
Abstract
Inflammation is a major contributor to the pathogenesis of cerebral ischemia and stroke. In the peripheral immune response, caspase-1 activation involves the formation of a macromolecular complex termed the inflammasome. We determined whether nucleotide-binding, leucine-rich repeat, pyrin domain containing 1 (NLRP1), molecular platform consisting of capase-1, apoptosis-associated speck-like protein containing a caspase-activating recruitment domain (ASC), and NLRP1, is expressed in the normal and postischemic brain. Mice underwent thromboembolic stroke to investigate the formation of the inflammasome and subsequent activation of downstream inflammatory responses. Western blot analysis showed expression and activation of interleukin (IL) IL-1β and IL-18 at 24 h after stroke. Size-exclusion chromatography and coimmunoprecipitation analysis showed protein association between NLRP1, ASC, caspase-1, and the X-linked inhibitor of apoptosis protein (XIAP). After ischemia, immunohistochemical analysis revealed inflammasome proteins in neurons, astrocytes, and microglia/macrophages. The potential of the inflammasome as an antiinflammatory target was showed by interference of inflammasome activation resulting in reduced cytokine levels in mice treated after ischemia with a neutralizing antibody against NLRP1. These findings show that the inflammasome complex forms after focal brain ischemia and may be a novel therapeutic target for reducing the detrimental consequences of postischemic inflammation.
Introduction
Ischemic events initiated by thromboembolic processes activate complex pathophysiologic mechanisms that result in neurologic deficits and neuronal cell death (Dietrich et al, 1993; Zhang et al, 2005). The initial vascular responses to embolic events also lead to secondary injury mechanisms including inflammation that contributes to the damaging cellular and molecular responses of ischemic injury (Danton and Dietrich, 2003; Tan et al, 2003). The inflammatory response after cerebral ischemia is mediated by inflammatory cytokines released by infiltrating inflammatory cells crossing the dysfunctional blood—brain barrier and by resident cells of the central nervous system (CNS; del Zoppo et al, 2000; Allan and Rothwell, 2001). The molecular mechanisms by which inflammatory cytokines mediate the deleterious effects initiated after cerebral ischemia are poorly understood and remain an active research focus (Rothwell, 2001).
Classic inflammatory cells, such as microglia, macrophages, and astrocytes, contribute to the postischemic inflammatory response through secretion of inflammatory mediators including various cytokines (Allan and Rothwell, 2001; Hedtjarn et al, 2002). The proinflammatory cytokines interleukin (IL)-1β and IL-18 are activated by proteolytic cleavage of inactive precursors by caspase-1 (Martinon and Tschopp, 2004; Dinarello, 2005). After an ischemic insult, caspase-1 expression is upregulated (Rothwell, 2003), leading to the generation of the active protein by oligomerization of procaspase-1 molecules and autoproteolytic cleavage into the active subunits (Martinon et al, 2002; Martinon and Tschopp, 2004; Ogura et al, 2006; Faustin et al, 2007). Oligomerization of procaspase-1 molecules involves the formation of a macromolecular complex known as the inflammasome (Martinon et al, 2002; Trendelenburg, 2008). To date, three different inflammasomes have been described including the nucleotide-binding, leucine-rich repeat, pyrin domain containing 1 (NLRP1), the NLRP3 and the nucleotide-binding, leucine-rich repeat, CARD domain containing 4 (NLRC4) inflammasome (Tschopp et al, 2003; Agostini et al, 2004; Dinarello, 2005; Miao et al, 2006; Ting et al, 2008). In humans, the NLRP1 inflammasome is comprised of NLRP1, the adaptor protein apoptosis-associated speck-like protein containing a caspase-activating recruitment domain (ASC), caspase-1, and caspase-5 (Kummer et al, 2007). However, reconstitution studies using recombinant proteins have challenged this interpretation (Faustin et al, 2007).
Recently, de Rivero Vaccari et al (2008) reported that spinal cord neurons contain a caspase-1, pro-ILβ, and pro-IL-18 activating complex that is involved with the innate CNS response to spinal cord injury. In that study, the inflammasome complex was shown to be an important therapeutic target to improve functional outcomes by inhibiting caspase-1 activation with a neutralizing antibody against ASC. The purpose of this study was to determine whether focal cerebral ischemia would also lead to the formation of the inflammasome complex in vulnerable brain regions. The cellular distribution of various inflammasome proteins was determined under control and postischemic conditions. To clarify the importance of this molecular platform in the pathophysiology of thromboembolic stroke, we also tested the value of therapeutic neutralization of NLRP1 on caspase-1 activation and cleavage of the proinflammatory cytokine IL-1β in injured animals. Here we show that a cerebral ischemic insult induced a rapid processing of IL-1β and IL-18, activation of caspase-1, and promoted activation of the NLRP1 inflammasome. Finally, neutralization of NLRP1 prevented activation of the inflammasome, reduced caspase-1 processing and decreased cleavage of IL-1β, leading to reduced histopathologic damage in this stroke model.
Methods
Animals
Male mice (C57BL/6J; Charles River Laboratories, South Carolina, Charleston, SC, USA) weighing 19 to 25 g and 12 to 16 weeks old were used in this study. Mice were provided with a standard diet and tap water
Mouse Model of Thromboembolic Stroke
Common carotid artery thrombosis (CCAT) was performed as described by Lozano et al (2007). Briefly, mice were anesthetized and a 32-gauge catheter connected to an infusion pump (PHD2000, Harvard) was inserted into the left femoral vein. The right common carotid artery (CCA) was dissected by blunt technique and the external carotid artery was ligated. Total occlusion of the CCA was obtained by photoactivation of Erythrosin B 190449 (ICN Biomedicals Inc., Costa Mesa, CA, USA). Mice were perfused with Erythrosin B (35 mg/kg) by the femoral vein at a rate of 17.5 mg/kg per min. Simultaneously, a tunable argon laser (Innova 70-4; Coherent, Santa Clara, CA, USA) was focused onto the right CCA and the artery was irradiated for 10 mins. Local vascular thrombosis and subsequent occlusion of the CCA was verified with a Transonic Doppler flow probe (Model 0.5 VB; Transonic Systems, Ithaca, NY, USA) coupled with a temperature probe placed on the distal right CCA (Lozano et al, 2007). Sham-operated animals were performed by perfusion with Erythrosin B but laser irradiation was omitted to avoid activation of the dye. In a separate series of CCAT mice, physiologic variables were assessed including arterial blood pressure, PCO2, PO2, and pH. These values were found to be within normal ranges as previously described (Lozano et al, 2007).
Protein Extraction
At various times after CCAT, brains were removed and placed on ice. Regions of the right cerebral cortex corresponding to the middle cerebral artery (MCA) territory were dissected in a glass petri dish on ice and stored at −80°C. The tissue was homogenized using a Dounce homogenizer (35 strokes) in 1.5 ml of lysis buffer: 15 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid pH 7.6, 0.25 mol/L sucrose, 1 mmol/L MgCl2, 2.5 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, and 1 × protease inhibitor cocktail set I (Calbiochem, La Jolla, CA, USA). Samples were assayed for total protein using the Coomassie Assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Samples were then heated with 1 × sample buffer and stored at −80°C.
Immunoblotting
Equal amount of proteins (25 μg) were electrophoresed on denaturing polyacrylamide gels (Bio-Rad precasted 10% to 20% SDS—polyacrylamide gel electrophoresis) and transferred by standard electroblotting techniques to a polyvinylidene difluoride membrane (Immobilon polyvinylidene difluoride; Millipore, Charlottesville, VA, USA), blocked with Tween 20 (0.1%) and incubated overnight at 4°C with the following antibodies: mouse anticaspase-1 monoclonal and rat antimouse ASC monoclonal were kindly provided by Dr Mariathasan (Genetech, CA, USA). Chicken anti-NLRP1 polyclonal was custom designed and produced by Aves Laboratories (sequence: CZYYTEIREREREKSEKGR). Mouse anti-IL-18 monoclonal was purchased from R&D Systems (Mab521; Minneapolis, MN, USA) and anti-X-linked inhibitor of apoptosis (XIAP) was obtained from BD Transduction Laboratories (Lexington, KY, USA). Rabbit anticleaved IL-1β polyclonal (Asp116) and secondary antibodies horseradish peroxidase-conjugated were obtained from Cell Signaling (Danvers, MA, USA). Visualization of the signal was performed by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ, USA). Quantification of bands was made by scanned densitometric analysis and Labwork 4.0 image analysis (UVP Bioimaging System).
Size-Exclusion Chromatography
Dissected ipsilateral cerebral cortices corresponding to the MCA territory were homogenized with the following protein extraction buffer: 20 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid-KOH (pH 7.5), 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L Na EDTA, 1 mmol/L Na EGTA, and 1 × protease inhibitor cocktail set I (Calbiochem). Samples were then centrifuged at 18,000
Coimmunoprecipitation
Fractions corresponding to the high molecular weight (>600 kDa) proteins in the size-exclusion chromatography analysis were pooled for coimmunoprecipitation studies (fractions 6 to 15, 2 ml total volume). Samples were precleared with the protein G-sepharose immunobeads (Amersham Biosciences) and then incubated with the monoclonal antibody anti-ASC overnight at 4°C. Protein G-sepharose immunobeads were added to the mixture and incubated for 2 h and then centrifuged at 12,000
Immunohistochemistry and Confocal Microscopy
Mice were anesthetized and perfusion-fixed with 4% paraformaldehyde for immunohistochemical analysis. Coronal floating sections (60 μm) were cut with a microtome and immunostained with the following antibodies using procedures previously established in our laboratory (Keane et al, 2001). Rabbit polyclonal anticaspase-1 (1:1,000) was obtained from Millipore whereas cell markers such as antiglial fibrillary acidic protein (GFAP) and antineuronal nuclei (NeuN) were purchased from Chemicon (Billerica, MA, USA). Secondary antibodies conjugated to Alexa-fluorochromes were obtained from Molecular Probes-Invitrogen (Carlsbad, CA, USA). In addition, sections were treated with Sudan black 0.3% in 70% ethanol to quench the autofluorescence produced by the injured tissue. Images were obtained with a LSM510 laser confocal microscope (Carl Zeiss Inc., Thornmood, NY, USA). At least three different sections were prepared from each animal; all animals in each group yielded similar results. Control labeling included omission of primary antibodies and labeling with preimmune serum and autofluorescence controls omitted the use of primary and secondary antibodies.
NLRP1 Antibody Treatment
Mice were subjected to CCAT as previously described. Fifteen minutes after CCAT, animals were placed in prone position and injected stereotactically into the right lateral ventricle with 5 μg of anti-NLRP1 chicken antibody. Control mice were injected with the same amount of preimmune chicken serum under identical conditions. For immunoblot analysis of caspase-1 and IL-1β, treated and control treated mice (
Infarct Volume Analysis
For histopathologic analysis of treated (
Statistical Analysis
Statistical comparisons for infarct volume and areas between uninjured and injured groups were made using Student's
Results
Thromboembolic Stroke Induces Processing of IL-1β and IL-18
The proinflammatory cytokines IL-1β and IL-18 are the downstream effectors of activated caspase-1 and directly mediate the inflammatory response (Martinon and Tschopp, 2004). First, we characterized the expression and activation of IL-1β and IL-18 after thromboembolic stroke using specific antibodies against the pro- and active forms of IL-1β and IL-18. Increased levels of pro- and active IL-1β and IL-18 were present in lysates of infarcted tissue at 24 h and remained elevated until 7 days after ischemia (Figure 1), and then decreased thereafter. Thus, the active forms of these inflammatory cytokines were increased acutely after ischemia and remained elevated during the first week after CCAT.
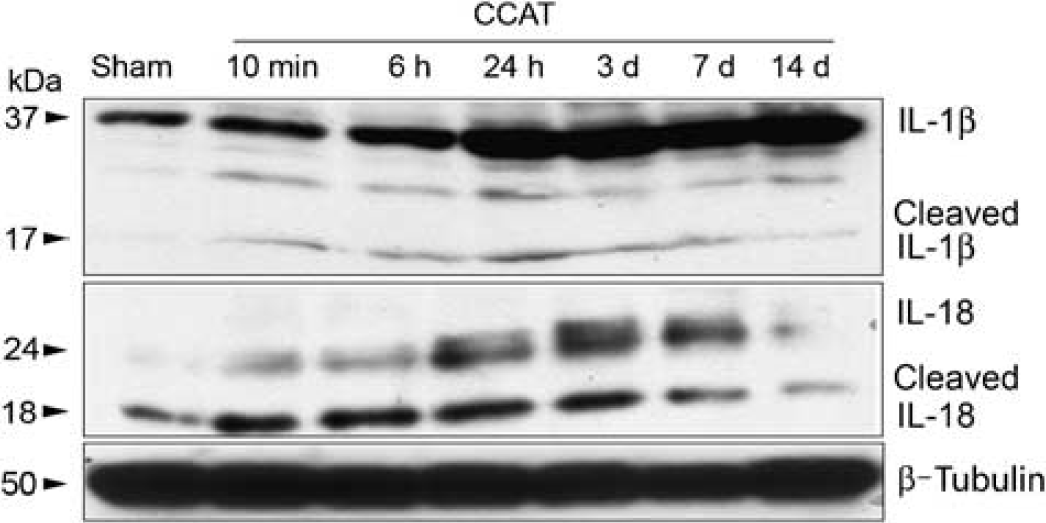
IL-1β and IL-18 are increased after ischemic brain injury. Representative immunoblots of IL-1β and IL-18 of cortical lysates of mice subjected to CCAT and killed at different time points show increased processing of the proinflammatory cytokines IL-1β and IL-18 after CCAT injury. β-Tubulin was used as a protein loading control (
Common Carotid Artery Thrombosis Induces Expression of Inflammasome Proteins
To investigate the profile of protein expression involved in the formation of the inflammasome complex, immunoblot analysis was performed on mice subjected to CCAT at different time points after ischemic insult (Figure 2). Caspase-1 activation, as detected by the presence of the cleaved 26-kDa fragment, was increased significantly at 3 days after CCAT. Seven days after ischemia, the adaptor protein ASC was also significantly increased. NLRP1 expression did not increase after ischemia as determined by densitometric analysis of immunoblots that showed no statistically significant differences when compared with sham-operated animals. These results show that CCAT stimulates the expression of inflammasome signaling molecules, suggesting a possible involvement of the inflammasome in the initiation of inflammation after thromboembolic stroke.
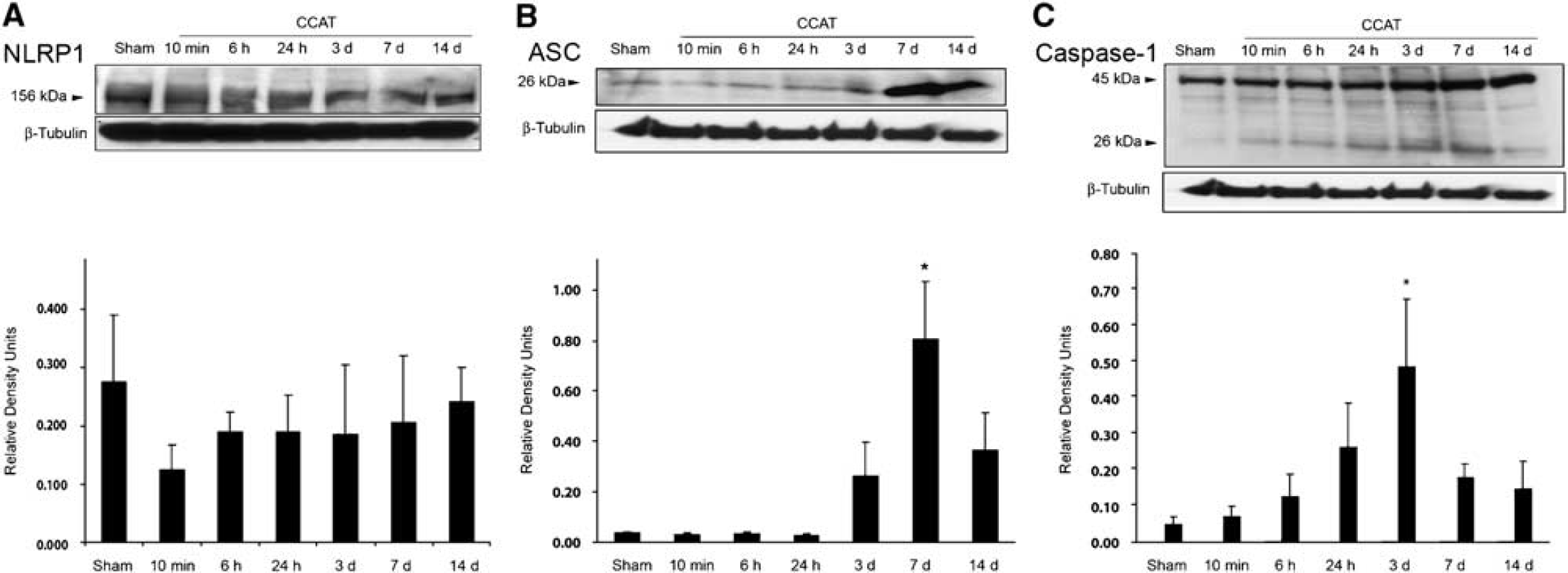
Inflammasome proteins are upregulated after CCAT. Representative immunoblot analysis of NLRP1 (
Common Carotid Artery Thrombosis Induces Formation of the NLRP1 Inflammasome Complex
We next analyzed the assembly of inflammasome proteins into the complex by size-exclusion chromatography in animals 24 h after CCAT (Figure 3A). Immunoblots were run on collected fractions and probed for caspase-1, ASC, XIAP, and NLRP1. NLRP1, ASC, caspase-1, the full-length 57 kDa XIAP, and 25 and 30-kDa cleaved fragments were present in the nonassociated low molecular weight fractions. The specific bands for caspase-1, NLRP1, ASC as well as the full-length XIAP, and the 30 kDa cleaved fragment were also detected in fractions corresponding to high molecular weight proteins (>600 kDa) termed the inflammasome fractions. Elution profile of caspase-6 was used as a control and showed that this protein was not present in the multiprotein complex in the inflammasome fractions. These data suggest that the NLRP1 inflammasome complex consisting of ASC, caspase-1, XIAP, and NLRP1 forms in the brain after a thromboembolic ischemic insult.
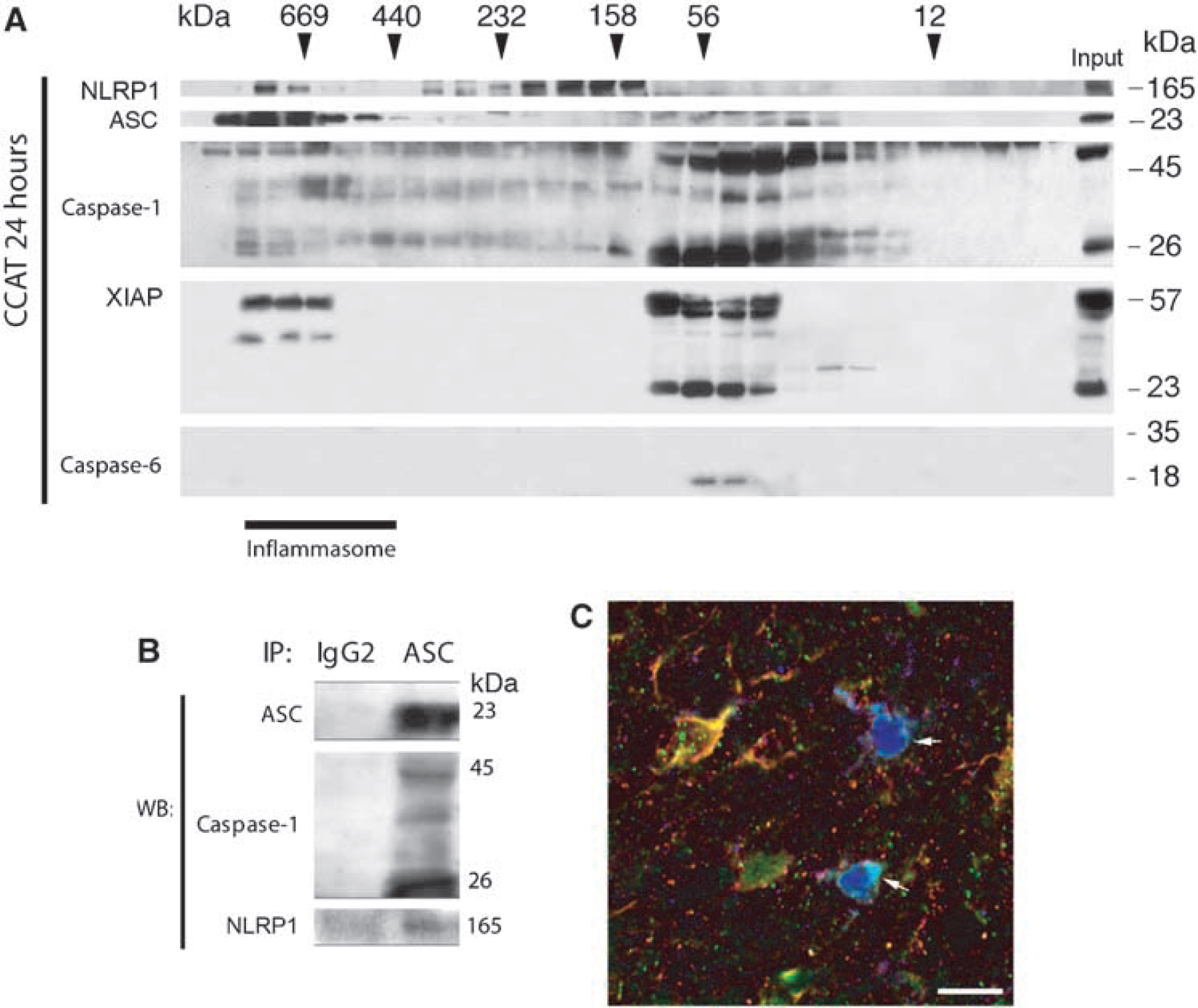
A NLRP1 inflammasome complex forms in the ischemic brain. Size-exclusion chromatography followed by immunoblotting of eluted fractions of brain lysates 24 h after CCAT (
Association of inflammasome proteins was confirmed by coimmunoprecipitation experiments of brain proteins lysates from mice subjected to CCAT and killed 24 h later (Figure 3B). Pooled fractions corresponding to the inflammasome molecular weight (>600 kDa) obtained by size-exclusion chromatography were immunoprecipitated with anti-ASC. Immunoprecipitates were blotted for ASC, caspase-1, and NLRP1. Anti-ASC immunoprecipitated ASC, NLRP1, and caspase-1. Normal rat IgG did not immunoprecipitate the inflammasome-associated proteins and was used as a control, showing antibody specificity.
NLRP1 Inflammasome Proteins are Present in Neurons, Astrocytes and Microglia/Macrophages
To determine the cell-type expression pattern of inflammasome proteins, immunohistochemical analysis was performed using triple labeling for caspase-1, NLRP1, and ASC on brain sections of animals killed 24 h after CCAT. Figure 3C shows that caspase-1 (green), NLRP1 (red), and ASC (blue) were present in cells surrounding the infarcted cortex (arrows). On the basis of morphologic criteria, cells appear to be microglia/macrophages, but other cells, probably astrocytes or neurons were also immunoreactive for caspase-1 and NLRP1 (yellow/orange). Although some inflammasome proteins were found in the nucleus, profile analysis showed that colocalization signals (white, yellow) were present in the cytoplasm of cells.
To determine whether other CNS cells express inflammasome proteins within the brain, immunohistochemical labeling was performed on normal/sham mouse brains and in animals subject to CCAT and killed at several survival periods (6, 24 h, and 7 days). Sections were double-labeled for the inflammasome proteins NLRP1, ASC, and caspase-1 and the cell markers NeuN, GFAP (astrocytes), and lectin (microglia/macrophages). Table 1 summarizes the cell-type expression patterns on inflammasome proteins in naive and injured animals. At 24 h and 7 days after CCAT, caspase-1, NLRP1, and ASC were detected in microglia/macrophages located within the core and periphery of the infarcted cerebral cortex. Caspase-1 and NLRP1 immunoreactivity was not detected in microglia in normal brain sections. However, caspase-1 staining was detected in cortical neurons of naive animals in the cytoplasm and the nucleus (Figure 4). At 24 h after ischemia, caspase-1 and NLRP1 immunoreactivity dramatically increased in the cytoplasm of neurons located in intact regions surrounding the infarcted cortex. ASC was expressed in both naive and ischemic cortical neurons (Table 1). Finally, GFAP-positive astrocytes were also immunoreactive for NLRP1 in the normal brain. Caspase-1 and ASC were only detected after the thromboembolic ischemic event in some astrocytes surrounding the infarct at 7 days after CCAT (Table 1). These findings together with the size-exclusion chromatography results suggest that CCAT triggers formation of the NLRP1 inflammasome in neurons, microglia/macrophages, and astrocytes.
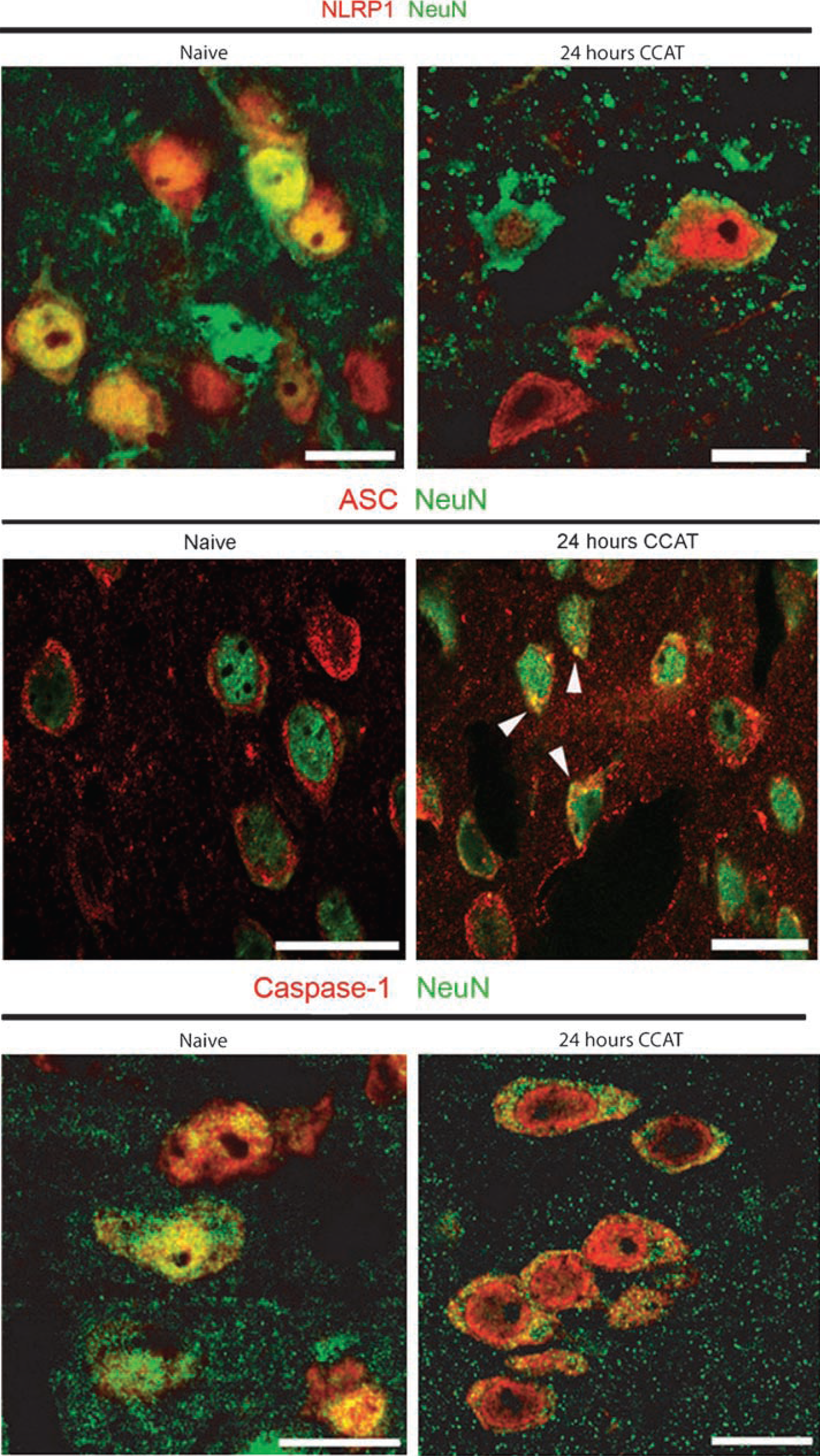
Cellular distribution of inflammasome components in the ischemic cerebral cortex. Representative coronal brain sections of mice subjected to CCAT (24 h) and naive animals. Sections were double-stained for NLRP1, caspase-1 and ASC (red), and NeuN (green). In the normal brain (naive), caspase-1 and NLRP1 are mainly present in the nucleus of neurons whereas ASC is mainly present in the cytoplasm. After injury inflammasome proteins are redistributed predominantly to the cytoplasm and ASC is present as specks in the cell cytoplasm (arrow heads). Scale bar=10 μm.
Summary of cellular localization of inflammasome proteins in the ischemic brain
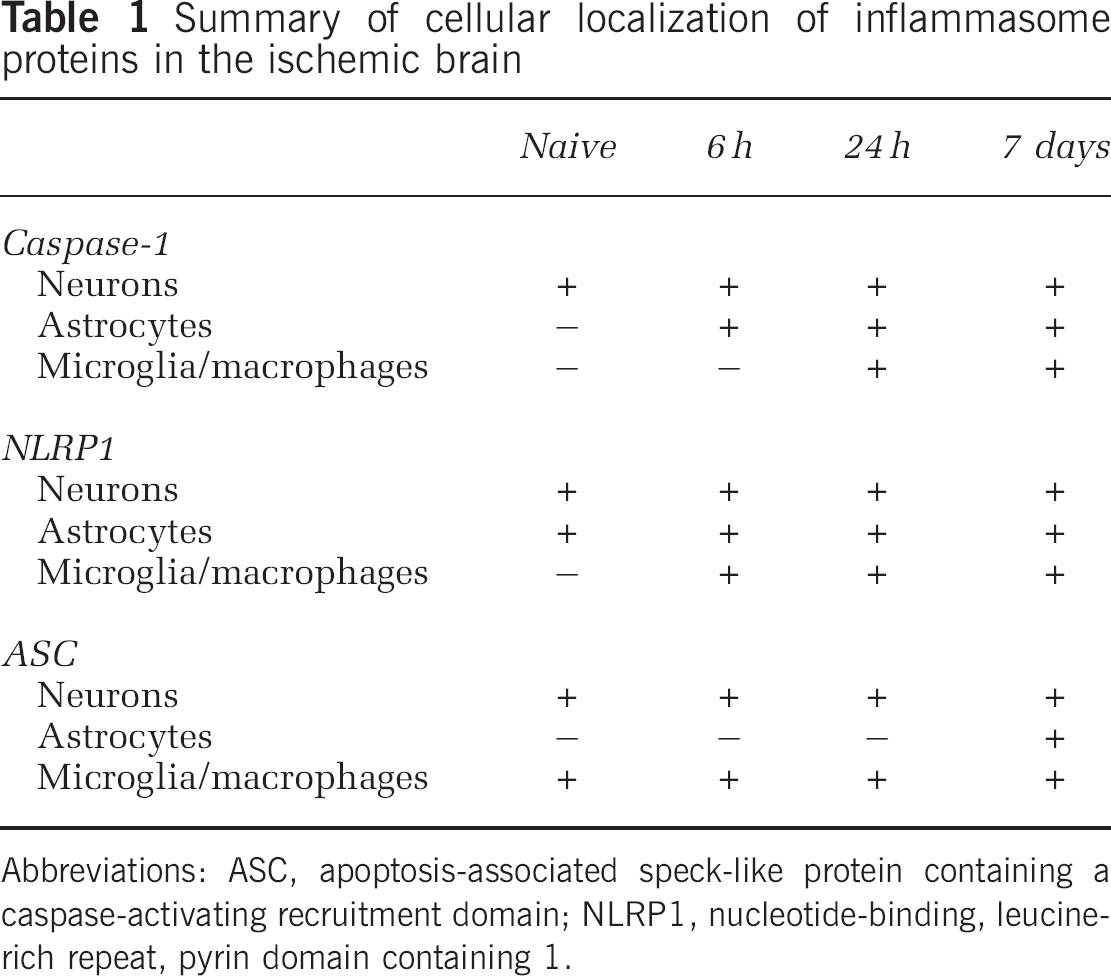
Abbreviations: ASC, apoptosis-associated speck-like protein containing a caspase-activating recruitment domain; NLRP1, nucleotide-binding, leucine-rich repeat, pyrin domain containing 1.
NLRP1 Neutralization Reduces Common Carotid Artery Thrombosis-Induced Activation and Processing of Caspase-1 and Interferes with the Assembly of the NLRP1 Inflammasome Complex
To determine whether inhibition of the inflammasome complex reduces the inflammatory response after CCAT, mice were injected intracerebroventricularly with a chicken polyclonal antibody against NLRP1 15 mins after CCAT. Animals were then killed at 24 h (
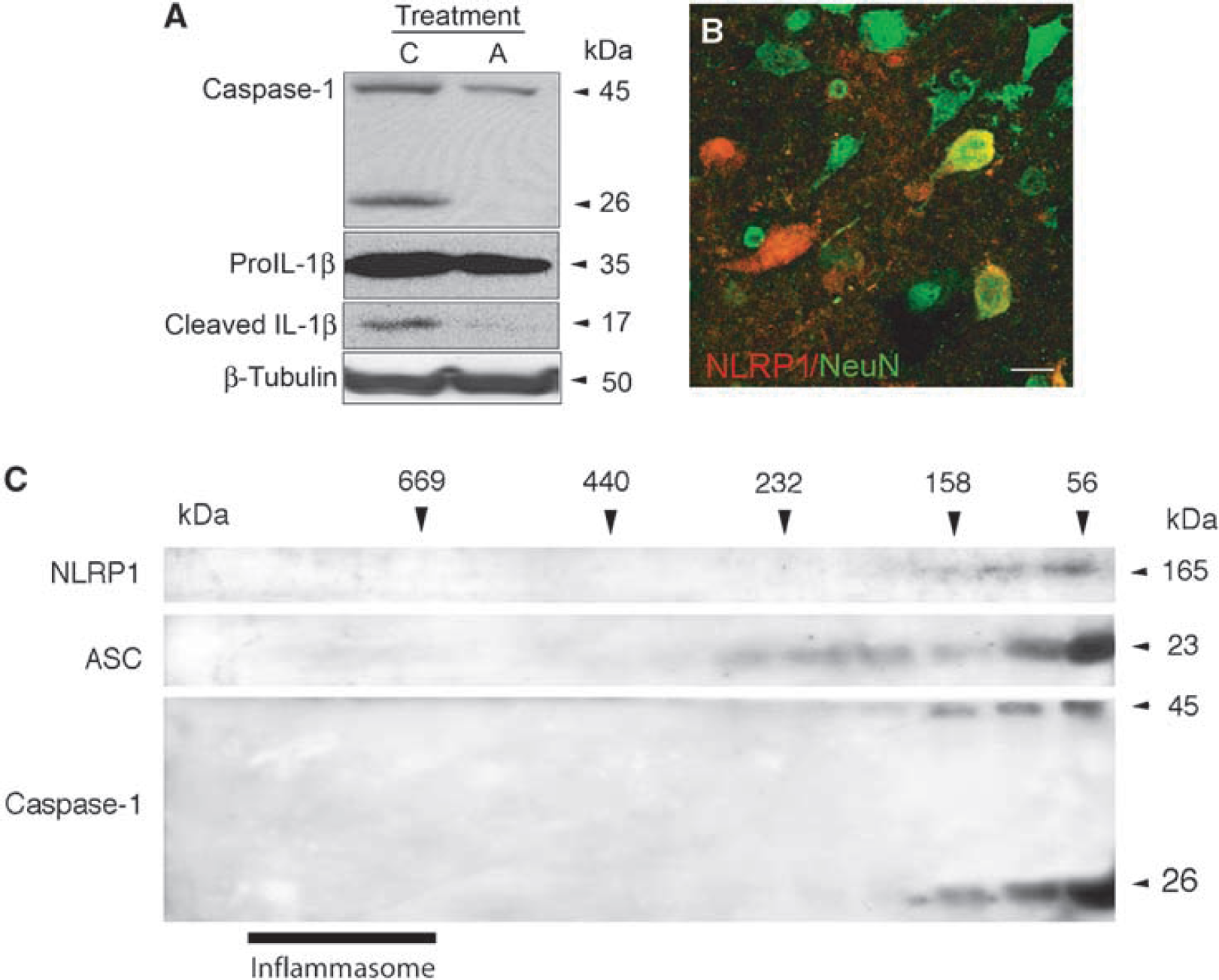
Intraventricular injections of anti-NLRP1 antibody reduced the activation of caspase-1 and the processing of IL-1β and interfered with assembly of the inflammasome complex. (
To determine whether anti-NLRP1 crossed the blood—brain barrier, anti-NLRP1 was injected into the right lateral ventricle 15 mins after CCAT and brains were processed for immunohistochemical analysis. Sections were then immunolabeled with antichicken-Alexa conjugated antibody to detect the chicken NLRP1 antibody. Double labeling with NeuN or GFAP antibodies were used to determine which CNS cells reacted with anti-NLRP1. Preimmune serum was used as a control. Figure 5B shows a confocal image of cortical brain tissue with antichicken-Alexafluor 594 conjugated NLRP1 antibody (red) and NeuN (green) double staining. The injected anti-NLRP1 antibody was detected in neurons and activated astrocytes (not shown) in brain regions within the ipsilateral cerebral cortex.
We next analyzed whether NLRP1 neutralization interfered with the assembly of inflammasome proteins into the complex by size-exclusion chromatography in animals 24 h after CCAT. Immunoblots were run on collected fractions and probed for NLRP1, ASC, and caspase-1. As shown in Figure 5C, NLRP1 neutralization completely blocked protein—protein interactions in the high molecular weight inflammasome fractions. However, these inflammasome components were detected in low molecular weight fractions (not shown) as depicted in Figure 3A. Thus, administration of anti-NLRP1 significantly reduced caspase-1 activation and interfered with assembly of inflammasome proteins into the complex.
Effect of NLRP1 Neutralization on Infarct Size
Studies were conducted to determine whether this anti-NLRP1 treatment strategy would also affect infarct size. Control CCAT mice showed a pattern of well-demarcated cortical infarcts within the right MCA territory as previously described (Lozano et al, 2007). Small infarcts were occasionally observed within subcortical areas, including the hippocampus, striatum, and thalamus. Although all CCAT mice showed some degree of cortical infarction, infarcts were somewhat variable in size and location from mouse to mouse. This variability is a characteristic of the CCAT model that has previously been described in both rats and mice (Dietrich et al, 1999; Lozano et al, 2007). In CCAT mice treated with the NLRP1 neutralizing antibody, infarct areas were not significantly smaller than nontreated CCAT mice (Figure 6) with overall infarct volume being reduced nonsignificantly by 18% compared to nontreated mice.
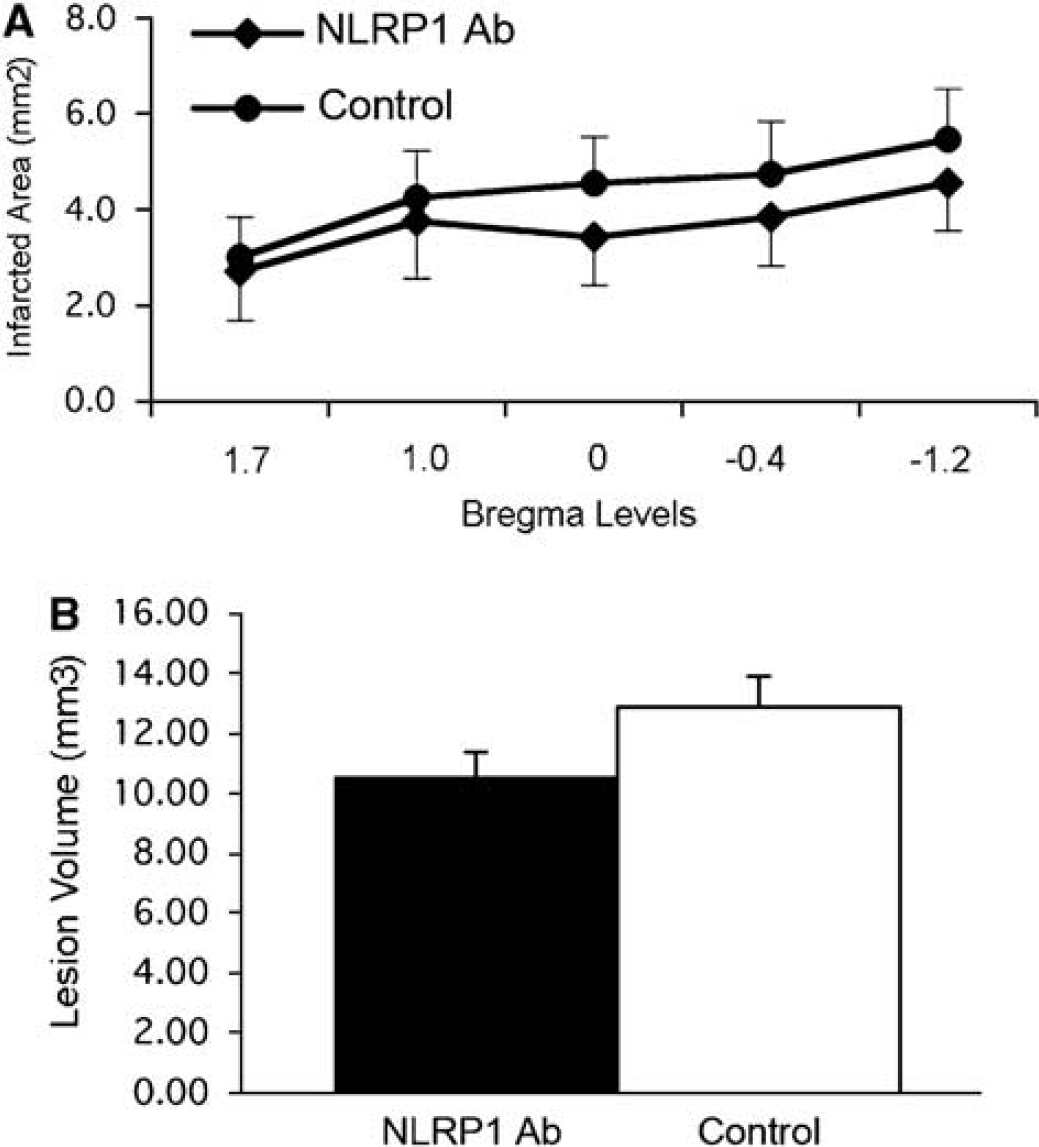
Effect of a single intraventricular injection of anti-NLRP1 antibody on infarct size. (
Discussion
In this study, we have described a role for the NLRP1 inflammasome complex in the inflammatory response induced in the ischemic mouse brain. Our data show that a thromboembolic stroke model induces the acute activation of the NLRP1 inflammasome, resulting in processing of inflammatory caspase-1, leading to maturation of IL-1β and IL-18. The NLRP1 inflammasome in the normal and ischemic brain was detected in multiple CNS cells including neurons, astrocytes, and microglia/macrophages. This multiprotein complex consists of inflammatory caspases-1, NLRP1, and the adaptor protein ASC. Furthermore, this is the first characterization of the mouse NLRP1 inflammasome
In this study, we used a thromboembolic model that produces focal ischemic insults and embolic infarcts (Dietrich et al, 1993; Lozano et al, 2007). The majority of animal models of focal ischemia generate ischemia by mechanical occlusion of large cerebral vessels (Danton and Dietrich, 2004). However, human thromboembolic stroke produces a variety of complex cellular events not necessarily observed in large vessel mechanical occlusion models (Albers et al, 2006). The thromboembolic model is induced by photoactivation of Erythrosin B that promotes local damage to the carotid vascular endothelial surface resulting in the accumulation of activated platelets and the formation of a large but unstable thrombus (Dietrich et al, 1993). Subsequently, the CCA thrombus breaks into a shower of small emboli leading to occlusion of downstream microvessels primarily localized within the MCA territory of the affected brain (Dietrich et al, 1993). This microvascular embolic insult produces multiple focal areas of severe ischemia, acute blood—brain barrier breakdown and the subsequent infiltration of inflammatory cells, resulting in multiple embolic infarcts (Dietrich et al, 1991, 1993, 1999). The model therefore mimics some of the clinical characteristics of transient ischemic events and thromboembolic stroke.
Focal cerebral ischemia leading to cerebral infarction promotes a cascade of inflammatory processes initiated by endothelial dysfunction, blood—brain barrier disruption, brain edema, and the expression of a variety of inflammatory mediators (Lucas et al, 2006). These include the generation of proinflammatory cytokines that induce the expression of chemokines and adhesion molecules with the subsequent recruitment of circulating immune cells (Rothwell, 2003). Macrophages are present within the ischemic brain by 24 h after the thromboembolic insult and remain for several days (Danton and Dietrich, 2003; Lozano et al, 2007). Resident microglia and astrocytes are also activated and may themselves release proinflammatory molecules that may promote the recruitment of blood-borne inflammatory cells into the ischemic area (Thery and Mallat, 1993). Inflammatory molecules such as IL-1β may amplify the inflammatory response by generating proliferation of microglia and astrocytes within the ischemic area (Rothwell, 2003). Interleukin-1β promotes increased endothelial permeability and the expression of a number of adhesion molecules (Rothwell and Luheshi, 2000). These effects further result in the recruitment of inflammatory cells into the brain parenchyma, amplifying the inflammatory reaction within the ischemic brain. In this study, confocal microscopy of the normal brain revealed that macrophage/microglia express the adaptor protein ASC, but do not express NLRP1 or caspase-1. After CCAT, inflammatory cells expressed caspase-1, NLRP1, and ASC and the temporal expression of activated microglia/macrophages correlated with the expression of the proinflammatory cytokines IL-1β and IL-18. In this focal ischemia model, the inflammasome is present in microglia/macrophages that represent the primary immune cells that contribute to the early inflammatory response to ischemia.
We also showed that the inflammasome in neurons is a protein complex containing NLRP1, caspase-1, ASC, and XIAP. A similar complex has recently been identified in spinal cord neurons by de Rivero Vaccari et al (2008). Spinal cord trauma resulted in the processing of IL-1β, IL-18, activation of caspase-1, cleavage of XIAP, and the promotion of the multiprotein complex. On the basis of the present ischemia work, it appears that a similar molecular platform is present in normal cortical neurons and that is activated after cerebral ischemia. In this study, alterations in intracellular ATP, potassium concentrations or other cellular regulations during the ischemic insult may trigger inflammasome activation as previously described (Mariathasan and Monack, 2007; Pétrilli et al, 2007). Thus, the NLRP1 inflammasome platform may represent an important component of the innate immune response of cortical neurons to both physiologic and pathologic conditions including cerebral ischemia and trauma (de Rivero Vaccari et al, 2008).
Although several treatments have been reported to promote neuronal survival, reduce infarct volumes and improve behavioral responses after experimental cerebral ischemia, recombinant tissue plasminogen activator is the only Food and Drug Administration approved therapy for ischemic stroke in humans (Gladstone et al, 2002; Legos et al, 2002; Hacke et al, 2004). Its narrow window of efficacy of 3 h after the onset of stroke and the risk of hemorrhages limits the use of this therapeutic approach to a low percentage of the stroke patient population. Because the inflammatory process is initiated early after an ischemic insult, antiinflammatory treatments have the potential to limit secondary damage in stroke patients (Rabuffetti et al, 2000; Rothwell and Luheshi, 2000; Hedtjarn et al, 2002; Danton and Dietrich, 2004; Lucas et al, 2006). For example, the intraventricular administration of the endogenous IL-1 receptor antagonist IL-1ra reduces neuronal injury after global and focal ischemia (Loddick et al, 1997; Rothwell, 2003). In clinical stroke IL-1ra has recently shown promise in an early clinical trial (Emsley et al, 2005). Attenuation of infarct size is also achieved by treatment with antiadhesion molecules in rats subjected to cerebral ischemia (Zhang et al, 1995; Goussev et al, 1998). Unfortunately, these strategies that have been shown to be efficacious in preclinical ischemia models have yet to be successfully translated to the clinic (Legos et al, 2002). In this study, we tested a novel upstream antiinflammatory therapeutic strategy that involves inhibition of caspase-1 activation by blocking the formation of the inflammasome complex. The anti-NLRP1 antibody was delivered intracerebroventricularly as a neutralizing antibody 15 mins after CCAT. Anti-NLRP1 treatment decreased the inflammatory response by interfering with assembly of the inflammasome complex and inhibiting the activation of caspase-1 and reducing the processing of pro-IL-1β into its mature form resulting in attenuated ischemia-induced NLRP1 inflammasome activation. Thus, neutralizing antibodies against the inflammasome are successful in inhibiting inflammasome activation in multiple models of CNS injury (de Rivero Vaccari et al, 2008).
Treatment with the neutralizing antibody was also tested on its effects on infarct volume in the present model. In treated mice receiving a single injection of anti-NLRP1, no significant difference between the treated and nontreated groups was showed in terms of infarct areas and volumes. The embolic nature of the thrombotic model, resulting in microvascular platelet occlusion and severe ischemic insults, may have contributed to these modest effects on infarct size (Dietrich et al, 1991, 1993). In this regard, evolving embolic infarcts commonly lack large ischemic penumbras as defined by perfusion—diffusion mismatch because of the microvascular occlusive nature of the thrombotic event (Thijs et al, 2001; Butcher et al, 2005). Thus, neuroprotective strategies including antiinflammatory strategies may not provide the same degree of protection seen in other ischemia models such as MCA occlusion. Alternatively, multiple injections of the antibody over a period of time may be necessary to provide histopathologic protection under the present experimental conditions. Nevertheless, the fact that anti-NLRP1 treatment in the present model reduced caspase-1 activation emphasizes the potential importance of this targeted treatment strategy on limiting proinflammatory processes that participate in the pathogenesis of thromboembolic stroke.
In summary, our findings suggest a novel upstream target for therapeutic interventions targeting ischemia-induced inflammatory events. We report the formation of an inflammasome complex after ischemia in neurons and inflammatory cells that is critical for the activation of the inflammatory modulator caspase-1. Recent evidence indicates that antiinflammatory treatments lead to improved outcomes in a number of neurologic disorders (Allen and Rothwell, 2003). The continued investigation into mechanisms underlying postischemic inflammatory processes should identify new strategies to limit secondary injury mechanisms and promote functional recovery in patients with stroke.
Footnotes
Acknowledgements
We thank Dr Helen Bramlett for technical assistance and Mr Jeremy Lytle for editorial support.
The authors declare no financial interest.