
Abstract
Carbamylerythropoietin (CEPO) does not bind to the classical erythropoietin (EPO) receptor. Nevertheless, similarly to EPO, CEPO promotes neuroprotection on the histologic level in short-term stroke models. In the present study, we investigated whether CEPO and other nonerythropoietic EPO analogs could enhance functional recovery and promote long-term histologic protection after experimental focal cerebral ischemia. Rats were treated with the compounds after focal cerebral ischemia. Animals survived 1, 7, or 60 days and underwent behavioral testing (sensorimotor and foot-fault tests). Brain sections were stained and analyzed for Iba-1, myeloperoxidase, Tau-1, CD68 (ED1), glial fibrillary acidic protein (GFAP), Fluoro-Jade B staining, and overall infarct volumes. Treatment with CEPO reduced perifocal microglial activation (P<0.05), polymorphomonuclear cell infiltration (P<0.05), and white matter damage (P<0.01) at 1 day after occlusion. Carbamylerythropoietin-treated rats showed better functional recovery relative to vehicle-treated animals as assessed 1, 7, 14, 28, and 50 days after stroke. Both GFAP and CD68 were decreased within the ipsilateral thalamus of CEPO-treated animals 60 days postoperatively (P<0.01 and P<0.05, respectively). Furthermore, behavioral analysis showed efficacy of CEPO treatment even if administered 24 h after the stroke. Other nonerythropoietic derivatives such as carbamylated darbepoetin alfa and the mutant EPO-S100E were also found to protect against ischemic damage and to improve postischemic neurologic function. In conclusion, these results show that postischemic intravenous treatment with nonerythropoietic EPO derivatives leads to improved functional recovery, which may be linked to their long-term effects against neuroinflammation and secondary tissue damage.
Introduction
Recombinant human erythropoietin (EPO) has shown widespread efficacy in animal models of stroke (Sakanaka et al, 1998; Brines et al, 2000; Calapai et al, 2000; Siren et al, 2001; Brines and Cerami, 2005). Despite its large size, recombinant human EPO administered peripherally crosses the blood—brain barrier to protect against brain injury (Brines et al, 2000). Erythropoietin may act against ischemic damage at multiple levels including attenuation of apoptosis (Siren et al, 2001; Villa et al, 2003), and reduction of brain inflammation (Villa et al, 2003). More recently, EPO treatment was shown to improve functional recovery, and enhance neurogenesis and angiogenesis after focal ischemia, suggesting a beneficial effect of EPO treatment on brain repair after stroke (Wang et al, 2004a). Translation of these research findings into therapeutic application looks promising because the use of erythropoietin in a small clinical trial in patients suffering from stroke improved neurologic scores and functionality (Ehrenreich et al, 2002).
In clinical situations that are likely to require multiple doses of EPO, a major limitation of the compound is that it would trigger unwanted overstimulation of the bone marrow, raise the hematocrit and induce a procoagulant state. For instance, a high hematocrit in transgenic mice overexpressing human EPO is associated with increased susceptibility to ischemic damage (Wiessner et al, 2001). To circumvent this side effect issue, various strategies to dissociate erythropoietic and tissue-protective activities of EPO have been developed. For instance, asialoerythropoietin, an EPO derivative with a very short half-life generated by enzymatic desialylation of EPO, is neuroprotective in models of focal ischemia (Erbayraktar et al, 2003) and neonatal hypoxia—ischemia (Wang et al, 2004b) without increasing the hematocrit. More recently, we have described chemically modified forms of EPO such as carbamylerythropoietin (CEPO) or EPO mutants that do not bind to the classical EPO receptor (EPOR). These retain their tissue-protective properties without effects on the bone marrow and hematocrit (Leist et al, 2004). Carbamylerythropoietin treatment reduced brain infarction after focal ischemia to the same degree as reported for EPO and with a broad therapeutic window (4 h) (Leist et al, 2004).
In the present study, we further explored the effects of CEPO and other nonerythropoietic derivatives of EPO on poststroke functional recovery, secondary tissue damage, and inflammation.
Materials and methods
Focal Ischemia Model
All experimental procedures were performed in accordance with the directives of the European Communities Council Directive #86/609 for care of laboratory animals and in agreement with national regulations on animal research in Italy and Denmark. Surgery was performed on male Crl:CD (SD)BR rats weighing 250 to 285 g (Charles River, Calco, Italy). Focal ischemic stroke within the distribution of the middle cerebral artery (MCA) was produced as described previously (Brines et al, 2000). Briefly, the right common carotid artery (CCA) was occluded by two sutures and cut. A burr hole adjacent and rostral to the right orbit allowed visualization of the MCA, which was cauterized distal to the rhinal artery. Animals were then positioned on a stereotaxic frame and the contralateral CCA was occluded for 1 h by using traction with a fine forceps. Body core temperature was thermostatically maintained at 37°C by using a heating pad and a rectal thermistor (Letica, Barcelona, Spain) for the duration of the anesthesia.
Reagents
Carbamylerythopoietin was synthesized from rhEPO (Dragon Pharmaceuticals, Vancouver, Canada) as described earlier (Leist et al, 2004). Carbamylated darbepoetin alfa (Caranesp) was synthesized from Aranesp (darbepoetin alfa; Amgen, Thousand Oaks, CA, USA) using the same protocol. Carbamylation of EPO and darbepoetin alfa transformed all lysines to homocitrulline resulting in products lacking bioactivity in the in vitro UT7 hematopoiesis assay and failing to bind to EPOR on these cells. Generation of mutant EPO-S100E was described previously (Leist et al, 2004).
Drug Treatments
The drug doses (CEPO, 50 μg/kg; Caranesp, 50 μg/kg; EPO-S100E, 50 μg/kg) were all equivalent with respect to the mass relation to approximately 5,000 IU/kg of EPO. Doses of nonerythropoietic derivatives were chosen based on the observation that equivalent doses of EPO and nonerythropoietic variants are required for neuroprotective effects (Erbayraktar et al, 2003; Leist et al, 2004; Wang et al, 2004b). Drugs or vehicle (0.05% human serum albumin in phosphate-buffered saline) were administered intravenously at different time points after MCA occlusion as described in the text and figures.
Neurologic Deficits
Animals were evaluated for neurologic deficits using the limb placing and the foot-fault tests at different times after occlusion. The limb-placing test developed by De Ryck et al (1989) evaluates sensorimotor integration in limb-placing responses to visual, vibrissae, tactile, and proprioceptive stimuli. For each test, limb placing scores were 0, no placing; 1, incomplete and/or delayed (>2 secs) placing; or 2, immediate and complete placing. Each test was repeated for each paw up to 10 times and for each body side; the maximum limb placing score was 16. The foot-fault test developed by Hernandez and Schallert (1988) measures the ability of the animal to integrate motor responses. The rats were placed on a grid with 2 cm spaces between 0.3 cm diameter metal rods and were observed for 2 mins. With each weight-bearing step, the paw may fall or slip between the wires and this is recorded as foot-fault. The number of foot-faults for the paws contralateral and ipsilateral to the infarction was recorded with the number of successful steps and the foot-fault index was calculated as the percentage of contralateral limb foot-faults per limb step minus the percentage of ipsilateral limb foot-faults per limb step. Baseline foot-fault index as acquired in nontreated nonoperated rats was usually <5 (data not shown).
Infarct Assessment
Infarct volumes were determined 24 h after MCA occlusion by quantitative image analysis of triphenyl tetrazolium chloride-stained 1-mm brain sections using a computerized image analysis system (AIS version 3.0 software, Imaging Research, St Catherine's, ON, Canada) as described previously (Brines et al, 2000). Alternatively, infarct volumes were measured at day 7 after occlusion. Sections selected from predetermined coronal planes (+5.2 to −7.4 mm from bregma) were stained with toluidine blue. Images of brain sections were captured and measurements of hemispheric damage to cortical neuronal perikarya was determined by summation of cortical infarct volumes measured in each brain slice using CAST software (Olympus, Denmark). Alternatively, the infarct volume was calculated as the percentage of infarct volume to the volume of the contralateral hemisphere (indirect volume calculation) as described previously (Zhang et al, 1997).
Immunohistochemistry and Fluoro-Jade B Staining
Animals were anesthetized with chloral hydrate and perfused transcardially with phosphate-buffered saline followed by 4% phosphate-buffered paraformaldehyde for 15 mins. Brains were cryoprotected in 30% sucrose, and sectioned into 20-μm coronal cryosections. Cryosections were processed as free-floating sections using the protocol based on the avidin—biotin—peroxidase technique as described previously (van Beek et al, 2000). Alternatively, triphenyl tetrazolium chloride-stained slices were postfixed in 4% paraformaldehyde fixative in phosphate buffer and paraffin-embedded. Four micron coronal sections were cut on a microtome and processed for immunohistochemistry using the same protocol as described above supplemented with antigen retrieval by microwaving in a citric acid buffer (10 mmol/L; pH 6). In all immunohistochemistry protocols, negative controls were performed by omitting the primary antibody, and this resulted in minimal detected signal in all cases. The following antibodies were used: goat anti-human Iba-1 (1:4,000; Abcam, Cambridge, UK; #Ab5076), mouse anti-rat CD68 (clone ED1; 1:50; Serotec, Oxford, UK), rabbit anti-human myeloperoxidease (1:4,000; DAKO, Glostrup, Denmark; #A 0398), mouse anti-cow Tau-1 (clone PC1C6; 1:5,000; Chemicon International, Temecula, CA, USA; #MAB3420), and rabbit anti-cow glial fibrillary acidic protein (GFAP) (1:4,000; DAKO; #Z 0334). Fluoro-Jade B staining was performed as described previously (Schmued et al, 1997).
Staining Quantification
Images were captured with a JenOptik ProgRes digital camera and image analysis was performed on an Openlab imaging station (Improvision, Coventry, UK). Images from brain areas were captured as follows: perifocal cortex for Iba1 and GFAP (1.00 mm relative to bregma), infarcted core for myeloperoxidase and CD68 (1.00 mm relative to bregma), ipsilateral internal capsule and corpus callosum for Tau1 (−3.14 mm relative to bregma). For examination of Fluoro-Jade B staining, images from whole ipsilateral striatum were captured (1.00 mm relative to bregma). For quantification of thalamic GFAP and CD68 staining, images from whole ipsilateral thalamus were captured (−3.14 mm relative to bregma). Density slicing of regions of interest under standardized conditions was used to detect the area of staining (Staining Index).
In Vitro Neuroprotection
Primary neuronal cultures were prepared from new born rat hippocampi by trypsinization, and cultured as described (Leist et al, 2004). On day 14, the cultures were challenged with 300 μmol/L N-methyl-
Hematopoietic Bioactivity
To test the hematopoietic bioactivity in a proliferation assay, the EPO-dependent human leukemia cell line UT7 was obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). The assay was performed as described previously (Erbayraktar et al, 2003) over 48 h. Compounds were tested at 0.2 pmol/L to 20 nmol/L and proliferation was quantified using WST-1 reduction (Roche Applied Science, Indianapolis, IN, USA).
Statistical Analysis
Data are presented as mean values±s.e.m. Cortical infarct distribution data at 7 days after occlusion were analyzed using repeated-measures analysis of variance followed by Bonferroni tests. For histopathologic data and comparison between vehicle- and CEPO-treated animals, a Student's t-test was used. The nonparametric Mann—Whitney and Kruskal—Wallis tests were used to determine significant differences in neurologic scores when two or more groups were compared, respectively.
Results
Protection Against Ischemic Damage In Vivo and Excitotoxic Injury In Vitro by Caranesp
Cortical infarct areas were significantly reduced by treatment with Caranesp and CEPO compared with vehicle in the 1-day survival group (31% and 28% reduction from control, respectively; P<0.05; Figures 1A and 1B). A significant (P<0.01) improvement in sensorimotor function was observed in Caranesp- and CEPO-treated animals compared with vehicle-treated rats (Figure 1C). Caranesp prevented N-methyl-
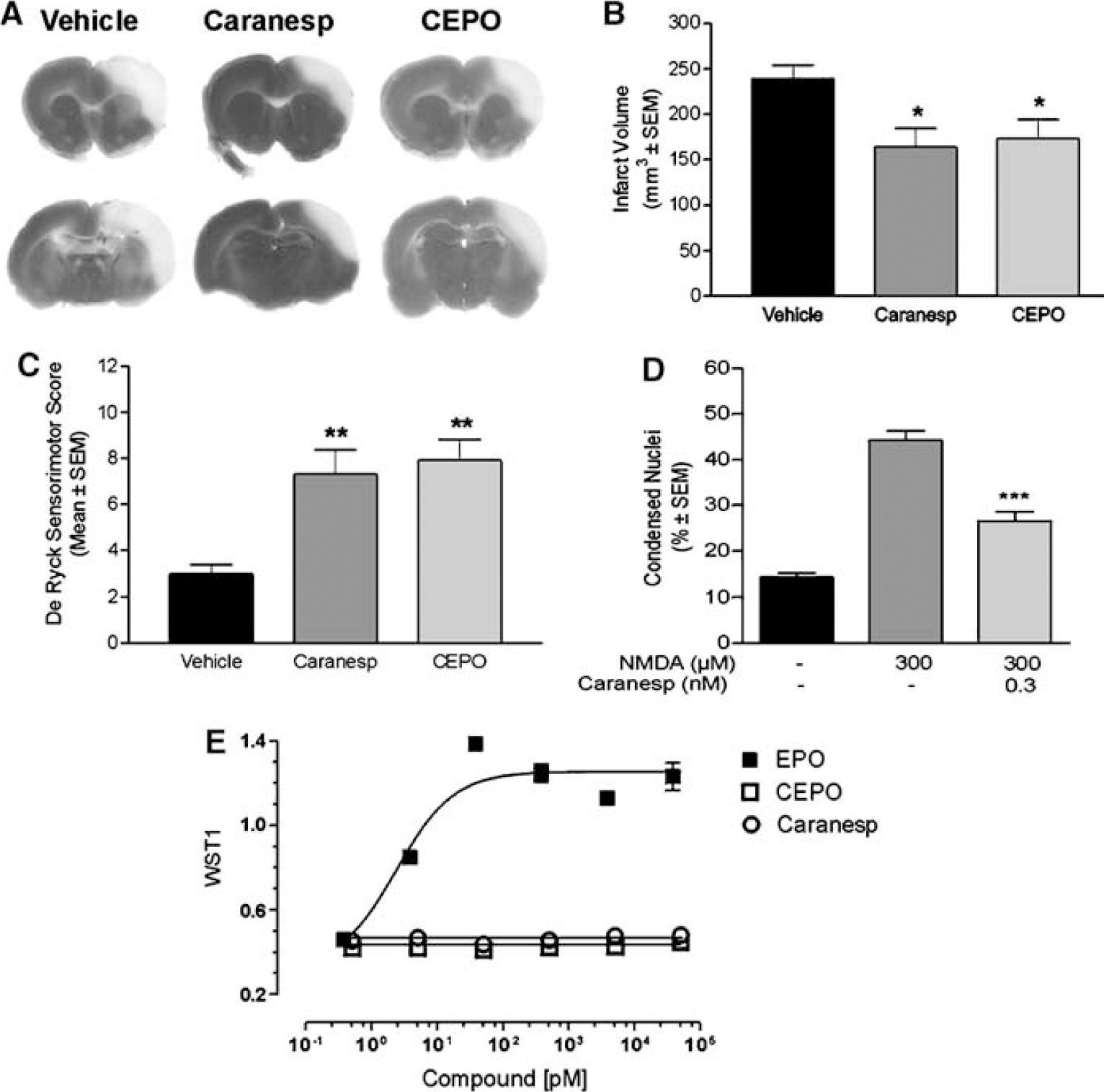
Neuroprotective properties of CEPO and Caranesp. (
Attenuation of Postischemic Perifocal Microglial Activation and Polymorphonuclear Leukocyte Infiltration by Carbamylerythropoietin Treatment
Triphenyl tetrazolium chloride-stained slices from vehicle- and CEPO-treated rats (1-day survival groups) were further processed for immunostaining for inflammatory markers including GFAP, Iba-1, and myeloperoxidase. At 1 day after occlusion, GFAP expression in the perifocal area was not significantly (P>0.05) increased compared with contralateral control area (data not shown). Glial fibrillary acidic protein expression was not affected by CEPO treatment (data not shown). Iba-1-positive microglia were observed in regions surrounding the ischemic core (Figure 2A) and numerous polymorphonuclear leukocytes stained for myeloperoxidase were seen in the ischemic core (Figure 2B). Very few CD68-positive macrophages were observed within the infarcted core (data not shown). Carbamylerythropoietin treatment was found to reduce perifocal microglial activation (P<0.05; Figure 2A) as well as polymorphonuclear leukocyte infiltration within the ischemic core (P<0.05; Figure 2B).
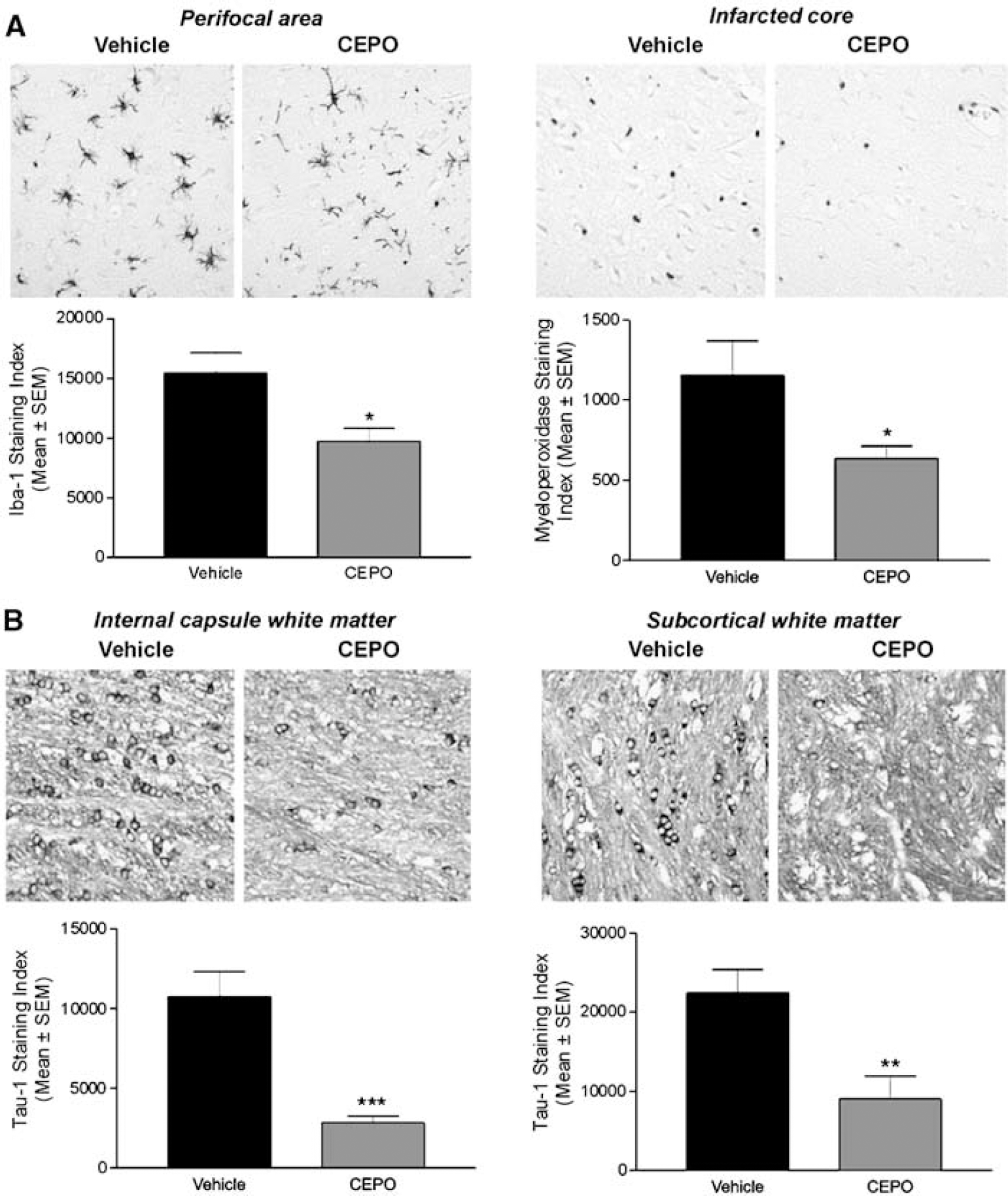
Reduction of postischemic microglial activation and polymorphonuclar leukocyte infiltration by CEPO. (
Reduction of Ischemic White Matter Ischemic Damage by Carbamylerythropoietin Treatment
Ischemic insult to oligodendrocytes was assessed by Tau-1 immunostaining as described previously (Valeriani et al, 2000). After 24 h occlusion, cells positive for Tau-1 with the characteristic morphology of oligodendrocytes, featuring a thin rim of cytoplasm and small soma, were present throughout ipsilateral gray and white matter, as described previously (Dewar and Dawson, 1995; Valeriani et al, 2000). In particular, Tau-1-positive oligodendrocytes were consistently observed in the ipsilateral internal capsule (Figure 2C) and subcortical white matter (Figure 2D). The extent of oligodendrocyte pathology in the internal capsule (Figure 2C) and subcortical white matter (Figure 2D) ipsilateral to the occluded MCA was significantly (P<0.001 and P<0.01, respectively) reduced in the CEPO-treated group compared with the vehicle-treated group.
Reduced Striatal Damage by Carbamylerythropoietin Treatment 7 Days After Occlusion
Fluoro-Jade B staining revealed extensive neurodegeneration in the ipsilateral but not the contralateral striatum, as assessed at 7 days postoperatively (Figure 3A). Intravenous treatment with CEPO (50 μg/kg) administered 3, 24, and 48 h after occlusion significantly (P<0.01) attenuated striatal (subcortical) damage (86% reduction from control; Figure 3B). Nevertheless, cortical infarct volume, as assessed by either direct (Figure 3C) or indirect (data not shown) calculation methods, was not significantly (P>0.05) reduced in rats treated with CEPO administered 3 h or 3, 24, and 48 h after occlusion compared with the vehicle group (Figure 3C).
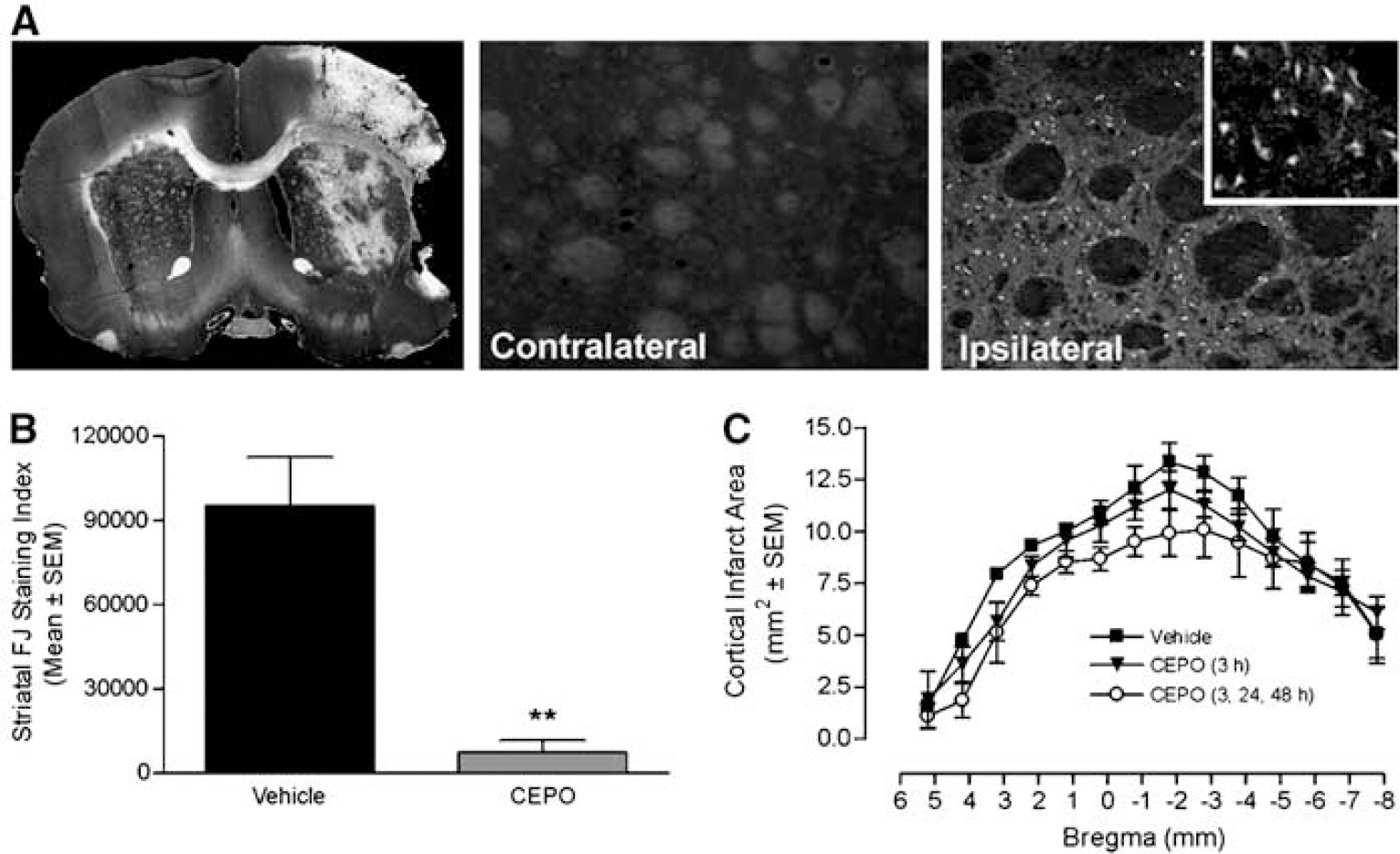
Protection against delayed striatal injury by CEPO. (
Improvement of Neurologic Outcome After Erythropoietin Treatment 7 Days After Occlusion
We further assessed the effect of EPO treatment on cortical infarct volume and behavioral outcome. Cortical infarct volume was not significantly (P>0.05) reduced in rats treated with EPO, as assessed by either direct (Figure 4A) or indirect (data not shown) calculation methods. Animals subjected to ischemia showed an increase in contralateral (left) limb placing deficits on the De Ryck sensorimotor test (Figure 4B) as well as in contralateral forelimb foot-faults on the Hernandez—Schallert foot-fault test (Figure 4C). No deficits in ipsilateral limb placing in animals with cerebral ischemia were observed (data not shown). Sham-operated animals had no impairment in limb behavior at any time periods and their score was 16 (data not shown). Treatment with EPO significantly improved neurologic outcome on the De Ryck (Figure 4B) and the foot-fault (Figure 4C) tests at days 1 and 7 after stroke.
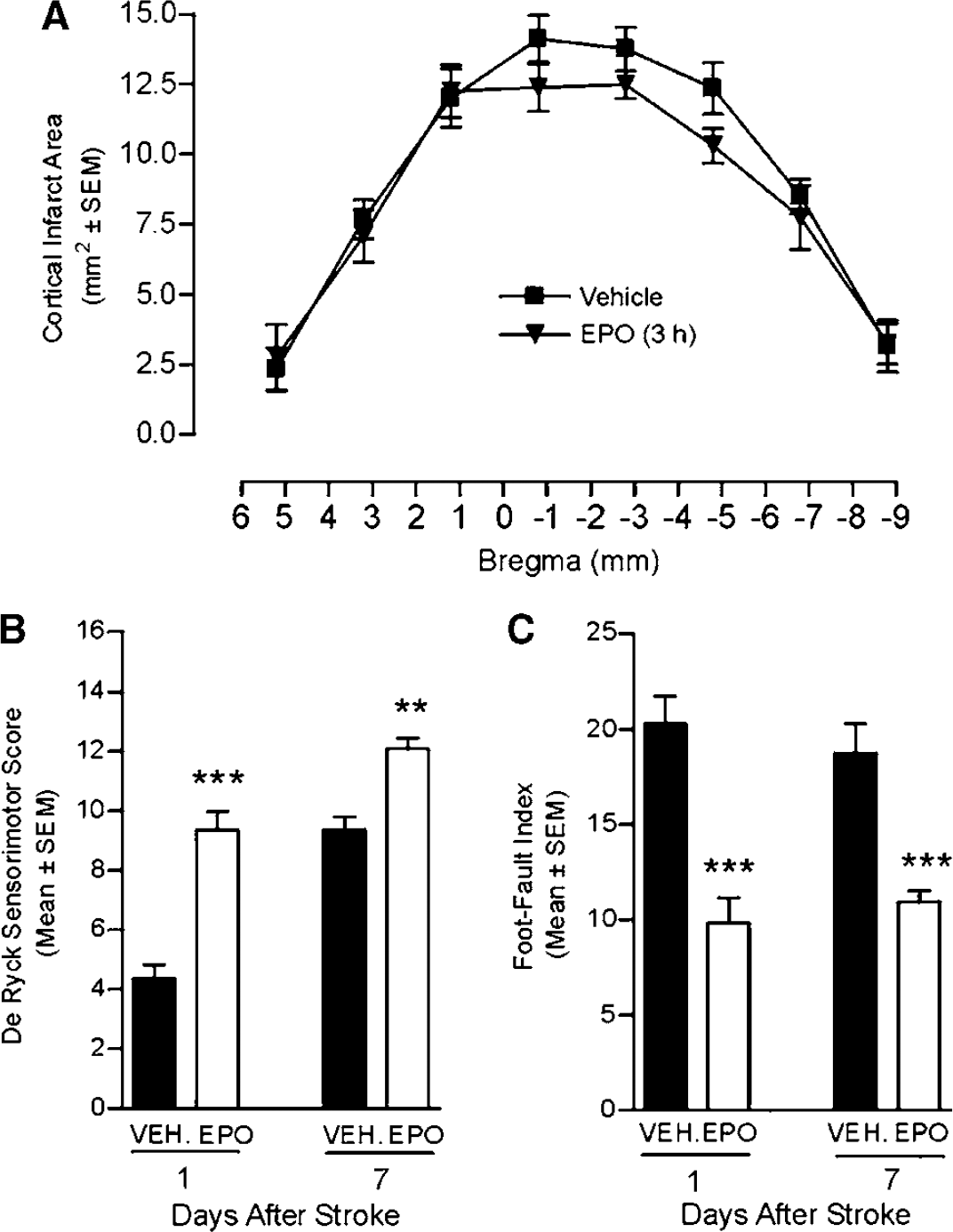
Rescue of neurologic function by EPO treatment. (
Rescue of Neurologic Function by Carbamylerythropoietin Treatment
Carbamylerythropoietin-treated rats showed a significant (P<0.05) enhancement in recovery of contralateral limbs at 28 and 50 days after occlusion (Figure 5A). Moreover, CEPO-treated rats had a significantly (P<0.05) better contralateral forelimb performance on the Hernandez—Schallert foot-fault test than the vehicle-treated animals within 7 days of treatment. This effect was sustained at every observational point throughout the survival period (Figure 5B). The two dosing regimes used (3 h versus 3, 24, and 48 h after occlusion) provided identical beneficial effects on functional deficits (Figures 5A and 5B).
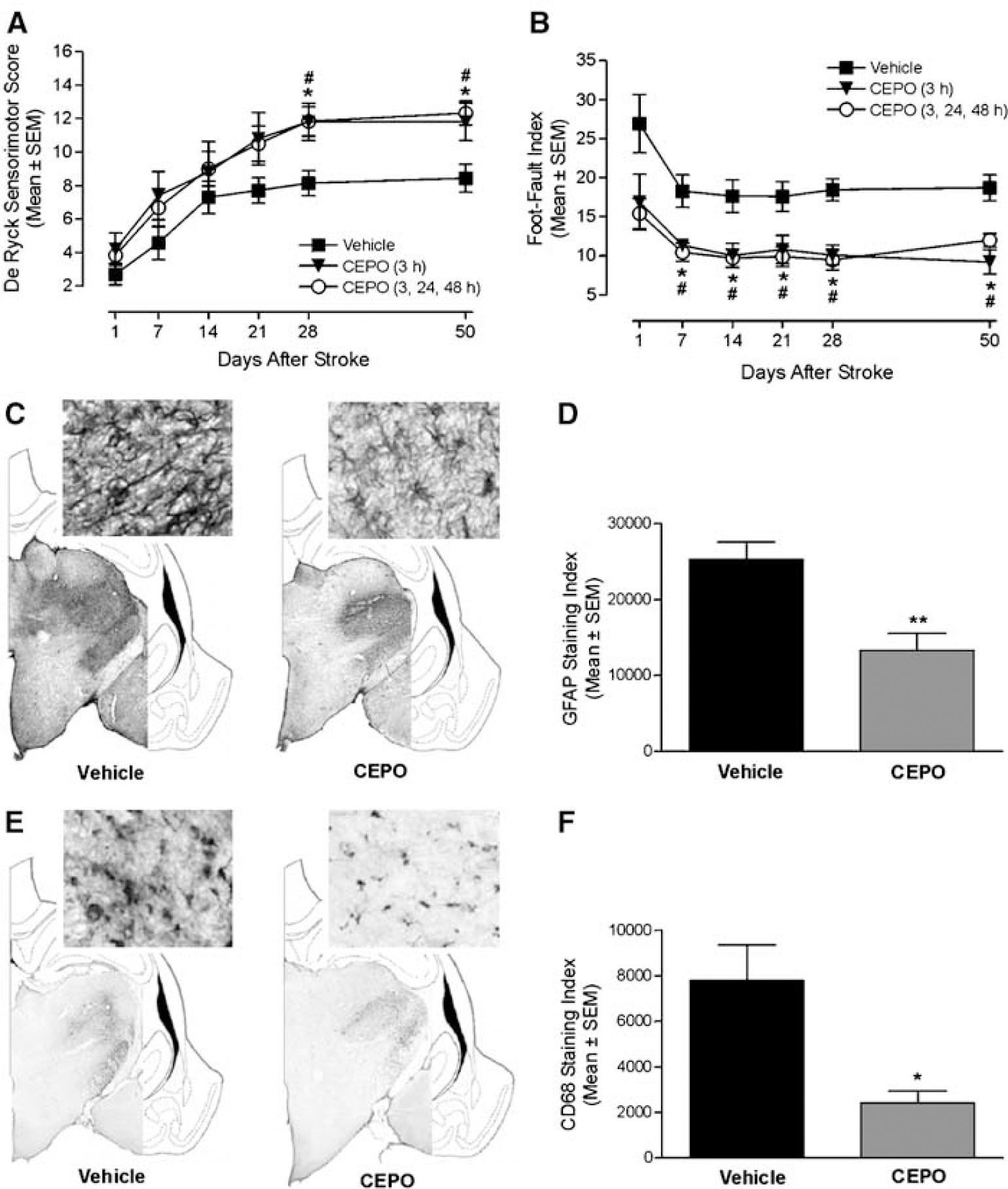
Facilitation of long-term recovery and attenuation of delayed thalamic glial activation by CEPO. (
Reduction of Delayed Postischemic Thalamic Gliosis by Carbamylerythropoietin
In addition to changes in the cortical infarct region, a dense homogeneous astrogliosis occurred in fiber tracts connecting cortex and thalamus and in the corresponding thalamic nuclei at 60 days after occlusion as assessed by GFAP immunostaining (Figure 5C). The extent of thalamic GFAP immunostaining significantly (P<0.05) correlated to behavioral impairment in the foot-fault test at day 50 after stroke, whereas the correlation analysis did not reach significance for the De Ryck sensorimotor test (data not shown). Carbamylerythropoietin treatment significantly (P<0.01) reduced GFAP density in the thalamic nuclei ipsilateral to the ischemic insult (Figures 5C and 5D). Glial fibrillary acidic protein-positive astrocytic cell bodies and processes in the ipsilateral thalamus were consistently thicker in vehicle-treated animals compared with CEPO-treated rats (Figure 5C). Microglia/macrophage activation was prominent within the ipsilateral thalamus, as assessed by CD68 immunostaining (Figure 5E). Microglia/macrophage activation significantly (P<0.001) correlated with functional deficit as measured in the foot-fault test 50 days after stroke. In contrast, the outcome of the De Ryck sensorimotor test did not significantly (P>0.05) correlate to microglia/macrophage activation. Carbamylerythropoietin treatment significantly (P<0.01) reduced CD68 immunostaining (Figures 5E and 5F), with CEPO-treated animals showing less retracted and thinner CD68-positive microglia/macrophages (Figure 5E).
Efficacy of Carbamylerythropoietin Treatment with Extended Time-to-Treatment Window
We further assessed whether animals treated with CEPO at a later time point after stoke, that is, 1 day, would exhibit improved functional recovery. Animals were administered CEPO intravenously at 1 and 2 days after occlusion. A significant (P<0.01) recovery of sensorimotor function was observed at 7 days after occlusion in CEPO-treated animals compared with vehicle-treated rats (Figure 6A). The effect was sustained at 14, 21, and 28 days postoperatively (P<0.01; Figure 6A). Additionally, CEPO-treated rats exhibited better contralateral forelimb performance on the Hernandez—Schallert foot-fault test than the vehicle-treated animals 7 (P<0.01) and 28 days (P<0.05) after stroke (Figure 6B).
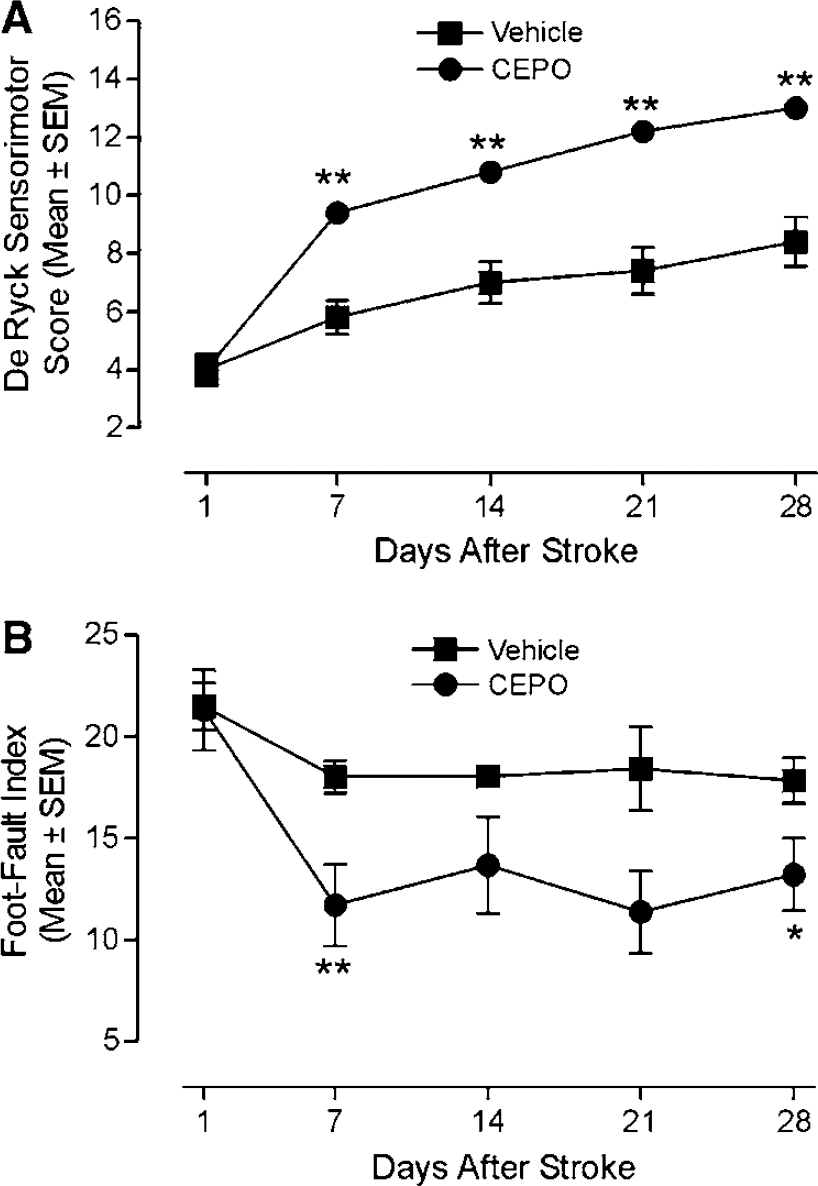
Improvement of postischemic motor function by CEPO with extended time-to-treatment window. (
Improvement of Functional Motor Recovery by Treatment with the Nonhematopoietic Mutant Erythropoietin S100E
Some mutants generated by site-directed mutagenesis of the human EPO encoding sequence lack affinity for the EPOR homodimer, but retain their tissue-protective property (Leist et al, 2004). We further tested the ability of the mutant EPO-S100E to improve neurologic function after stroke. When administered 3 h after ischemia, EPO-S100E significantly improved the sensorimotor score in the De Ryck test at 1 (P<0.01) and 14 days (P<0.05) postoperatively (Figure 7A). Moreover, EPO-S100E treatment resulted in reduced foot-faults compared with vehicle treatment at days 7 (P<0.05) and 14 (P<0.05) after stroke (Figure 7B). Erythropoietin-S100E had no hematopoietic bioactivity as measured in the UT7 EPO-dependent human leukemia cell line proliferation assay (Figure 7C).
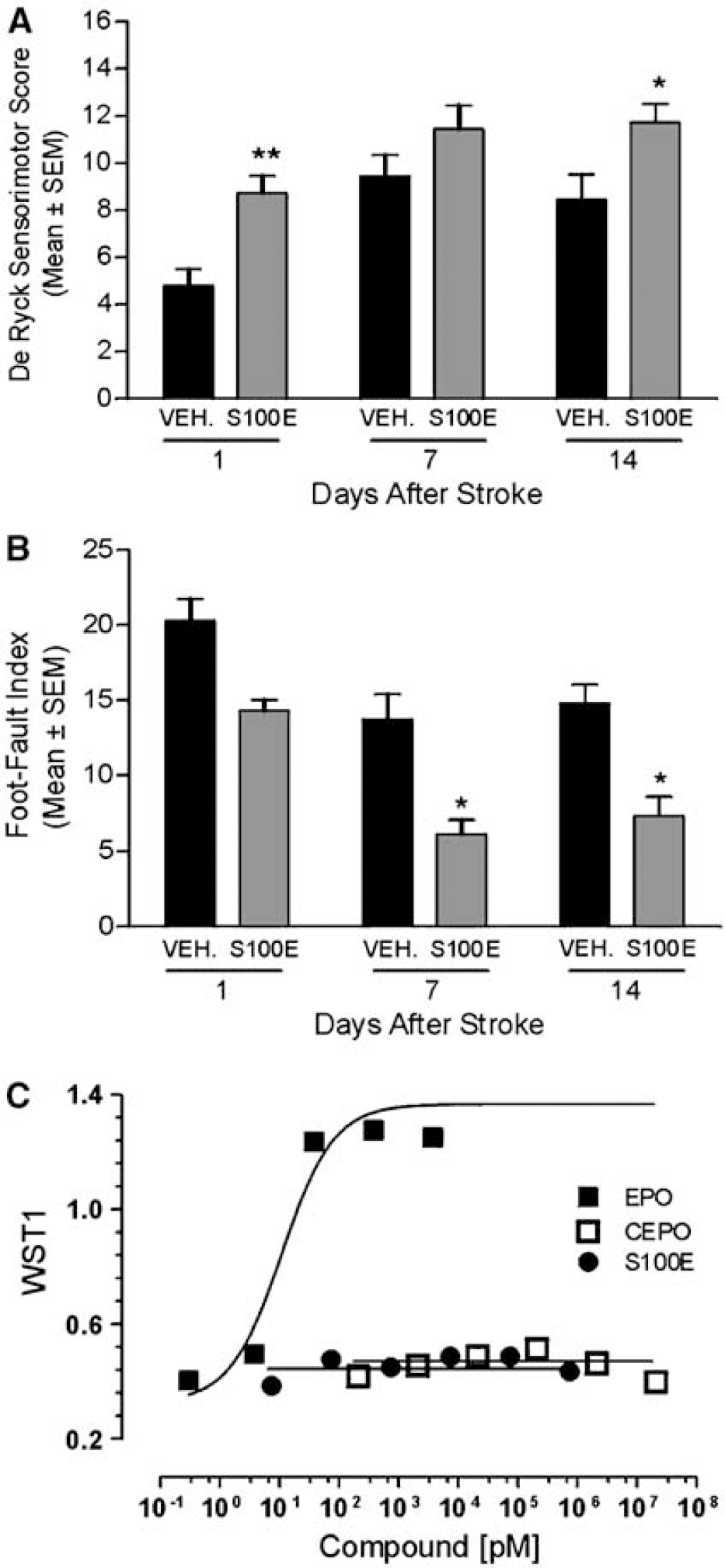
Improvement of motor function after stroke by the nonhematopoietic mutant EPO-S100E. (
Discussion
Our results show that postischemic intravenous treatment with CEPO elicits histologic protection and promotes recovery. CEPO treatment inhibited microglia activation and neutrophil infiltration, protected against ischemic white matter injury, reduced delayed striatal injury and thalamic glial activation, and ameliorated sensorimotor function. The time-to-treatment window with CEPO was extended to 24 h after stroke. Moreover, other nonerythropoietic derivatives such as Caranesp and the mutant EPO-S100E were also found to protect against ischemic damage and to improve postischemic neurologic function.
Carbamylerythropoietin has been described previously to decrease postischemic cortical infarct volume 1 day after occlusion (Leist et al, 2004). We further assessed whether the neuroprotective effect of CEPO was sustained 7 days after occlusion. We found that CEPO treatment had no effect on the apparent cortical infarct volume at 7 days postoperatively as assessed by toluidine blue staining. One potential explanation is that infarct volume measurements in rodents become inexact and influenced by many confounding factors (tissue shrinkage, glial scarring, cell infiltrates) at periods of more than 3 days after the ischemic insult. These effects may have obscured a potential tissue protective effect, and the issue needs to be addressed in the future by a longitudinal study based on magnetic resonance imaging technology. Another potential explanation is that this might reflect differential effects of CEPO on acute and delayed infarct expansion, as observed for other compounds (Tateishi et al, 2002). In our stroke model, the temporary occlusion of the contralateral CCA produces a penumbra surrounding the fixed MCA lesion (Zimmerman et al, 1995) and a wide ischemic penumbra is a prerequisite for the occurrence of a delayed infarct expansion (Hossmann, 1994). It can thus not be refuted that CEPO treatment might delay cortical infarct expansion without affecting final cortical infarct volume. In line with this view, we further observed that cortical infarct volume at day 7 after occlusion was not affected in rats treated with EPO. The separation between behavioral outcome and infarct size after EPO treatment has been described before (Renzi et al, 2003; Wang et al, 2004a), indicating that dosing regimen may be critical for long-term histologic effect of EPO and EPO derivatives. Nevertheless, EPO, CEPO, and related analogs all elicited robust and long-lasting improved functional recovery, suggesting a poor correlation between final cortical infarct volume and behavioral outcome in our model, as described before in other models. For instance, intravenous administration of a subneuroprotective dose of brain-derived neurotrophic factor was found to improve functional outcome without affecting final infarct size (Schabitz et al, 2004). Because infarct volume correlates only moderately with clinical outcome of stroke patients, it was suggested to constrain the use of infarct volume as a surrogate (or auxiliary) end point in ischemic stroke clinical trials (Saver et al, 1999).
Cerebral infarction induced by tandem permanent occlusion of the right MCA and ipsilateral CCA followed by temporary occlusion of the contralateral CCA has been shown to be confined to the cortical zone (Zimmerman et al, 1995). However, using Fluoro-Jade B, a polyanionic fluorescein derivative which sensitively and specifically binds to degenerating neurons (Schmued et al, 1997), we were able to show extensive neuronal degeneration in the ipsilateral but not the contralateral striatum 7 days after occlusion. Furthermore, we found that CEPO dramatically protected animals against postischemic delayed striatal damage. Delayed degeneration of fiber tracts in the striatum after focal ischemia, as evidenced using Fluoro-Jade staining has been documented before (Butler et al, 2002). Fluoro-Jade was proposed as a useful alternative to tedious (e.g., suppressed silver staining) or nonspecific staining methods (e.g., toluidine blue) for the evaluation of postischemic damage. Moreover, because Fluoro-Jade labelling is not specific to a particular mechanism of injury or type of cell death, the method broadens the opportunities to assess neuroprotective effect of compounds.
Recently, Belayev et al (2005) reported that treatment of experimental focal stroke with darboetin alfa, a novel EPO-derived protein, resulted in behavioral and histologic neuroprotection. Our observation that Caranesp is neuroprotective in vitro and in vivo (same degree as observed with CEPO) broadens the proof-of-concept for carbamylation of other EPO-derived erythropoiesis-stimulating agents. The carbamylation of EPO-derived agents thus may have potential utility in treating stroke in the clinical setting.
Focal ischemia elicits a profound inflammation response that is believed to contribute to cell death (Dirnagl et al, 1999). Although clinical trials undertaken with compounds inhibiting cellular inflammation have not shown efficacy so far, further development of strategies to modulate postischemic inflammatory events remain attractive (Legos and Barone, 2003; Dirnagl, 2004). In addition to its neuroprotective effect, EPO administration is also associated with decreased production of proinflammatory cytokines within the ischemic tissue after focal stroke (Villa et al, 2003). Similarly, tissue protection by CEPO has been correlated with reduced inflammatory mediators (interleukin-6 and membrane cofactor protein-1 levels) in ischemic tissue (Leist et al, 2004). We herein further show that CEPO inhibits perifocal microglial activation and reduces polymorphonuclear leukocyte infiltration within the ischemic core, possibly leading to decreased damage. However, the exact mechanisms underlying the antiinflammatory properties of CEPO treatment after stroke remain to be elucidated.
Few compounds have been examined for their ability to protect against ischemic white matter damage in preclinical models before reaching clinical trials. Nevertheless, functional recovery after an ischemic insult will be improved not only by protection of cortical gray matter but also protection of associated white matter (Dewar et al, 1999). A reason why stroke clinical trials have, so far, proved disappointing might reside in the inability of the tested drugs to protect white matter, specifically axons and oligodendrocytes, against ischemic damage (Dewar et al, 1999). Accordingly, ability of drugs to protect white matter damage was recently proposed as an additional read-out to the STAIR recommendations for preclinical evaluation of compounds before progression to clinical trials (Green et al, 2003). Our current observation that white matter damage, as reflected by Tau-1 immunostaining index, was reduced by CEPO treatment thus may have important clinical implications.
The present study further shows that CEPO is not only a neuroprotectant but also mediates functional recovery after stoke. The administration of CEPO 3 h after stroke improved functional neurobehavior, as assessed by sensorimotor and foot-fault placing tests. This beneficial effect was maximal within the first week after treatment and persisted throughout the 50-day survival period. The mutant EPO-S100E, which lacks affinity for the EPOR homodimer but retains its neuroprotective activity in vitro (Leist et al, 2004), improved postischemic behavioral outcome to a similar extent to that observed with CEPO treatment. This interesting observation further supports the existence of a second cognate receptor mediating neuroprotective activities of EPO. Several studies using various models of ischemic stroke have reported beneficial effect of EPO on postischemic behavioral outcome (Sadamoto et al, 1998; Wang et al, 2004a; Chang et al, 2005; Spandou et al, 2005). We herein provide further evidence that CEPO treatment ameliorates the functional recovery even if administered 24 h after stroke. Similarly, delayed administration of CEPO by up to 48 or 72 h after spinal cord injury resulted in enhanced functional recovery (Leist et al, 2004). This information is critical from the clinical point of view when treating patients in subacute or even long-term dosing regimes and distinguishes CEPO as a potential treatment of stroke from many other drugs that failed in clinical trials.
Functional improvement elicited by CEPO treatment after stroke could be caused by modulation of long-term tissue inflammation. The outcome of behavioral impairment in the foot-fault test significantly correlated with the extent of both microglia/macrophage and astrocyte activation in the ipsilateral thalamus. Moreover, we found that the beneficial effect of CEPO treatment on neurobehavioral effect is associated with reduced thalamic glial inflammation. Increased astrocytic and microglial reactivity is a common feature of neurologic disorders, but whether beneficial or adverse effect on neuronal function predominate is unclear. Recent studies have suggested that reactive astrocytes secrete neurotrophic factors at the lesion site in response to injury (Clarke et al, 2001), providing a permissive substrate for axonal regrowth (Ridet et al, 1997). However, at later stages, scar-type astrocytes may be an obstacle to axonal regrowth (Fawcett and Asher, 1999). Our observation that reduced glial activation at late stage (e.g., 2 months after stroke) after CEPO treatment is associated with diminished behavioral impairment corroborates recent findings by Badan et al (2003) demonstrating that increased postischemic glial reactivity in aged rats correlates with reduced functional recovery.
In the time frame of 60 days after stroke, long-term neurorestorative effects of CEPO may also be considered, such as angiogenesis and neurogenesis. Erythropoietin, in addition to a direct protective effect on neuronal cells during cerebral ischemia, has been reported to promote brain vessel growth in vivo and in vitro (Marti et al, 2000). Recently, Wang et al (2004a) showed that treatment with EPO significantly improved poststroke functional recovery along with increased density of cerebral microvessels and number of neuroblasts in the perifocal area. Erythropoietin receptor conditional knockdown were further found to lead to deficit in poststroke neurogenesis through impaired migration of neuroblasts to the peri-infarct cortex, suggesting that both EPO and EPOR are essential for migration of regenerating neurons during postinjury recovery (Tsai et al, 2006). Studies to evaluate the effect of CEPO and other nonerythoropoietic EPO derivatives on postischemic angiogenesis and neurogenesis are warranted.
In conclusion, our present findings add to the accumulating evidence that engineered derivatives of EPO that are tissue protective without stimulating erythropoiesis could have significant clinical application for the treatment of stroke.
Footnotes
Acknowledgements
The authors thank the excellent technical assistance of Kirsten Jørgensen, Pia Carstensen, and Bo Albrechtslund. Jacob Nielsen, Pekka Kallunki, Lone Helboe, and Thomas Sager are acknowledged for valuable discussion.