
Abstract
[11C]PK11195 is used in positron emission tomography (PET) studies for imaging brain inflammation in vivo as it binds to the peripheral-type benzodiazepine receptor (PBR) expressed by reactive glia and macrophages. However, features of the cellular reaction required to induce a positive [11C]PK11195 signal are not well characterized. We performed [11C]PK11195 PET and autoradiography in rats after transient focal cerebral ischemia. We determined [3H]PK11195 binding and PBR expression in brain tissue and examined the lesion with several markers. [11C]PK11195 standard uptake value increased at day 4 and grew further at day 7 within the ischemic core. Accordingly, ex vivo [3H]PK11195 binding increased at day 4, and increases further at day 7. The PET signal also augmented in peripheral regions, but to a lesser extent than in the core. Binding in the region surrounding infarction was supported by [11C]PK11195 autoradiography at day 7 showing that the radioactive signal extended beyond the infarcted core. Enhanced binding was preceded by increases in PBR mRNA expression in the ipsilateral hemisphere, and a 18-kDa band corresponding to PBR protein was detected. Peripheral-type benzodiazepine receptor immunohistochemistry showed subsets of ameboid microglia/macrophages within the infarcted core showing a distinctive strong PBR expression from day 4. These cells were often located surrounding microhemorrhages. Reactive astrocytes forming a rim surrounding infarction at day 7 also showed some PBR immunostaining. These results show cellular heterogeneity in the level of PBR expression, supporting that PBR is not a simple marker of inflammation, and that the extent of [11C]PK11195 binding depends on intrinsic features of the inflammatory cells.
Keywords
Introduction
Focal cerebral ischemia leads to an inflammatory process involving glial reactivity and leukocyte infiltration. Resting microglial cells respond to alterations in their environment after brain injury by becoming reactive. This cellular reactivity takes place through acquisition of a distinct phenotype, which implies considerable morphological alterations and changes in the pattern of gene expression (Kreutzberg, 1996). In this manner, reactive microglia attain new functions in the injured tissue, including the capacity to phagocyte dead cells. However, reactive microglia/macrophages may include a heterogeneous population of cells, as shown by the selective expression of certain antigens (Jander et al, 1998, 2002; Schroeter et al, 1999; Reichmann et al, 2002; Beschorner et al, 2002).
Cells of the mononuclear phagocyte lineage such as macrophages and also reactive microglia express the peripheral-type benzodiazepine receptor (PBR) (Stephenson et al, 1995; Myers et al, 1991a). However, the expression of this receptor is very low in the brain under physiological conditions (Benavides et al, 1983). Thus, PBR is a potential target to identify reactive microglia/macrophages in the brain. Increased binding of several PBR ligands has been reported after brain injury, including focal (Myers et al, 1991a, b ) and global (Stephenson et al, 1995) cerebral ischemia. The isoquinoline carboxamide derivative PK11195 (1-(2-chlorophenyl)-N-methyl-N-(1-methyl-propyl)-3-isoquinolinecarboxamide) selectively binds to PBR and has an antagonistic action on this receptor (Benavides et al, 1983; Le Fur et al, 1983). [3H]PK11195 has been extensively studied in rodent brain tissue after ischemia by autoradiography (e.g., Benavides et al, 1983; Myers et al, 1991a, b ). In addition, [11C]PK11195 has been used for brain imaging with positron emission tomography (PET). Studies with this tracer have been performed in patients with neurological diseases, such as multiple sclerosis (Banati et al, 2000), Alzheimer's disease (Cagnin et al, 2001), Rasmussen's encephalitis (Banati et al, 1999), idiopathic Parkinson's disease (Gerhard et al, 2006), and stroke (Ramsay et al, 1992; Gerhard et al, 2000, 2005; Pappata et al, 2000; Price et al, 2006). Recent PET studies in stroke patients report increased binding as early as 3 days after the onset of stroke, and that binding was not limited to the magnetic resonance imaging (MRI) lesion but was also found in peripheral areas (Gerhard et al, 2005; Price et al, 2006). Here we examined the cellular correlates of the [11C]PK11195 PET signal in the rat brain after ischemia.
Materials and methods
Animals and Surgery
Adult male Sprague-Dawley rats (280 to 320 g body weight; Harlan, Spain) (n = 70) were used. Animal work was conducted in compliance with the Spanish legislation on the ‘Protection of Animals used for Experimental and other Scientific Purposes’, and in accordance with the Directives of the European Union. Transient focal ischemia was produced by 1-h intraluminal occlusion of the middle cerebral artery with a three-vessel occlusion/reperfusion model, as described elsewhere (Justicia et al, 2006). Rats were anesthetized with 4% isofluorane in a mixture of 70% N2O and 30% O2, and after tracheal intubation, anesthesia was maintained using 1 to 1.5% isofluorane. During surgery, body temperature was maintained at 37.5°C using a heating pad connected to a rectal probe. A 2.6-cm length of 4 to 0 monofilament nylon suture, which had been heat-blunted at the tip was introduced into the right external carotid artery up to the level where the middle cerebral artery branches out. Both common carotid arteries were clamped to minimize collateral circulation. After 50 mins of ischemia, the clip on the left common carotid artery was released. Ten minutes later, the filament was gently removed and the clip on the right common carotid artery was released to allow reperfusion. Nonoperated rats were used as controls (n = 14). Animals were killed under anesthesia at 1 (n = 12), 2 (n = 6), 6 (n = 7), 4 (n = 14), and 7 (n = 17) days after ischemia. These time points correspond to 24, 48, 72, 96, and 168 h, with a maximal deviation of 2 h for the first 2 days and of 4 h for days 3 to 7.
Radiochemistry
(R)-[1-(2-chlorophenyl-N-methyl-N-(1-methyl-propyl)-3-isoquinolinecarboxamide] ((R)-PK11195) and (R)-[1-(2-chlorophenyl-N-(1-methyl-propyl)-3-isoquinolinecarboxamide] ((R)-N-desmethyl-PK11195) were obtained from Advanced Biochemical Compounds (ABX, Radeberg, Germany). [11C]PK11195 was synthesized according to the procedure described by Camsonne et al (1984), with slight modifications. Briefly, 1 mg of (R)-N-desmethyl-PK11195 was dissolved in 350 μL dimethyl sulfoxide, containing 8 mg of potassium hydroxide. After trapping of [11C]CH3I, the vial was heated at 90°C for 3 mins. Purification was performed by high-performance liquid chromatography on a RP-C18 column (9.6 × 250 mm, 5 μm particle size) using ethanol/water (75/25) as mobile phase. The purified fraction was evaporated to dryness and reconstituted with 7 mL physiological saline solution. Average radiochemical yield and specific activity were 40% and 25 GBq/mmol, respectively. Radiochemical purity was higher than 99%.
Positron Emission Tomography
Positron emission tomography studies were performed in an animal dedicated camera (microPET R4; Concorde, Siemens, Knoxville, TN, USA). Animals (n = 16) were anesthetized with isofluorane and received an intravenous bolus injection of [11C]PK11195 (1.8 mCi/rat), and emission data were acquired for 35 mins. Positron emission tomography data were reconstructed with an OSEM3D + MAP algorithm (matrix size: 256 × 256 × 63, two OSEM3D iterations, 18MAP iterations, b = 0.005), yielding scans with a spatial resolution of 1.5 mm full-width at half-maximum. Positron emission tomography studies were carried out at 1 (n = 3), 2 (n = 3), 3 (n = 3), 4 (n = 5), or 7 (n = 6) days after ischemia, and in controls (n = 2). Two rats were repeatedly studied at 1, 4, and 7 days, whereas the remaining rats underwent one scan only. Two hours after the last PET scan, rats were anesthetized with halothane, killed by decapitation, and the brain was removed for further evaluation of the infarction, and histological and immunohistochemical analyses.
Image Analysis
Positron emission tomography images were coregistered with MRI images (T1-3D) obtained from ischemic rats of the same gender and body weight. Positron emission tomography images were also coregistered to anatomical data of a rat brain atlas (Rubins et al, 2003) to verify the anatomical location of the signal. Image coregistration was performed by maximizing their mutual information using the SPM2 programme (http://www.fil.ion.ucl.ac.uk/spm/). In addition, for each animal, we used the information of brain infarction on the tissue sections (stained with 2,3,5-triphenyltetrazolium chloride (TTC), see below) to draw regions of interest (ROIs) in the corresponding PET image. For this purpose, we manually aligned the image of a coronal TTC-stained brain tissue section at the level of Bregma with the corresponding MRI image. Regions of interest were manually defined over the TTC image and then applied over the PET image (see Figure 1). Visual inspection was carried out to verify ROI placement in the PET image. Regions of interest were defined for each rat on the region of infarction (ROI-i1) and the peripheral region (ROI-i2) in the ipsilateral hemisphere, and symmetrical ROIs (ROI-c1 and ROI-c2) were drawn in the homologous contralateral regions. The mean value and the standard deviation of each ROI in the PET images were measured using ImageJ (http://rsb.info.nih.gov/ij/) software. The standard uptake value was calculated for each ROI, dividing ROI activity by the radioactive dose and body weight.
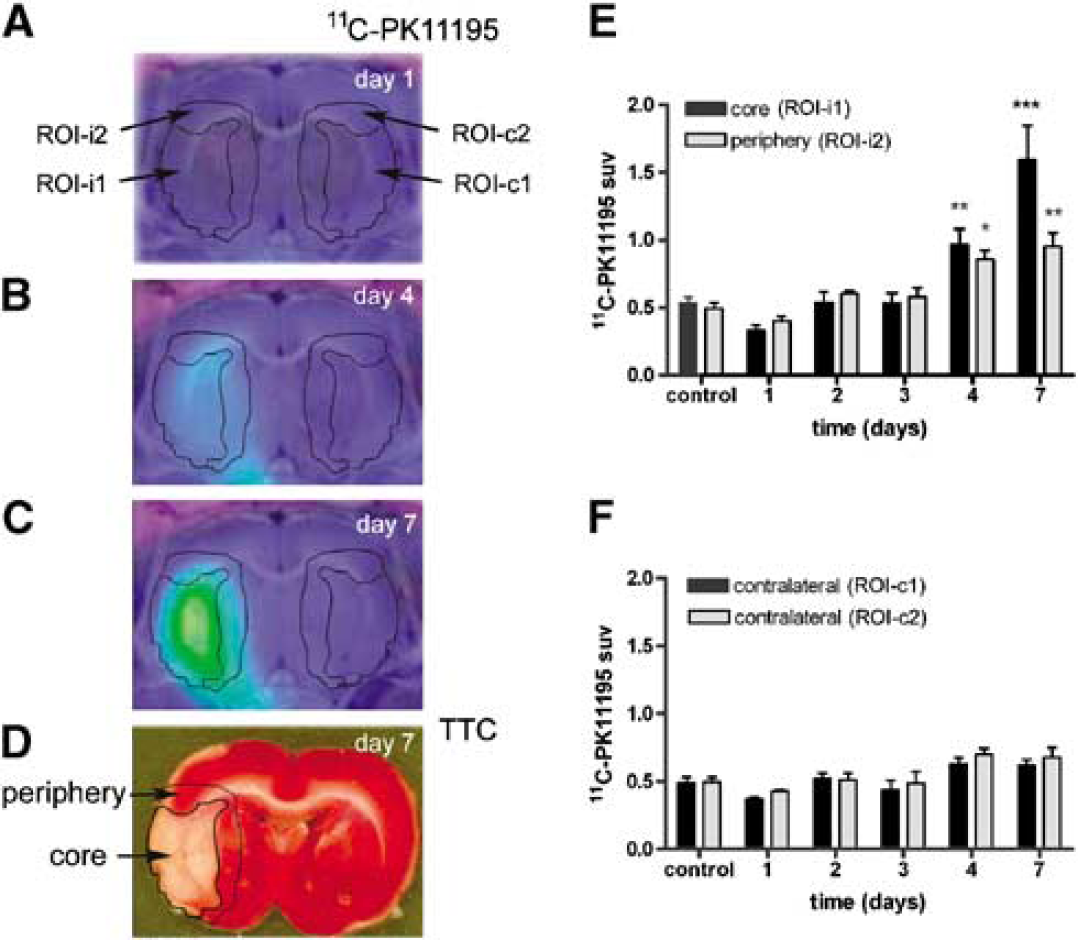
Progression of the [11C]PK11195 PET signal after ischemia. Images are from one representative rat showing [11C]PK11195 signal at various time points after ischemia (
[11C]PK11195 Autoradiography
An additional group of rats at 7 days after ischemia (n = 3) was anesthetized and received an intravenous injection of [11C]PK11195, as above. After 30 mins, rats were killed, and the brain was removed from the skull and sliced in 2-mm coronal sections, which were immediately used for autoradiography (Fujifilm BAS-5000, FUJIFILM Europe GmbH, Dusseldorf, Germany). After 30 mins exposure, the autoradiographic image was obtained and the sections were then stained with TTC (see below) to visualize tissue infarction. For each section, the autoradiographic image was superimposed to the corresponding TTC image (Adobe Photoshop software) to relate the region giving a positive radioactive signal with the region of infarction. Then, each section was embedded in paraffin, sliced in sections with a microtome, and used for immunohistochemistry (see below).
Evaluation of Brain Damage
At 24 h after ischemia, a neurological test in a 9-point scale (0 = absence of functional impairment) was performed to evaluate the functional lesion, as reported (Justicia et al, 2006). Animals scoring 2 points or less were removed from the study. Controls and rats subjected to ischemia were anesthetized and killed at different times (1, 2, 3, 4, and 7 days; n = 3 to 5 per time point). The brain was removed and sliced in 2-mm-thick coronal sections. These were then stained with 1% solution of TTC for 10 mins at 37°C. Sections were then immersed overnight in a 4% paraformaldehyde in phosphate buffer and washed in this buffer. Pale areas in each section were taken as the infarcted zones.
[3H]PK11195 Binding
A separate group of rats (n = 18) was used to study [3H]PK11195 binding following a method reported previously (Pubill et al, 2003). We studied controls (n = 4), and rats at day 1 (n = 4), day 4 (n = 4), or day 7 (n = 6) after ischemia. The ipsilateral striatum was dissected out and immediately frozen. We chose to study the striatum as this region always showed a large zone of infarction using this experimental model. Tissue was homogenized in 2 mL cold homogenization buffer (5 mmol/L Tris—HCl, 320 mmol/L sucrose, containing the protease inhibitor cocktail (Complete; Roche Pharma, Basel, Switzerland) and 1 mmol/L sodium orthovanadate, pH 7.4) with a polytron for 15 secs. All reagents, unless otherwise stated, were from Sigma. The homogenates were centrifuged at 15,000g for 30 mins at 4°C. The pellets were resuspended in homogenization buffer, washed twice, and centrifuged under the same conditions. The final pellets were resuspended in 50 mmol/L Tris-HCl buffer (pH 7.4) with protease inhibitors. Protein content was determined by the Bradford method (Bradford assay; Bio-Rad Laboratories Headquarters, Hercules, CA, USA). Assays were carried out with 150 μg of protein in each sample in a final volume of 0.25 mL buffer and 2 nmol/L [3H]PK11195 (83.5 Ci/mmol/L). Specific binding was defined as the difference between the radioactivity bound in the absence (total binding) and in the presence of 10 μmol/L unlabelled PK11195 (nonspecific binding). After 2 h of incubation on ice, samples were filtered under vacuum on to Whatman GF/B glass fiber filters presoaked in 0.5% polyethyleneimine. The radioactive concentration was measured using 5 mL of scintillation liquid (Optiphase ‘HigSave’ 2 scintillation cocktail, Perkin Elmer, Waltham, MA, USA) in a Wallac WinSpectral 1414 for scintillation spectroscopy.
Real-Time Detection of the Expression of Peripheral-Type Benzodiazepine Receptor mRNA in Brain
Another group of rats (controls, n = 6; and ischemic rats at day 1, n = 3; day 2, n = 3; day 3, n = 4; day 4, n = 4; day 7, n = 4, postischemia) was used to study PBR mRNA expression by real-time polymerase chain reaction. Brain tissue was sliced in 2-mm sections. All sections but one were rapidly dissected out to remove ipsilateral and a contralateral tissue, and the tissue was immediately frozen and kept at −80°C until further procedures. The remaining section was fixed with 4% paraformadehyde overnight, stained with TTC to verify the presence of infarction, and cut in a vibratome to obtain 50-mm sections that were used for immunohistochemistry, as above. RNA was extracted from the ipsilateral (ischemic) and contralateral hemispheres using guanidinium thiocyanate (Chomczynski and Sacchi, 1987). Two micrograms of RNA were used for cDNA synthesis using the AMV First-Strand cDNA Syhthesis Kit (#12328-040; Invitrogen, Carlsbad, CA, USA). Two micrograms of cDNA synthesis reaction were used in a 25 mL real-time polymerase chain reaction with FAM-labelled Taqman® assays for PBR (Rn00560892_m1) and β-actin (Rn00667869_m1) (Applied Biosystems, Foster City, CA, USA). The amplification conditions were 10 mins at 95°C followed by 40 cycles of 15 secs at 95°C, 1 min at 60°C. CT values were analyzed using the 2−δδCT method (Livak and Schmittgen, 2001).
Peripheral-Type Benzodiazepine Receptor Protein Expression by Western Blotting
Another group of rats (n = 12) was used to study the expression of PBR protein by Western blotting, using a method previously reported (Justicia et al, 2006) with modifications. Briefly, the striatum was dissected and homogenized in radioimmunoassay precipitation buffer, centrifuged at 2,000g, and the supernatant was used for protein determination as above. Thirty micrograms of protein was loaded in a 15% polyacrylamide gel and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were then transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). After blockade, membranes were incubated overnight at 4°C with anti-PBR antibody (Ab1, #6361; R&D Systems, Inc., Minneapolis, MN, USA) diluted 1:2,000, followed by incubation with a peroxidase-linked secondary antibody (Amersham Biosciences, Piscataway, NJ, USA). The reaction was developed with a chemiluminiscence method. Membranes were reprobed with a monoclonal antibody anti-β-tubulin (Boheringer, Ingelheim, GmbH, Ingelheim, Germany) diluted 1:100,000 to verify correct amount of protein loading in each lane.
Immunohistochemistry
One of the 2-mm brain sections (at the level of Bregma) was embedded in paraffin and cut into 5-μm-thick sections with a microtome. A set of paraffin sections was stained with hematoxylin and eosin for microscopic examination of the lesion. Other paraffin sections were subjected to antigen retrieval by immersing them in a sodium citrate solution and boiling for 10 mins. Sections were used for immunohistochemistry with two different rabbit polyclonal antibodies recognizing rat PBR (Ab1 #6361 and Ab2 #6362; R&D Systems) diluted 1:100. After incubation with a goat anti-rabbit secondary antibody (Bio-Rad) and the ABC kit (Vector Laboratories, Burlingame, CA, USA), the reaction was visualized with 0.05% diaminobenzidine and 0.1% hydrogen peroxide. After PBR immunostaining, a set of sections was counterstained with hematoxylin. Double immunoreactions were carried out for PBR and markers of either microglia/macrophages, with biotin-labeled lectin from Lycopersicon Esculentum (2.5 μg/mL; Sigma), or astroglia, with a monoclonal antibody against glial fibrillary acidic protein (GFAP) diluted 1:500 (Boehringer, Mannheim, Germany). After the first immunoreaction, sections were incubated with the second primary antibody, followed by the secondary antibody and the avidin-biotin complex, washed with 0.01 mol/L sodium phosphate buffer (pH 6), and preincubated for 10 mins with 0.01% benzidine dihydrochloride and 0.015% sodium nitroferricyanide in 0.01 mol/L sodium phosphate buffer (pH 6). The reaction was developed with this solution containing 0.005% H2O2. Immunoreaction controls included omission of the primary antibody. Preparations were examined under the bright field microscope and also under a fluorescent light using a FITC filter to assess for the presence of autofluorescence from erythrocytes.
A consecutive 2-mm brain section was cut into 50-μm-thick sections in a vibratome, and immunohistochemistry against antimacrophage complement receptor 3 (OX42 antibody; AbD Serotec, Kidlington, UK) diluted 1:500 was carried out free floating to characterize the time course of the microglial/macrophage reaction.
Statistical Analyses
Differences at the various time points after ischemia versus control were analyzed with one-way analysis of variance, and two-way analysis of variance was used to compare by region and time versus control. The Bonferroni's multiple comparison test was used for post hoc analysis. Statistical analyses were carried out with GraphPad Prism software.
Results
[11C]PK11195 Positron Emission Tomography Signal
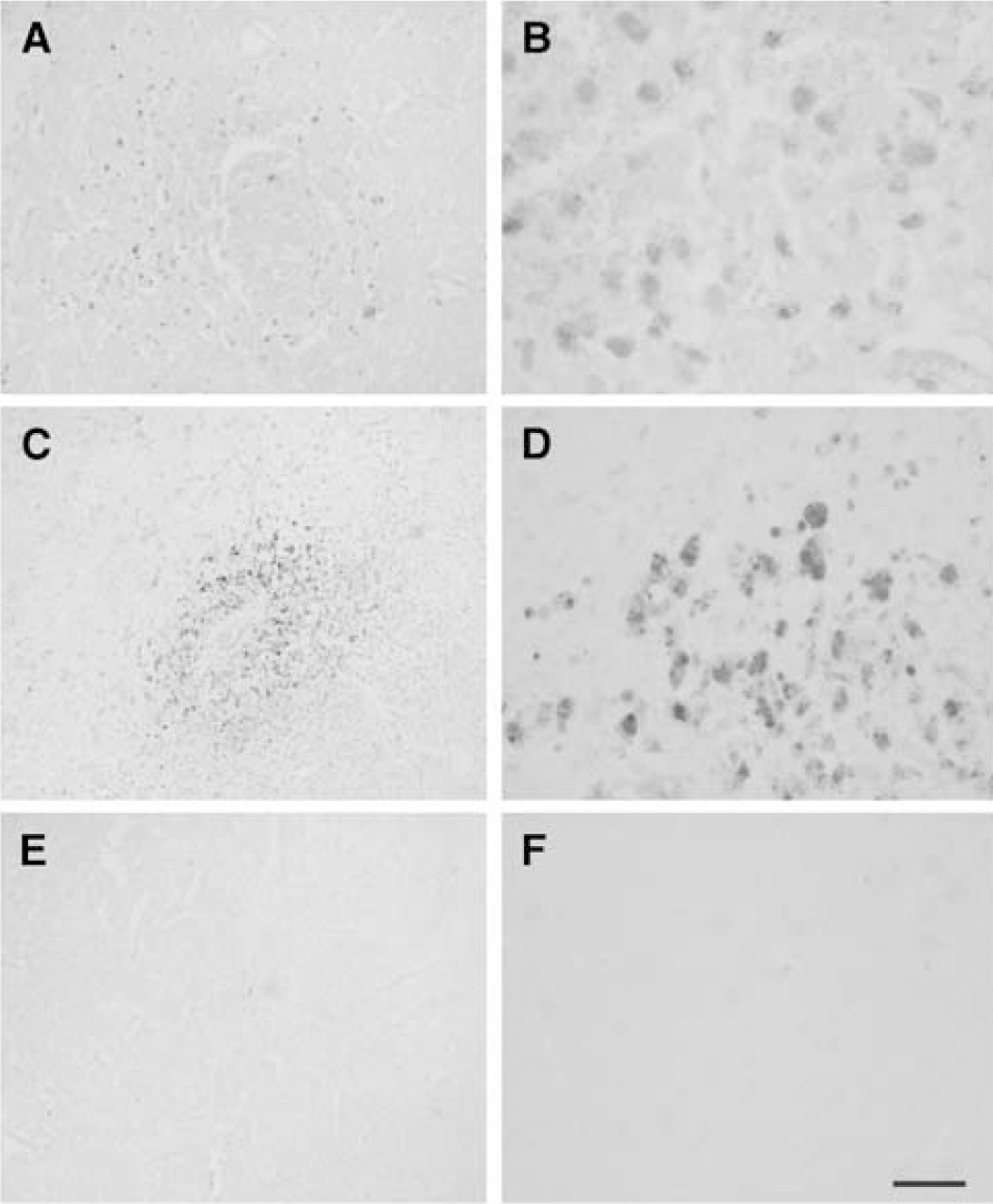
Increased [11C]PK11195 binding was detected at 4 and 7 days in the ipsilateral hemisphere (Figures 1A to 1C). Two ROIs were defined in the ipsilateral hemisphere corresponding to the region of infarction (ROI-i1) and the surrounding region (ROI-i2). These ROIs were defined for each rat on the basis of the regional location of infarction, as assessed in the post-mortem tissue (see Methods; Figure 1D). Two ROIs corresponding to the homologous contralateral regions (ROI-c1 and ROI-c2) were also defined. The standard uptake value in the infarcted region (ROI-i1) was significantly higher than the control at 4 (P < 0.01) and 7 (P < 0.001) days after ischemia (Figure 1E). Also, standard uptake value showed moderate increases in the peripheral region surrounding infarction (ROI-i2) at 4 (P < 0.05) and 7 (P < 0.01) days versus control (Figure 1F).
Binding of [3H]PK11195 in the Zone of Infarction
We performed ex vivo studies with [3H]PK11195 in the region of infarction to verify that the enormous increase in binding observed by PET in this zone was not due to hemodynamic features associated to the in vivo study with [11C]PK11195. The results showed no changes in [3H]PK11195 binding at 24 h in relation to the control, a high increase at 4 days (P < 0.05), followed by a further increase at 7 days (P < 0.001; Figure 2A), thus validating in the tissue the results found in vivo with PET.
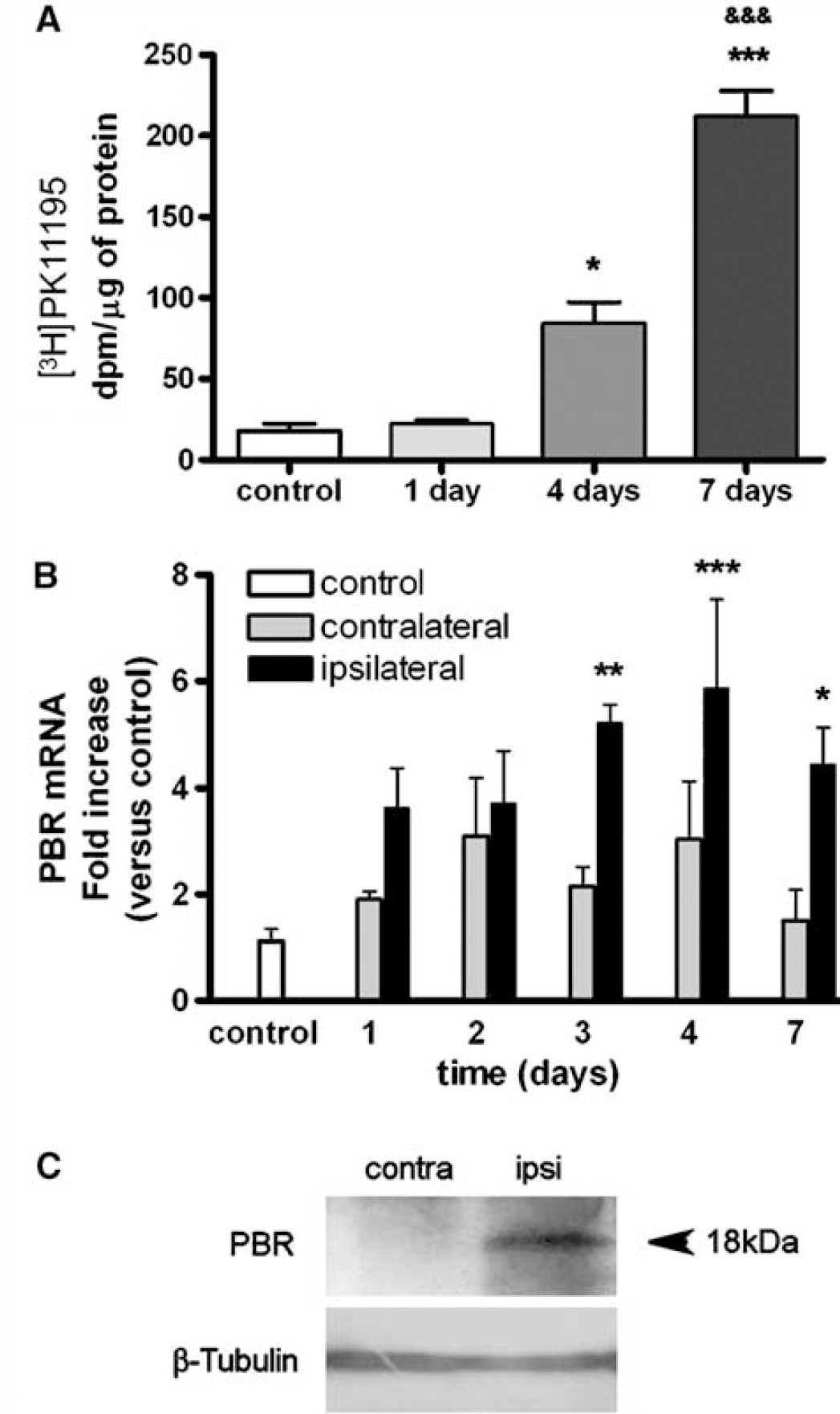
Measures of [3H]PK11195 tissue binding and PBR mRNA and protein expression after ischemia. (
Peripheral-Type Benzodiazepine Receptor mRNA and Protein Expression
Expression of PBR mRNA increased significantly in the ipsilateral hemisphere after ischemia. Two-way analysis of variance by region (ipsilateral and contralateral) and time (days after ischemia) followed by post hoc test showed significant increases at day 3 (P < 0.01), day 4 (P < 0.001), and day 7 (P < 0.05) after ischemia in the ipsilateral hemisphere in relation to control (Figure 2B), whereas changes in the contralateral hemisphere did not become statistically significant despite a tendency to increase at days 2 and 4.
Peripheral-type benzodiazepine receptor protein was detected by Western blotting as a 18-kDa band (corresponding to the molecular weight of PBR) in the ipsilateral hemisphere at 7 days after ischemia (Figure 2C).
Time Course of the Microglial/Macrophage Reaction
Microglial reactivity was evidenced by increased intensity of immunostaining with OX42 antibody (Figure 3) and of lectin (data not shown) staining in relation to controls, which show low intensity of staining with these markers. Ramified stellate-reactive microglial cells were detected at the margins of infarction and in the surrounding corpus callosum at day 1 after ischemia (Figure 3B). The reactive phenotype of the cells progressed in these areas at 2 days (data not shown), but they were rarely seen within the infarction during this time period (data not shown). By 3 days, reactive microglial cells were more abundant in the periphery (Figure 3D), and were also detected within the infarcted core (Figure 3C). At 4 days, cells in the core showed a higher intensity of OX42 immunoreactivity, immunoreactive cells were more abundant than at 3 days, and most showed an ameboid phenotype that was indistinguishable from macrophages (Figure 3E), whereas in the periphery, cells with a rather ramified morphology were predominant (Figure 3F). By 7 days, reactive microglia/macrophages were abundant in the core of infarction (Figure 3G), whereas they were scarce in surrounding regions (Figure 3H), which mainly contained reactive astrocytes (see Figure 7).
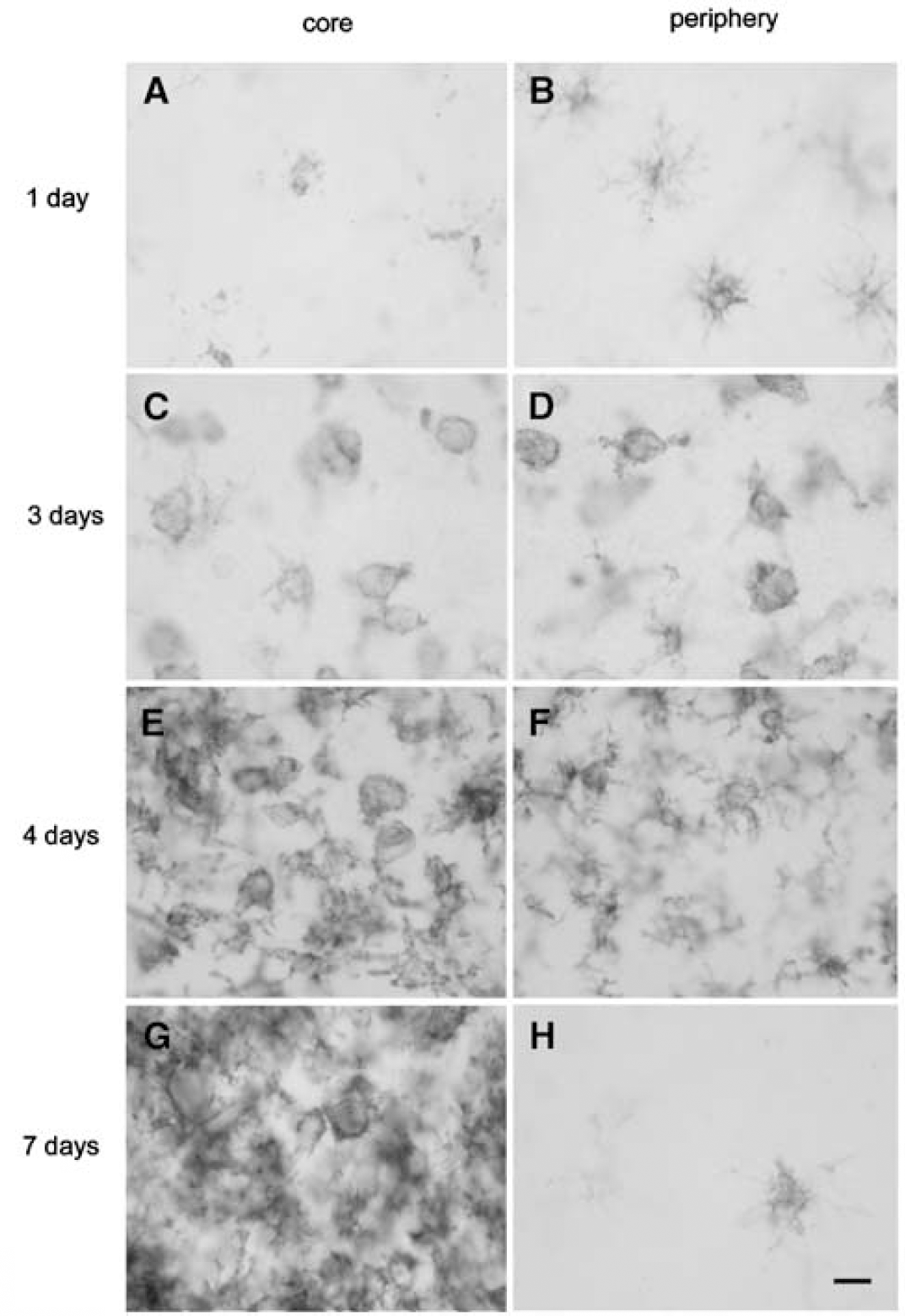
Temporal evolution of the cellular microglia/macrophage reaction. Microglia and macrophages stain positive with the OX42 antibody. (
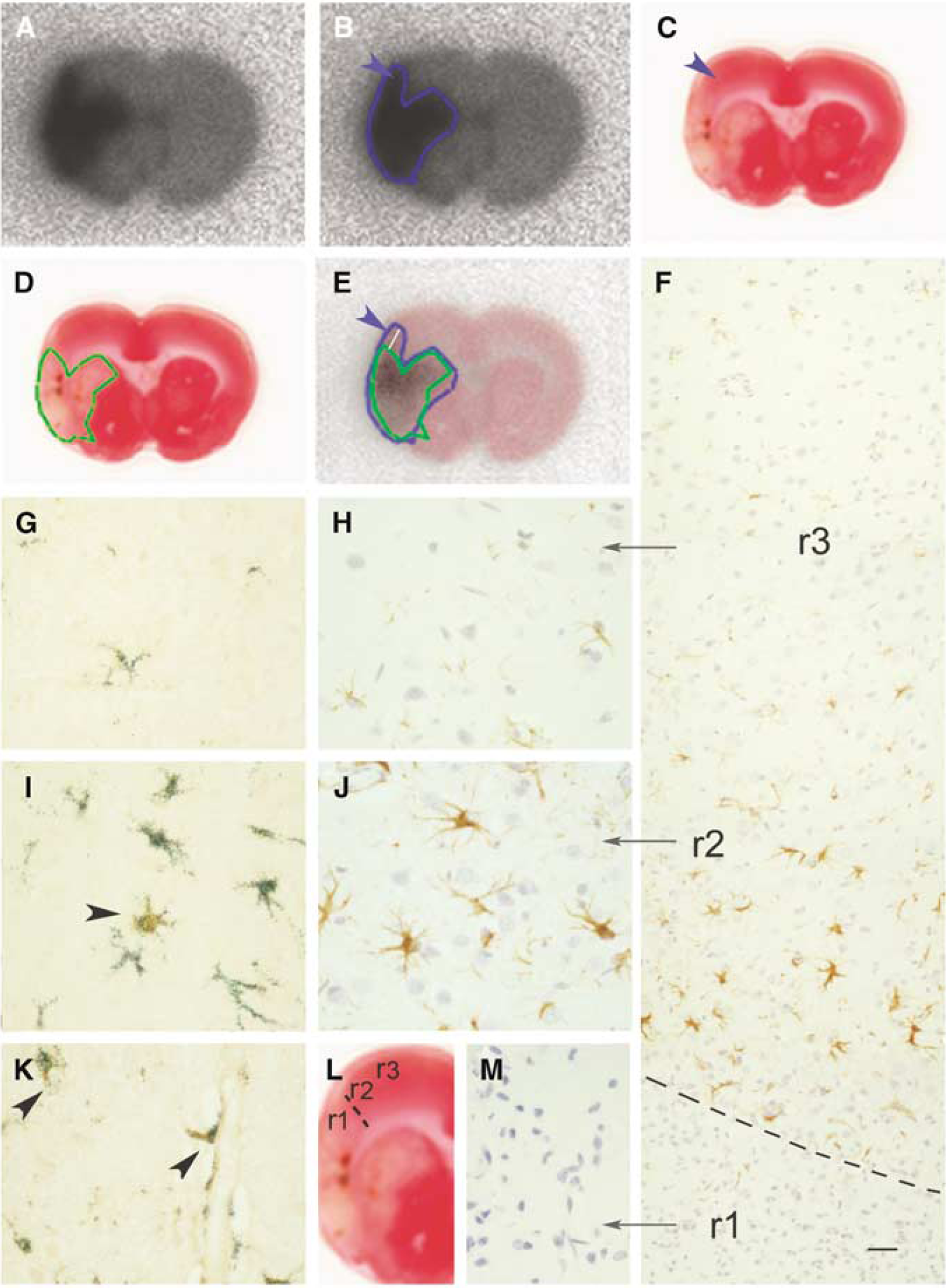
[11C]PK11195 autoradiography. At 7 days after ischemia, rats received [11C]PK11195 in vivo and were killed 30 mins later. The brain was sliced and autoradiography was carried out on the sections. After 30 mins exposure, the sections were stained with TTC and further processed for immunohistochemistry. Images in this figure correspond to one single rat as representative of this experiment. (
Ramified microglial cells showing a higher intensity of OX42 staining than control were also seen in the contralateral hemisphere during the first days after ischemia (data not shown), indicating a nonspecific microglial response through the brain. This contralateral reaction was not associated to local cell death and was not accompanied by leukocyte infiltration or the presence of round phagocytic microglia, and it was completely resolved by 7 days, thus indicating the presence of a mild and transient remote inflammatory response.
Cellular Expression of Peripheral-Type Benzodiazepine Receptor
Immunohistochemistry to PBR revealed low levels of immunostaining in the control brain (Figure 4A). From day 4 after ischemia, certain cells with the morphology of ameboid reactive microglia/macrophages showed a positive reaction in the core of infarction (Figures 4B to 4E), as evidenced with two different antibodies against PBR (Figure 4). At 4 and 7 days after ischemia, we observed the presence of microhemorrhages within the infarcted core, and weak PBR immunoreactivity was detected in erythrocytes (Figures 4B and 4C). The ameboid cells showing intense PBR immunoreactivity were seen grouped in certain zones of the core at day 4 (Figures 5A and 5B) and 7 (Figures 5C and 5D), and they were identified as microglia/macrophages by double staining with lectin (Figures 4H and 4I). These cells often surrounded areas of erythrocyte extravassation to the parenchyma (Figure 6), as showed by examining erythrocyte autofluorescence in the preparations. Furthermore, the presence of erythrocytes inside PBR-positive ameboid cells was frequently observed (Figure 6) evidencing that this particular population of macrophages had the ability to phagocyte erythrocytes. In addition, cells with morphological features of reactive astrocytes were occasionally detected in peripheral areas (Figure 4F). The astroglial nature of these latter cells was showed by double immunohistochemistry against PBR and GFAP (Figures 4K and 4L).
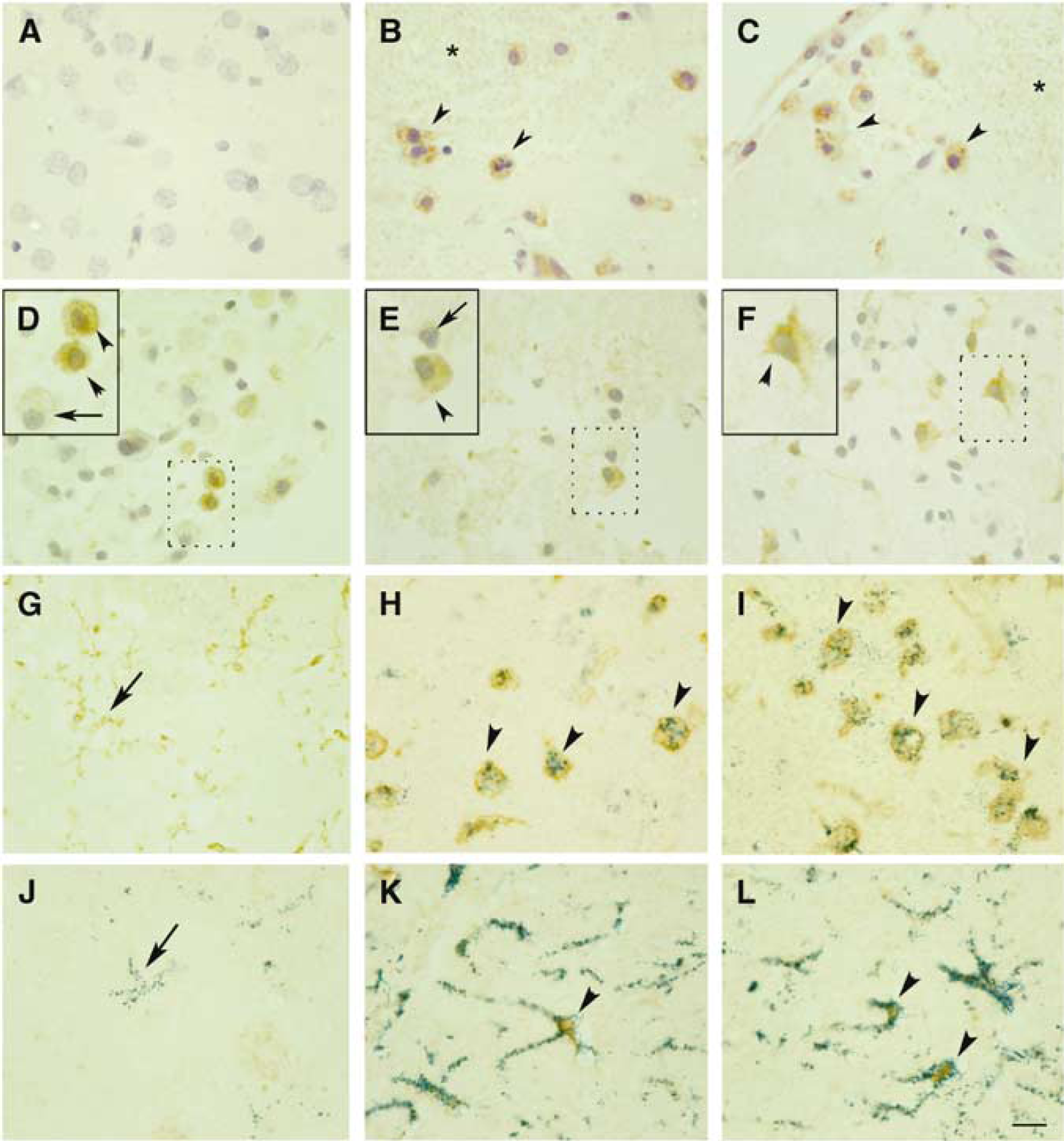
Cellular expression of PBR after ischemia. (
Peripheral-type benzodiazepine receptor immunoreactivity is not homogeneously distributed. Peripheral-type benzodiazepine receptor immunoreactivity (Ab1) is low in the control brain (
Peripheral-type benzodiazepine receptor-positive reactive microglia/macrophages often surround erythrocytes. (
[11C]PK11195 Autoradiography
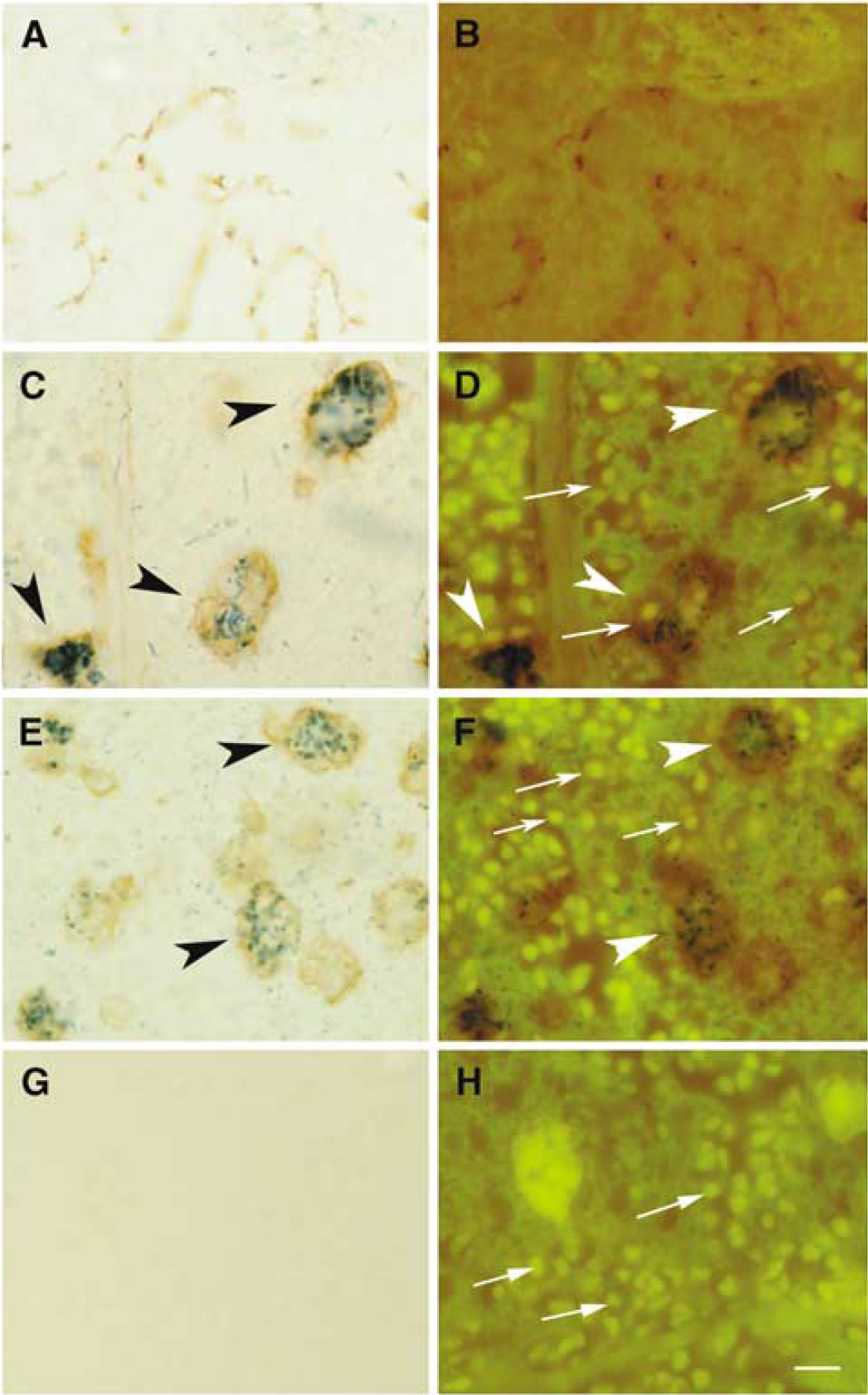
The autoradiographic image obtained after in vivo administration of [11C]PK11195 (Figure 7A) was compared with the corresponding TTC image (Figure 7C) from the same section. This experiment was performed in three animals at day 7 after ischemia (one representative rat is shown in Figure 7). The area showing a radioactive signal in the autoradiographic image (black area in Figure 7A that was delimitated with a blue line in Figure 7B) was compared with the area of infarction (pale area in Figure 7C that was delimitated with a green line in Figure 7D). Superimposition of images revealed that the region showing intense radiotracer signals was in good agreement with the infarction core, except for tracer accumulation in small areas neighboring the infarction (arrowhead in Figure 7). In Figure 7E, the maximal distance between the green and blue line in the cortex was around 1 mm. Under the microscope, we took sequential pictures across a cortical zone surrounding infarction (as shown in Figure 7L) and reconstructed an image (Figure 7F) from infarction (r1 in Figure 7F) to the nonaffected cortex (r3 in Figure 7F). Immunohistochemistry against GFAP revealed the presence of a rim of astroglial cells (r2 in Figure 7F, and higher magnification in Figure 7J) surrounding the infarcted core (r1 in Figure 7F, and higher magnification in Figure 7M). The latter was full of macrophagic cells but devoid of any positive GFAP signal. We estimated that the thickness of the rim of reactive astrocytes was around 1 mm. In addition, double immunohistochemistry with antibodies against PBR and GFAP revealed the expression of this receptor by the reactive astrocytes surrounding infarction (Figures 7I and 7K), but not by resting astrocytes (Figure 7G).
Discussion
Transient focal cerebral ischemia induced a strong [11C]PK11195 PET signal at 4 and 7 days after ischemia in the infarcted hemisphere. This temporal pattern of in vivo [11C]PK11195 uptake was validated in tissue by ex vivo binding studies with [3H]PK11195. Enhanced radioactive-PK11195 binding was coincidental with the temporal pattern of PBR expression in inflammatory cells, as expected, and it was particularly associated with the presence of ameboid microglia/macrophages. These results are consistent with previous studies using autoradiographic techniques, which reported that [3H]PK11195 binds to activated microglia after brain ischemia (Banati et al, 1997), particularly to cells with macrophagic phenotype (Myers et al, 1991 a). In addition, here we show a heterogeneous expression of PBR in the population of reactive microglia/macrophages, with selective high expression in subsets of the ameboid cells located within the core of infarction. This observation is consistent with the view that the population of reactive microglia/macrophages found in brain after ischemia is heterogeneous regarding the expression of several antigens (Jander et al, 1998, 2002; Schroeter et al, 1999; Reichmann et al, 2002; Beschorner et al, 2002). However, the reason why the level of PBR expression in this cellular population is heterogeneous is unknown, and putative specific functions remain to be identified. Nonetheless, our findings show that strongly PBR-immunoreactive ameboid cells were located surrounding areas of microhemorrhages, and that these cells carried erythrophagocytosis. The high expression of PBR that we observed in this particular subset of cells might have contributed to the enhanced intensity of the [11C]PK11195 PET signal in the infarcted core at 4 and 7 days after ischemia. These findings indicate that the extent of [11C]PK11195 uptake depends on the presence of specific reactive cells highly expressing PBR. In addition, we detected some low PBR immunoreactivity in erythrocytes within the ischemic parenchyma, which is consistent with the report of peripheral-type benzodiazepine binding sites on erythrocyte membranes (Olson et al, 1988).
In addition to the strong PET signal at the core of infarction we also found an increased signal in the peripheral region, although the value of this result is unclear. First, it is expected that partial volume effect will contribute to the signal at the periphery of infarction, and, second, the rim of reactive astrocytes surrounding the infarcted core (a putative source of signal in the periphery), is very narrow. Indeed, we estimated that at 7 days after ischemia the rim of reactive astrocytes had a thickness of around 1 mm, which is below the spatial resolution of our PET camera. Therefore, whether there is intrinsic [11C]PK11195 binding in this peripheral region cannot be precisely evaluated in rats by PET. As far as [11C]PK11195 is used for ex vivo autoradiography, it is difficult to clearly detect the extension of PBR in the periphery. However, in humans, there is evidence in favor of [11C]PK11195 uptake in peripheral regions after recent studies showing [11C]PK11195 binding outside the MRI ischemic lesion at time points after the onset of stroke, comparable to those in our study (Gerhard et al, 2005; Price et al, 2006). In addition, we found that reactive astrocytes surrounding the core of infarction at 7 days after ischemia can express PBR, unlike resting astrocytes. This finding agrees with previous reports showing that astrocytes show [3H]PK11195 binding and/or PBR expression under certain circumstances (e.g., Chen et al, 2004; Kuhlmann and Guilarte, 2000; Moynagh et al, 1994).
One limitation of our PET study is that we could not perform MRI to each animal and for this reason we had to coregister the PET images to MRI images of ischemic rats of the same weight and gender. This is likely to cause a higher degree of regional imprecision than if we had used the corresponding MRI of each rat. Also, we noticed some positive PET signal outside the brain, which could be due to extracerebral 11C-PK1195 uptake. Nonetheless, these effects should not alter the general findings in our work.
The expression of PBR mRNA and protein strongly increased in the ipsilateral hemisphere, as expected. However, in the contralateral hemisphere, we detected that PBR mRNA expression had a tendency to increase during the first days after ischemia, but this had subsided completely by day 7. This observation mirrored a mild, diffuse, and transient microglial reaction in this zone, as assessed with OX42 immunoreactivity. Nevertheless, this contralateral microglial reaction was not sufficient to induce detectable levels of PBR protein expression or 11C-PK11195 binding. It feasible that microglial cells from remote sites became ‘aware’ of the ischemic lesion and reacted to it in a mild and transient manner, through a nonidentified mechanism that might involve spreading depression.
Peripheral-type benzodiazepine receptor overexpression has been related to increased cellular resistance to hydrogen peroxide cytotoxicity, thus suggesting that PBR might protect against oxidative stress (Carayon et al, 1996). However, PBR ligands generate free radicals in a process that may involve the mitochondrial permeability transition pore (Jayakumar et al, 2002). Peripheral-type benzodiazepine receptor is located mainly in the mitochondrial outer membrane in close association with the voltage-dependent anion channel (McEnery et al, 1992; Papadopoulos et al, 2006; Veenman and Gavish, 2006). Moreover, PBR ligands induce mitochondrial permeability (Chelli et al, 2001), and under certain circumstances they are involved in mitochondrial apoptosis (Castedo et al, 2002; Jorda et al, 2005). In contrast, in other studies, PBR upregulation has been associated with cell survival (Rey et al, 2000). In addition to its possible role in mitochondrial function and mitochondrial-driven apoptosis, PBR participates in a wide variety of functions such as synthesis of steroids, transport of cholesterol, porphyrin transport and heme synthesis, and immunomodulation (see reviews by Papadopoulos et al, 2006; Veenman and Gavish, 2006). The multiple functions of PBR, the lack of knowledge about its exact role in reactive glia and macrophages, and the present finding showing heterogeneous levels of cellular expression after cerebral ischemia indicate that PBR remains an intricate target for imaging brain inflammation.
In brief, here we show that [11C]PK11195 PET uptake is mainly associated with increased expression of PBR in reactive ameboid microglia/macrophages in the core of infarction. However, the level of PBR expression in the population of reactive inflammatory macrophagic cells was heterogeneous, and in addition, reactive peripheral astrocytes showed some PBR expression. Therefore, these results suggest that the extent of [11C]PK11195 uptake reflects intrinsic properties of the inflammatory reaction that remain to be further characterized.
Footnotes
Acknowledgements
We thank Ms N Montoya, Mr F Gil, and Ms E Gómez for technical assistance, and Dr D Pubill for help and advice on the ex vivo binding experiments.