral ischemia strongly suggest that these molecules participate in intrinsic tissue protection by providing neuroprotection as a mimic of cyclosporin A; by recruiting microglia/macrophages to remove potentially deleterious debris, promote tissue repair by secreting growth factors, and facilitate the return to tissue homeostasis; and by suppressing proinflammatory cytokines in response to ischemia. Preservation of these effects could represent a novel pharmacological approach to counter the inflammatory reaction triggered by cerebral ischemia. In addition, overexpression of CyCAP in the penumbra suggests a role in cell survival, an intriguing possibility that deserves further investigation.
Footnotes
Acknowledgements
We thank Dr Kenjiro Konno of Gunma University Graduate School of Medicine for helpful discussion and Ms Miyuki Umemura and Ms Mitsue Maniwa for technical support.