
Abstract
Osteopontin (OPN) is a secreted extracellular phosphoprotein involved in diverse biologic functions, including inflammation, cell migration, and antiapoptotic processes. Here we investigate the neuroprotective potential of OPN to reduce cell death using both in vitro and in vivo models of ischemia. We show that incubation of cortical neuron cultures with OPN protects against cell death from oxygen and glucose deprivation. The effect of OPN depends on the Arg–Gly–Asp (RGD)-containing motif as the protective effect of OPN in vitro was blocked by an RGD-containing hexapeptide, which prevents integrin receptors binding to their ligands. Osteopontin treatment of cortical neuron cultures caused an increase in Akt and p42/p44 MAPK phosphorylation, which is consistent with OPN-inducing neuroprotection via the activation of these protein kinases. Indeed, the protective effect of OPN was reduced by inhibiting the activation of Akt and p42/p44 MAPK using LY294002 and U0126, respectively. The protective effect of OPN was also blocked by the protein synthesis inhibitor cycloheximide, suggesting that the neuroprotective effect of OPN required new protein synthesis. Finally, intracerebral ventricular administration of OPN caused a marked reduction in infarct size after transient middle cerebral artery occlusion in a murine stroke model. These data suggest that OPN is a potent neuroprotectant against ischemic injury.
Introduction
Osteopontin (OPN) is a multifunctional cytokine and adhesion protein that has been linked to a variety of physiological and pathological processes (Lee et al, 2003; Mazzali et al, 2002; O'Regan and Berman, 2000). It is a secreted glycosylated phosphoprotein found in all body fluids and exists as an immobilized extracellular matrix protein in mineralized tissues (Mazzali et al, 2002). Stimuli known to regulate OPN expression include pro-inflammatory mediators, hormones, growth factors and mechanical stressors (reviewed in Mazzali et al, 2002). Osteopontin acts on a number of cell types (e.g. endothelial cells, fibroblasts, smooth muscle cells, macrophages) via an RGD (arginine–glycine–aspartic acid) motif that binds several distinct integrins αv (β1, β3 or β5) and (α4, α5, α8, or α9) β1, and various isoforms of CD44 (Denhardt et al, 2001; Malyankar et al, 2000). The outcome of the signal delivered by OPN depends on the particular receptors engaged, their cellular context and the specific signal transduction pathway used. Thus, the downstream effects of OPN signaling are complex and have made difficult the elucidation of precise biological roles for OPN. Nevertheless, recent studies using OPN knockout mice or the administration of recombinant OPN suggest that OPN may have important functions in cell survival, inflammation and the maintenance of tissue integrity in the setting of injury (Ellison et al, 1999; Mazzali et al, 2002).
Several observations suggest that OPN might be involved in the cellular response to ischemic injury (Ellison et al, 1999; Noiri et al, 1999; Persy et al, 1999; Wang et al, 1998). The net effect of OPN in conditions of ischemia has been difficult to ascertain because, in addition to effects that appear to promote cell survival, OPN has actions that are both proinflammatory and ant-iinflammatory. In renal ischemic injury, a protective role for OPN is supported by the finding that increased OPN expression occurs in the distal tubules, which contain cells that are resistant to ischemic injury (Padanilam et al, 1996; Persy et al, 1999). In contrast, proximal tubules, which are less tolerant to ischemic injury, show very little OPN expression after ischemia. Furthermore, a renoprotective effect of OPN is supported by the finding that OPN knockout mice display greater renal damage and dysfunction after acute renal ischemia (Persy et al, 1999).
The response to ischemic brain injury is also associated with increased OPN expression (Lee et al, 1999; Wang et al, 1998), which is observed primarily in microglia and macrophages found in the infarct and periinfarct regions. Osteopontin expression parallels the time course of macrophage infiltration into the infarct and expression of CD44 (an OPN receptor) (Wang et al, 1998). In addition, astrocyte expression patterns of another receptor for OPN, integrin αVβ3, parallel the temporal course of OPN upregulation in ischemia (Ellison et al, 1999, 1998). It has been proposed that OPN is a chemoattractant that recruits microglia, macrophages and astrocytes to assist the formation of a glial scar after ischemic injury (Ellison et al, 1999, 1998). This latter finding is in keeping with the previously recognized ability of OPN to act as a migration stimulus via integrin binding and downstream signaling (Liaw et al, 1995).
Osteopontin and integrin receptors have both also been ascribed functions in cell survival. Such functions might be attributed to effects that oppose apoptotic pathways (Liaw et al, 1995; Ophascharoensuk et al, 1999, 1998; Persy et al, 2003; Scatena et al, 1998) or enhance cell growth. Osteopontin acts through multiple intracellular pathways and downstream effectors, leading to diverse outcomes that could confer protection against ischemic injury. Examples of such pathways are: the reduction of nitric oxide synthase expression, activation of the phosphatidylinositol 3-kinase (PI3K) and NF-κB pathways, and increased expression of prosurvival genes (Das et al, 2003; Denhardt et al, 1995; Hwang et al, 1994; Khan et al, 2002). Integrin receptors promote cell survival in both neuronal and nonneuronal cells through antiapoptotic mechanisms. Recently, activation of integrin receptors (containing the β1 subunit) on neurons was shown to protect neurons against glutamate toxicity—an effect mediated through the protein kinase Akt and increased expression of the prosurvival protein Bcl-2 (Gary and Mattson, 2001; Gary et al, 2003).
We postulated that OPN may promote neuronal survival in the setting of injury because OPN is increased in the brain after ischemia and can act through β1 integrins, which are linked to cell survival pathways. Accordingly, we investigated the neuroprotective potential of OPN using in vitro and in vivo models of ischemia. We find OPN reduces cell damage after injurious ischemia in vitro and in vivo. Using modeled ischemia in vitro, we show that the neuroprotective effect of OPN involves the activation of the PI3K and P42/44 MAPK pathway. In an in vivo model of stroke, OPN administration provided a marked reduction in infarct size. These data suggest an important function for OPN in protection against ischemic damage.
Materials and methods
Neuronal Cell Culture
Cortical neuronal cultures were prepared from 1- to 3-day-old Sprague–Dawley rat pups using the method of Goslin (1998), as described previously (Stenzel-Poore et al, 2003). Briefly, cortices were dissected from 10 to 12 rat pups, dissociated with papain (Worthington Biochemicals, Lakewood, NJ, USA) and grown in Neurobasal-A/B27 media (Invitrogen, Carlsbad, CA, USA) for 7 to 10 days, when cultures consist of 80% to 90% neurons as assessed by NeuN versus GFAP staining (not shown). Cells were plated out at a density of 400,000 cells per coverslip for cell viability assays, or 5,000,000 cells per 10-cm culture dish (Primara; Becton Dickinson, San Jose, CA, USA) for Western blotting.
Oxygen–Glucose Deprivation
Oxygen–glucose deprivation (OGD) was performed by washing the cells with phosphate-buffered saline (PBS) (0.5 mmol/L CaCl2, 1.0 mmol/L MgCl2; pH 7.4) and placing culture dishes in an anaerobic chamber for 120 minutes (Forma Scientific, Marjetta, OH, USA) (85% N2, 5% H2, 10% CO2; 35°C). Anaerobic conditions in the chamber were monitored using Gaspack anaerobic indicator strips (Becton Dickinson, San Jose, CA, USA). Oxygen–glucose deprivation was terminated by removing cells from the chamber, replenishing with Neurobasal A media without B27 and placing them back in the normoxic incubator. Mouse recombinant OPN (Calbiochem, San Diego, CA, USA) (0.01 to 1.0 μg/mL) was dissolved in media and incubated with the cells for either 24 hours before 120-minute OGD, or 24 hours after 120-minute OGD. LY294002, U0126 (both 10 μmol/L) (Cell Signaling Technology, Beverly, MA, USA), cycloheximide (1.0 μmol/L) (Sigma, St Louis, MO, USA), and GRGDSP (10 nmol/L) (Sigma-Genosys, The Woodlands, TX, USA) were incubated with cells for 10 minutes before addition of OPN (0.1 μg/mL) to the cultures. Ac-DEVD-CHO (10 μmol/L) (Biomol Plymouth Meeting, PA, USA) was incubated with the cells for 24 hours before OGD.
Cell Viability Assay
For cell viability assays, coverslips containing cortical cells were incubated with propidium iodide (1.5 μg/mL) for 2 minutes, washed with PBS and fixed with 4% formaldehyde. Cells were permeabilized with 0.1% Triton X-100 and then mounted onto glass slides using Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs, Burlingame, CA, USA). Cell viability was determined as the ratio of propidium iodide-stained cells to the total number of DAPI-stained cells, visualized using a fluorescence microscope (Leica GMBH, Bannockburn, IL, USA).
Western Blotting
Western blotting was performed as described previously (Meller et al, 2002). Tissue samples were lysed in a nondenaturing buffer containing the protease inhibitors phenylmethylsulfonylfluride (100 μg/mL), aprotinin (1 μg/mL), leupeptin (1 μg/mL), pepstatin (1 μg/mL), NaF (50 mmol/L) and Na3VO4 (2 mmol/L). Protein concentration was determined by Bradford reagent spectrophotometrically at A595. Protein samples (50 μg) were denatured in a gel-loading buffer at 100°C for 5 minutes and then loaded onto 12% SDS-polyacrylamide gels. Proteins were transferred to polyvinylodene difluoride membranes and incubated with primary antibodies at 4°C overnight: anti-phospho-Akt, anti-phospho-p42/p44 MAPK, full-length and cleaved caspase 3 (Cell Signaling Technology, Beverly, MA, USA) and α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were incubated with anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (Cell Signaling Technology, Beverly, MA, USA) followed by chemiluminescence detection (NEN Life Science Products, Boston, MA, USA) and then exposed to Kodak film (Biomax). Images were captured using a Dage 72 camera and gel bands analyzed using gel-scanning integrated optical density software (Bioquant, Nashville, TN, USA).
Mice
C57Bl/6 mice (male, 8 to 10 weeks) were obtained from the National Cancer Institute for experiments with OPN administration. Osteopontin-deficient mice (OPN−/−) were obtained from Dr Lucy Liaw, Maine Medical Center Research Institute, Scarborough, Maine (Liaw et al, 1998). OPN−/− mice were backcrossed five generations onto C57Bl/6. Littermates were used as controls. All procedures met NIH guidelines with the approval of the Oregon Health & Science University Institutional Animal Care and Use Committee.
Surgery
Cerebral focal ischemia was induced by middle cerebral artery occlusion (MCAO) as published previously (Clark et al, 1997). Briefly, mice were anesthetized by halothane inhalation (4%/litre O2) and maintained with 1.5%/litre O2. The middle cerebral artery (MCA) was blocked by threading silicone-coated 7-0 monofilament nylon surgical suture through the external carotid to the internal carotid, and finally blocking its bifurcation into the MCA and anterior cerebral artery. The filament was maintained intraluminally for either 60 minutes (OPN-deficient mice) or 75 minutes, while the mice were maintained under anesthesia. The filament was then removed, thereby restoring blood flow. Cerebral blood flow (CBF) was monitored throughout the surgery by laser Doppler flowmetry (Periflow 5000; Perimed, Sweden). Band body temperature was monitored throughout surgery using a rectal thermometer (Hi-Lo Temp 8200 Patient Temperature Monitor, Sensortek Inc, Lake forest, CA, USA). After surgery, mice were kept on a thermal barrier pad with free access to soft food and were killed after either 24 or 96 hours. Body temperature of mice was measured at 3, 6, 24, 48, 72, and 96 hours after MCAO surgery. In a second group of animals, body temperature was measured at 1, 3, 6, 24, and 48 hours after administration of either artificial cerebrospinal fluid (aCSF) or OPN, but not subjected to MCAO. Blood glucose was measured (One Touch SureStep, Lifescan, Milpitas, CA, USA) 1 hour and 24 hours after i.c.v. administration of OPN or aCSF.
Intracerebral Ventricular Injections
Mouse recombinant OPN (Calbiochem, San Diego, CA, USA), human albumin (ZLB Bioplasma AG, Berne, Switzerland), or vehicle (aCSF) was injected into the left lateral ventricle (coordinates: 0 mm bregma, 1 mm lateral, and 2.5 mm ventral) using a 27-gauge needle. Injections (0.5 μL) of either aCSF, albumin (50 ng), or OPN (50 ng) were administered immediately before and immediately after surgery.
Infarct Measurement
Mice were deeply anesthetized with isoflurane, then perfused via the ascending aorta with saline containing 2 U/mL heparin at a flow rate of 9 mL/minute. Brains were rapidly removed, placed on a tissue slicer (Stoelting, Wood Dale, IL, USA) and covered with soft agarose. The olfactory bulb and cerebellum were removed and discarded. The remaining brain was sectioned into 1-mm slices beginning from the rostral end. To visualize the region of infarction, sections were placed in 1.5% 2,3,5-triphenyltetrazolium chloride (TTC) in 0.9% phosphate-buffered saline and stained for 15 minutes at 37°C (Bederson et al, 1986). After staining, the sections were transferred to 10% paraformaldehyde. Images of the sections were scanned, and the areas of the infarct and the ipsilateral hemisphere were measured using NIH image 1.62. The measurements were multiplied by the section thickness (1 mm) and then summed over the entire brain to yield volume measurements. The percent infarct was calculated as: (Infarct Volume)/(Ipsilateral Hemisphere Volume) × 100.
Statistical Analysis
Data are shown as means±s.d. of n determinations. Data from cell viability assays and in vivo stroke experiments were analyzed by one-way ANOVA followed by Bonferroni's multiple comparison test, using Graphpad Prism version 4.0 (Graphpad software, San Diego, CA, USA).
Results
Osteopontin is Protective in an In Vitro Model of Ischemia
We investigated whether OPN protects neurons from injury in an in vitro model of ischemia, OGD. Rat cortical neuron cultures (80% to 90% neurons) exposed to 120-minute OGD, showed ∼45% cell death (propidium iodide (PI)). The cell death was associated with an increase in TUNEL staining of nuclei and approximately 60% of PI-positive cells were TUNEL positive (Meller, unpublished observation). Some cells that were subjected to 120-minute OGD showed shrunken cell bodies and nuclei (Figure 1A). In contrast, other cells had ‘classical apoptotic features’, such as blebbing of nuclei and packaging of cellular contents (Figure 1A). Interestingly, cells with both types of morphology stain positive for propidium iodide (Figure 1A). We observed a robust increase in the cleavage of the cell death pathway-associated protein, caspase 3, after 120-minute OGD (Figure 1B). Finally, cell death after 120-minute OGD was reduced by the caspase 3 inhibitor Ac-DEVD-CHO (10 μmol/L) (Figure 1C). Hence, these data suggest that after 120-minute OGD, cells die by ‘apoptotic and necrotic’ cell death mechanisms.
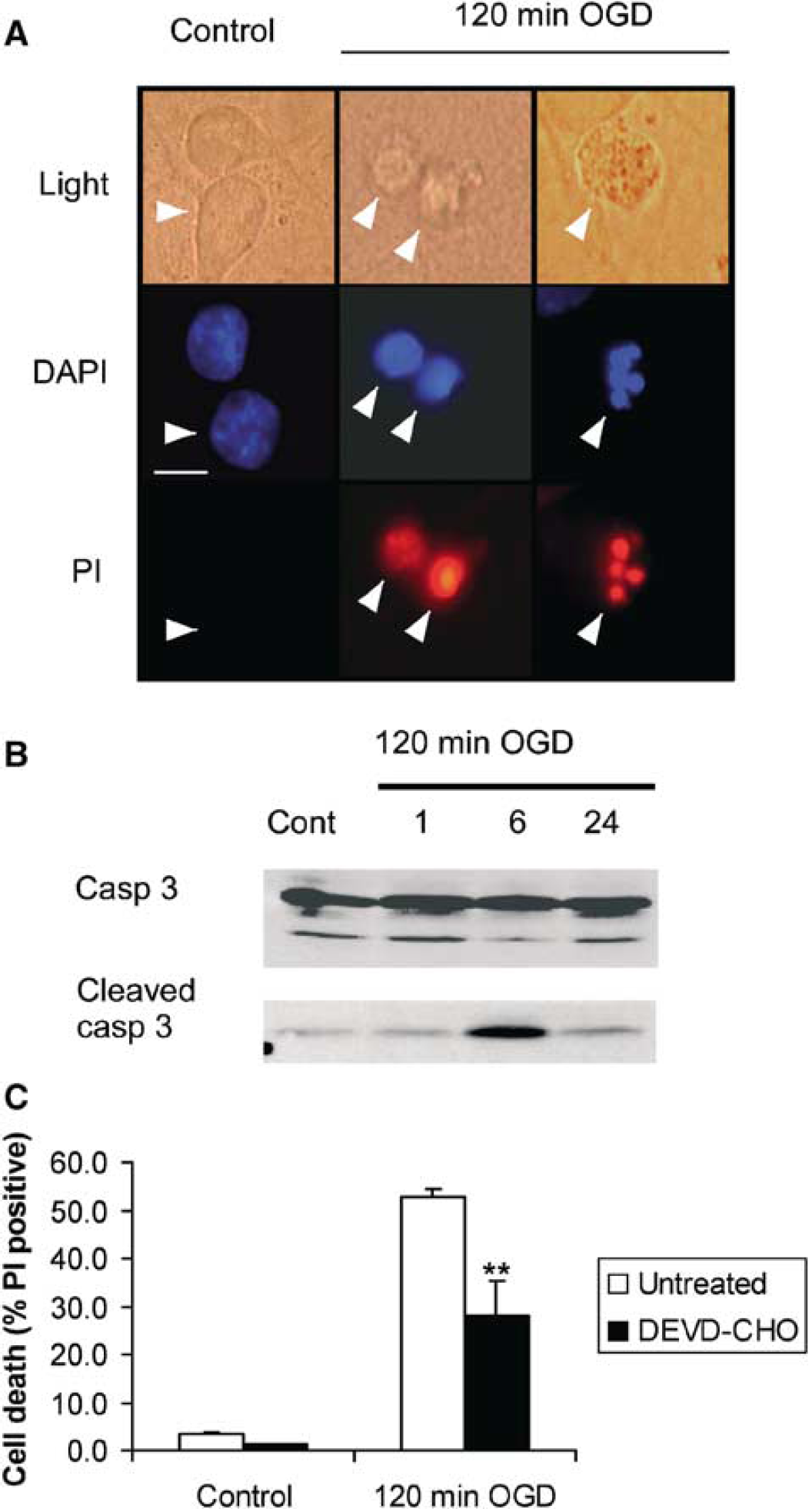
Effect of 120-minute OGD on neuronal cultures. (
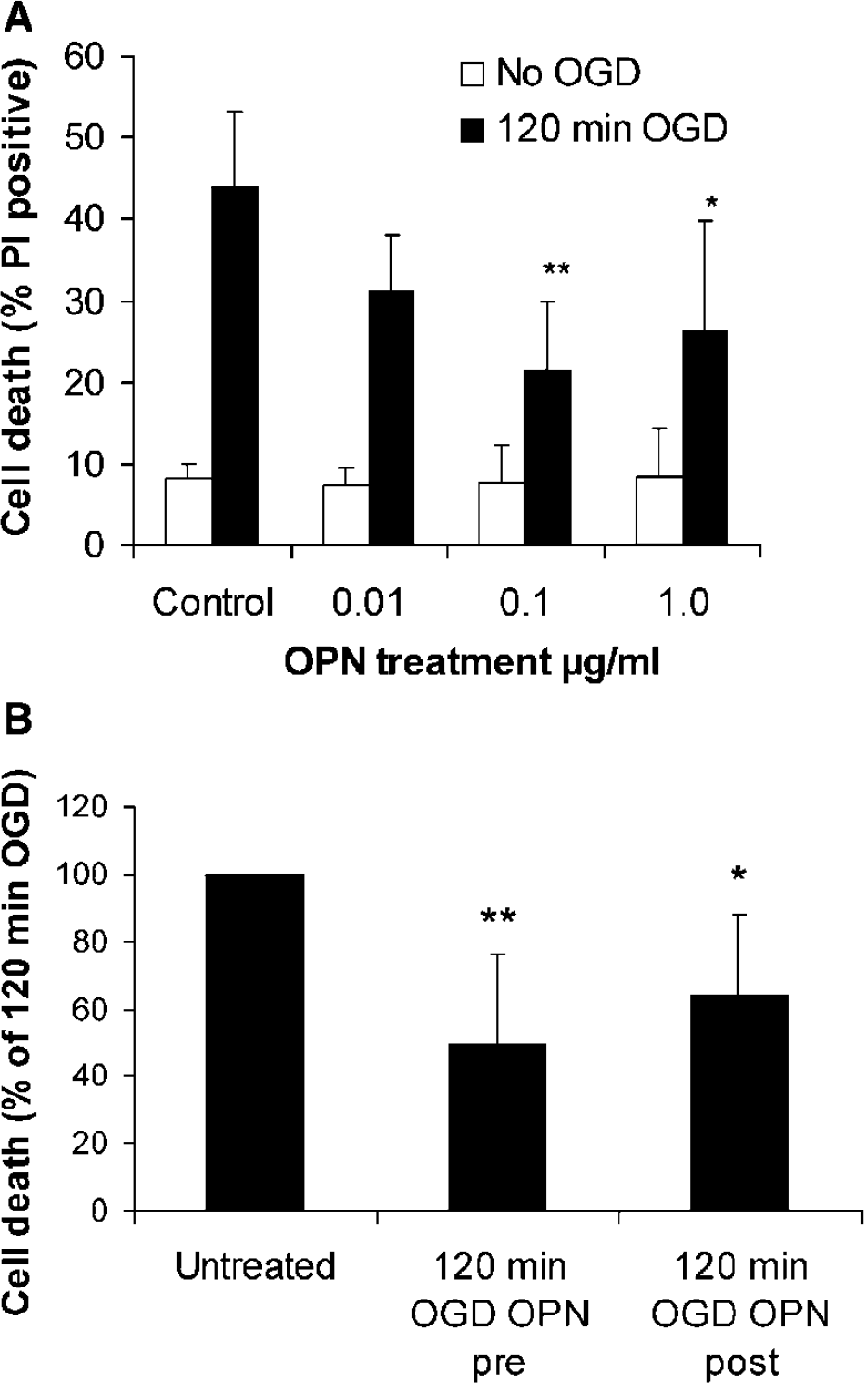
Pretreatment of cortical cultures with OPN (0.01 to 1.0 μg/mL) for 24 hours before exposure to OGD (120 minutes) significantly reduced cell death (∼2-fold). The effect of OPN was concentration-dependent and maximally protective at 0.1 μg/mL (Figure 2A). Thus, all further studies were performed with 0.1 μg/mL OPN. In the absence of OGD, there was no effect of OPN treatment on cell number or basal cell death. These results indicate that OPN treatment of neuronal cells before ischemic challenge provides marked protection against ischemic injury.
(
Osteopontin is Neuroprotective Administered After Ischemic Challenge In Vitro
Our data suggest that pretreatment with OPN prevents cell death from harmful ischemia. We next tested whether OPN confers neuroprotection if administered after exposure to ischemia, because such a finding would suggest an important potential for this molecule in therapeutic applications after stroke. Cortical cells were administered an ischemic challenge (120-minute OGD) followed by incubation with OPN for 24 hours. We found a reduction in cell death similar to that observed when cells were incubated with OPN before 120-minute OGD (Figure 2B). These data show that OPN treatment after ischemia is neuroprotective and suggest that OPN might have therapeutic benefits after an ischemic event.
The Neuroprotective Effect of Osteopontin is Blocked by an RGD-containing Peptide
To determine whether the neuroprotective effect of OPN in vitro involves an interaction with an integrin receptor, we tested whether a peptide antagonist that contains the RGD motif needed to bind to integrins could block the ability of OPN to reduce cell death due to ischemia. Cortical cultures were incubated with OPN in the presence of the competitor RGD-containing peptide, GRGDSP, which has been shown previously to competitively block OPN interactions with integrins (Guo et al, 2001). Cortical cultures treated with OPN in the presence of GRGDSP (10 nmol/L) before OGD showed reduced protection against cell death (Figure 3). Incubation of cortical cultures with the GRGDSP peptide (10 nmol/L) for 24 hours had no effect on cell viability compared with control untreated cells (5.2%±3.0% versus 4.9%±2% of PI-positive cells, respectively). Furthermore, GRGDSP peptide had no significant effect on 120-minute OGD-induced cell death (Figure 3). These data suggest that OPN exerts its neuroprotective effect through integrin receptors.
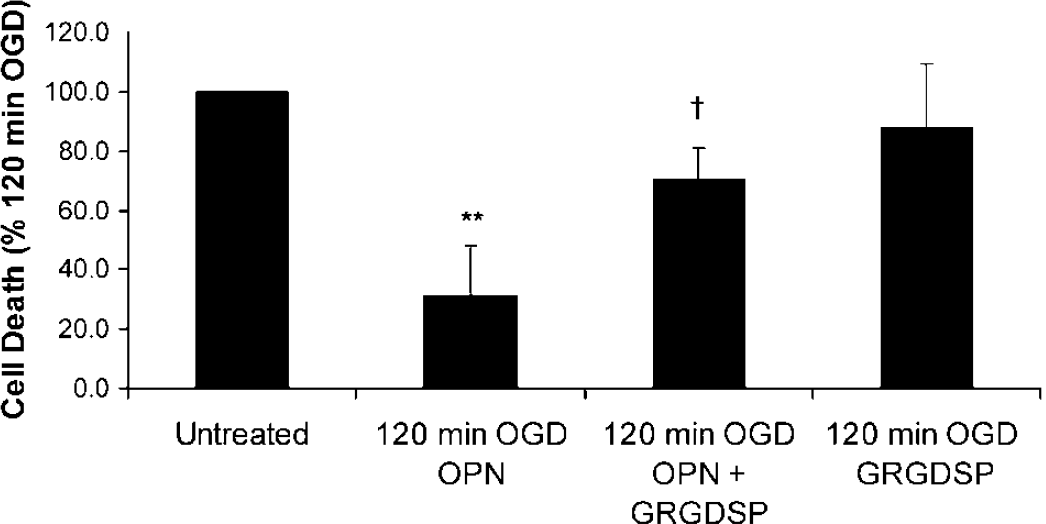
The neuroprotective effect of OPN is blocked by the RGD containing hexapeptide GRGDSP. Cells were treated with GRGDSP (10 nmol/L) for 10 minutes before addition of OPN (0.1 μg/mL) for 24 hours. Cells were then subjected to 120-minute OGD. Data shown are means±s.d. (n=4). ∗∗ denotes P<0.01 versus effect of 120-minute OGD and † denotes P<0.01 versus OPN-treated cells subjected to 120-minute OGD (one-way ANOVA with Bonferroni's post hoc test).
Activation of Akt and p42/p44 MAPK in Osteopontin-induced Neuroprotection
We tested whether the neuroprotective actions of OPN occur through PI3K, a known downstream mediator of OPN's effects in certain cell types, which, in turn, activates Akt (protein kinase B) (Das et al, 2003; Lin and Yang-Yen 2001; Zheng et al, 2000). Osteopontin treatment of cortical cells failed to protect against OGD-induced cell death in the presence of LY294002 (10 μmol/L), a PI3K inhibitor (Figure 4A). We also investigated the activation of the prosurvival protein kinase p42/p44 MAPK cascade after treatment with OPN. Osteopontin-induced neuroprotection was reversed in the presence of the Mek inhibitor U0126 (10 μmol/L) (Figure 4A), which blocks activation of p42/p44 MAPK. Incubation of cortical cells for 24 hours with either LY294002 (10 μmol/L) or U0126 (10 μmol/L) had no effect on basal cell viability or on cell death after 120-minute OGD (Figure 4A). These data suggest that OPN may mediate its neuroprotective effects via the activation of Akt or p42/p44 MAPK cascades.
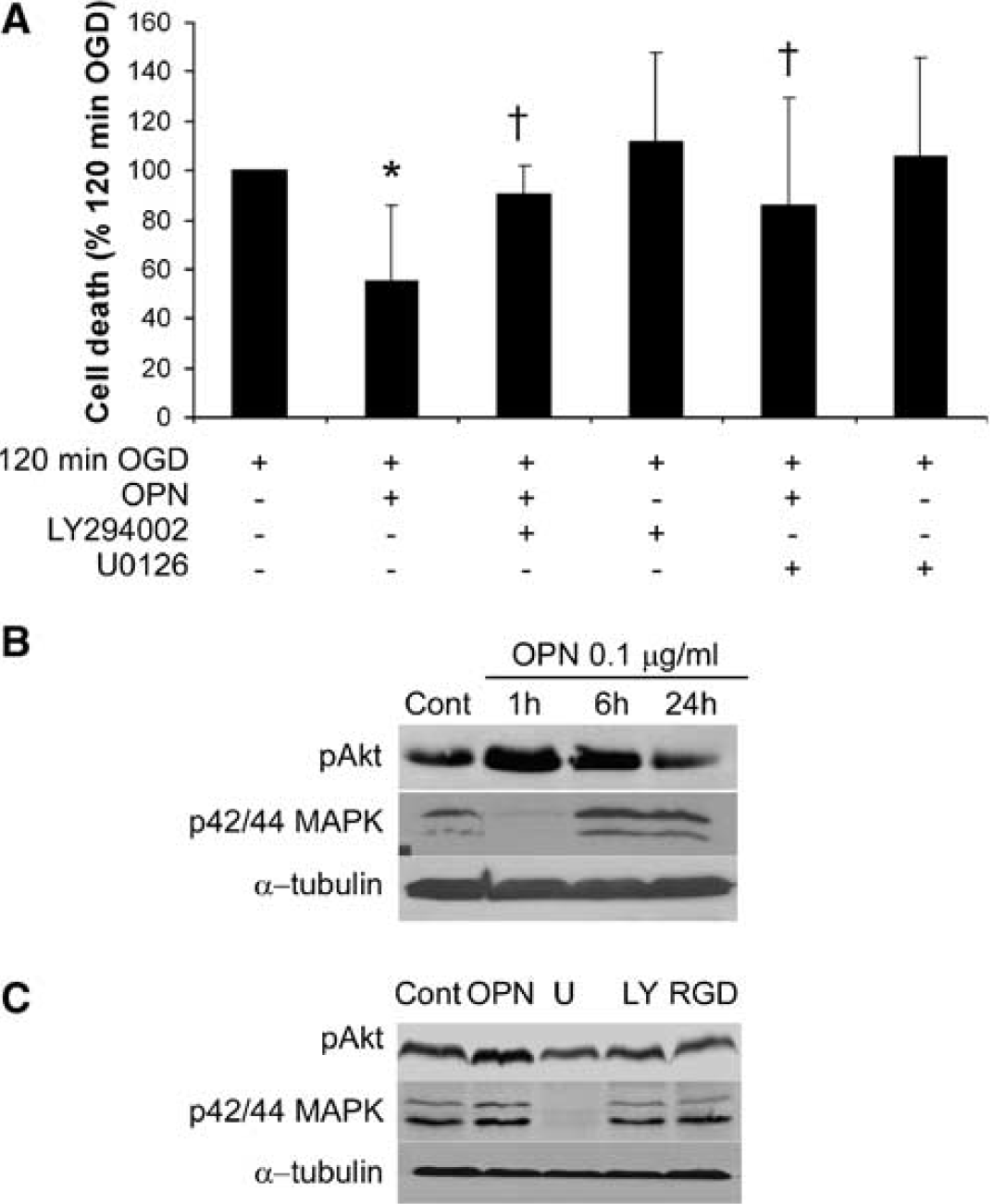
The neuroprotective effect of OPN is blocked by the PI3K inhibitor LY294002 and the Mek inhibitor U0126. (
To further test whether treatment of cells with OPN leads to activation of Akt or p42/p44 MAPK, we probed Western blots with antibodies specific for phosphorylated Akt (Ser 473) and phosphorylated p42/p44 MAPK. Cortical cultures treated with OPN display increased Akt phosphorylation (Figure 4B). Phosphorylation of p42/p44 MAPK by OPN was biphasic, with an initial reduction, followed by an increased phosphorylation (Figure 4B). Further, the effect of OPN on p42/44 MAPK phosphorylation was reduced by LY294002 and GRGDSP peptide and abolished by U0126, whereas the effect of OPN on Akt phosphorylation was reduced by LY294002 and GRGDSP peptide (Figure 4).
Finally, the neuroprotective effect of OPN against OGD-induced cell death was blocked when cortical cultures were treated with OPN in the presence of the protein synthesis inhibitor cycloheximide (data not shown). These data suggest that the neuroprotective effects of OPN require the activation of the Akt and p42/p44 MAPK cascades, as well as new protein synthesis.
Exogenously Administered Osteopontin Protects Against Focal Ischemic Injury
Given the neuroprotective effects of OPN that we observed in vitro, we tested whether the absence of endogenous OPN increased susceptibility to focal ischemia in a mouse model of MCAO. Mice with a targeted disruption in the OPN gene (Liaw et al, 1998) (OPN−/−, n=7) had similar infarct sizes compared with wild-type mice (OPN+/+, n=8). After 60-minute MCAO, OPN−/− mice had infarcts measuring 31.7±13% compared with wild-type infarcts, which measured 26.5%±9%; P=0.4 (Figure 5). These results indicate that the absence of endogenous OPN does not predispose the brain to greater injury in the setting of ischemia.
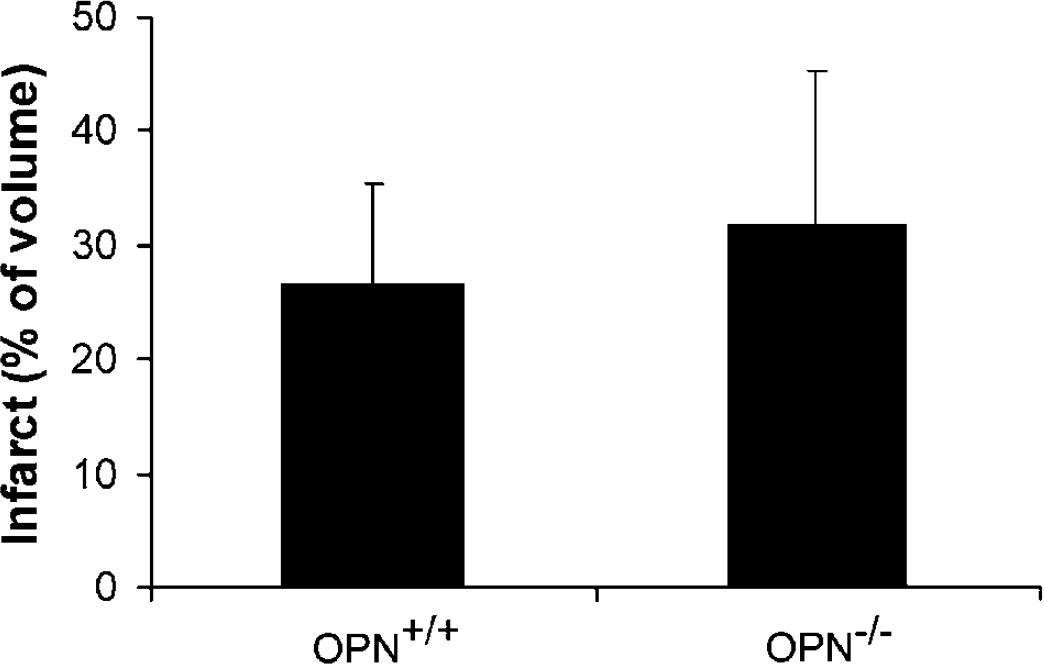
Osteopontin-deficient mice show similar infarct size to wild-type littermate controls. OPN−/− (n=7) and wild-type littermates (n=8) were subjected to 60-minute MCAO. Infarct volume was measured 24 hours after stroke using TTC staining. Data shown are mean±s.d.; P=0.4.
To determine if administration of exogenous OPN conferred protection against ischemic injury, we injected OPN (2 × 50 ng) or aCSF into the lateral ventricle just before and immediately after MCAO (75 minutes). Mice treated with OPN had significantly smaller infarcts (∼2-fold) compared with mice treated with aCSF at 24 and 96 hours after MCAO (Figure 6). As a control, we injected albumin, a protein with an equivalent weight to OPN, (2 × 50 ng) into the lateral ventricle before and after MCAO (75 minutes). Infarct size in mice injected with albumin did not differ from aCSF-treated animals (data not shown).
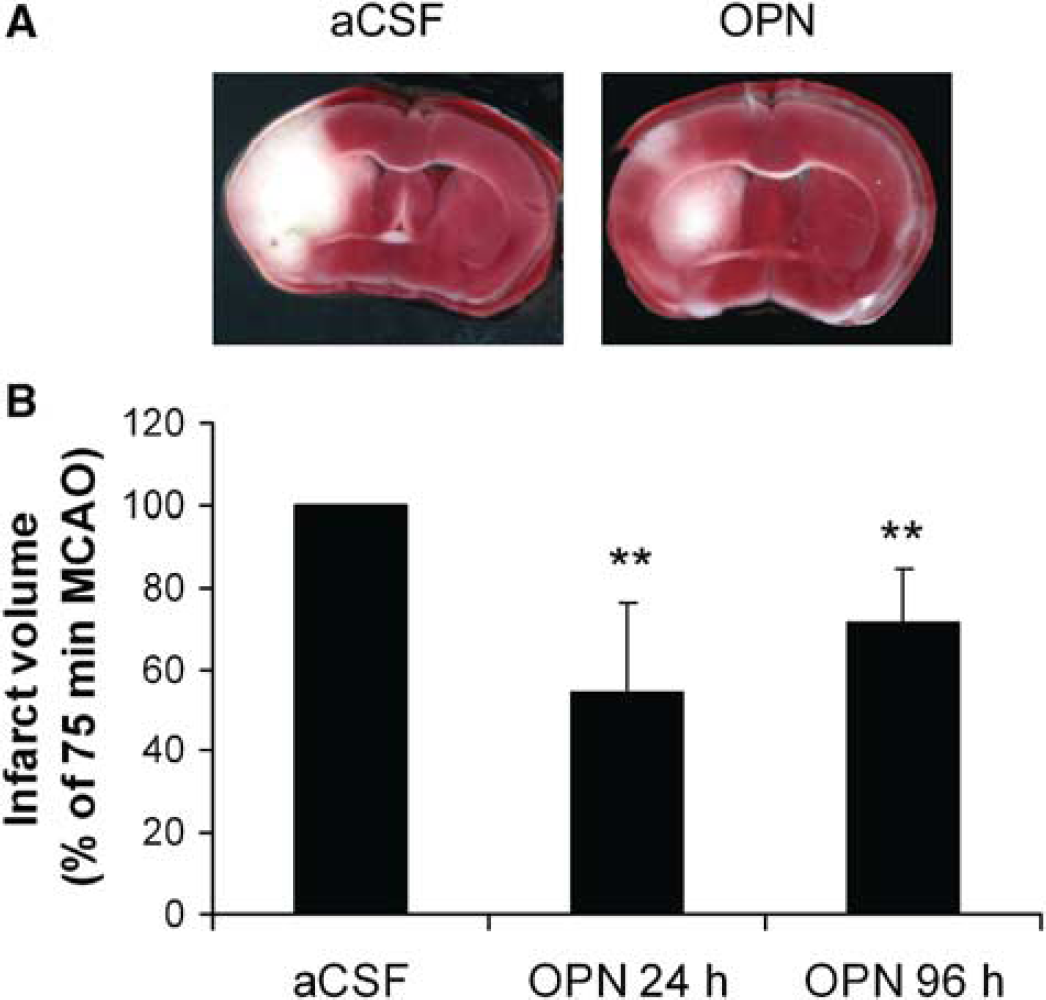
Osteopontin reduces infarct volume after stroke. Mice were administered OPN (2 × 50 ng) or CSF (n=15 and 8) and subjected to 75-minute MCAO. Infarct volume was measured 24 hours (OPN, n=9; aCSF, n=15) and 96 hours (OPN, n=8; aCSF, n=8) after stroke using TTC staining. (
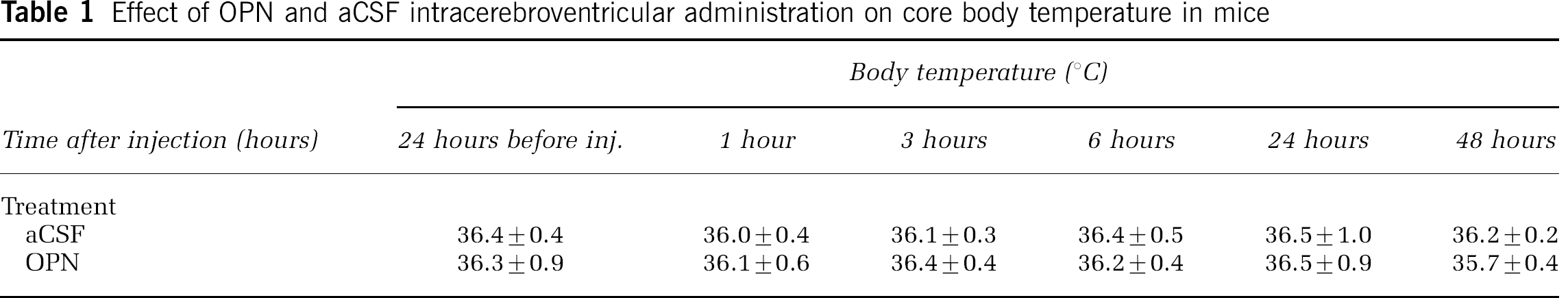
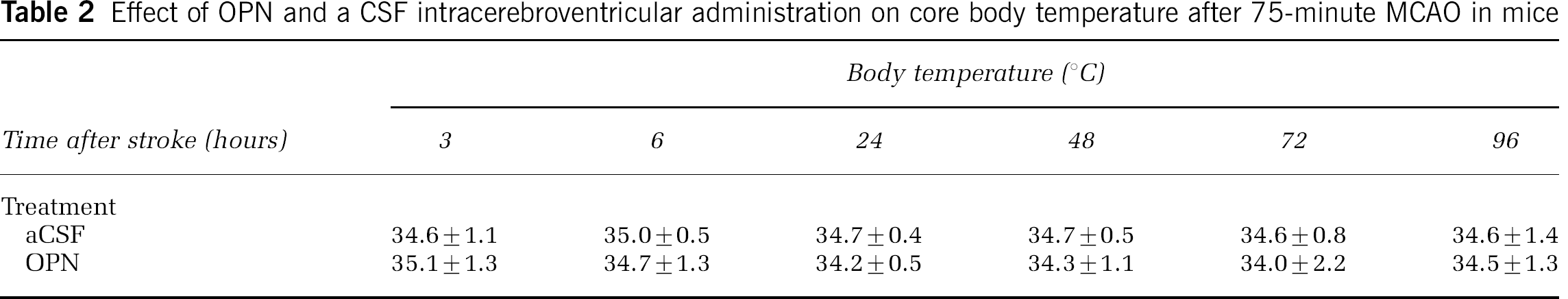
The effect of OPN and aCSF on body temperature was measured in the presence or absence of focal ischemia and found not to differ (Tables 1 and 2). Furthermore, no differences in blood glucose levels were observed after OPN administration (data not shown); hence, the protective effect of OPN is unlikely to be due to changes in body temperature and blood glucose availability. Thus, OPN administered at the time of stroke acts as a potent neuroprotectant in focal ischemia.
Effect of OPN and aCSF intracerebroventricular administration on core body temperature in mice
Effect of OPN and a CSF intracerebroventricular administration on core body temperature after 75-minute MCAO in mice
Discussion
There is a dearth of effective neuroprotective therapies for the treatment of stroke. Here, we have investigated the potential of OPN as a neuroprotective therapy for acute brain ischemia. Osteopontin reduced ischemic cell death significantly in in vitro and in vivo models of stroke. Osteopontin increased cell survival in vitro after ischemia when administered either before or after ischemia. Blockade of integrin receptors with the RGD-containing peptide inhibitor, GRGDSP, reduced the neuroprotective effect of OPN, which indicates that OPN's neuroprotective actions are likely to occur via binding to integrin receptors. We showed that the neuroprotective effect of OPN is mediated via the PI3K/Akt pathway as OPN increased Akt phosphorylation and OPN-induced neuroprotection was blocked by the PI3K inhibitor, LY294002. Osteopontin also activated the p42/44 MAPK cascade, and blocking this cascade reduces OPN-induced neuroprotection. Finally, while the absence of endogenous OPN in OPN-knockout mice does not increase the susceptibility of these mice to ischemic injury, OPN administered to mice intracerebrally induced marked protection against ischemic injury in a model of stroke. Collectively, these data suggest that OPN might be neuroprotective and thereby offer therapeutic benefit in the treatment of stroke injury.
We investigated the neuroprotective potential of OPN on the basis of its increased expression in the brain after ischemia (Stenzel-Poore et al, 2003; Wang et al, 1998). We hypothesized that increased expression of OPN after ischemia may affect neuronal survival in brain ischemia. Given that OPN contains an integrin receptor-binding motif (Denhardt et al, 2001; Hwang et al, 1994; Malyankar et al, 2000), it was conceivable that OPN acts on neurons via interactions with integrin receptors. It has been shown previously that activation of integrin receptors on neurons renders them tolerant to glutamate toxicity (Gary and Mattson, 2001). Thus, OPN may induce a neuroprotective effect via integrin receptor activation. In our studies, OPN was neuroprotective after injurious 120-minute OGD in cultured rat cortical cells. Osteopontin also reduced infarct volume after MCAO-induced ischemia in mice. Taken together, these data suggest OPN has therapeutic potential as a stroke therapy. A protective role of OPN in ischemia has been suggested in the kidney (Kleinman et al, 1995; Persy et al, 1999) because of its increased presence after ischemia and preferential localization with cells destined to survive (Padanilam et al, 1996). Further, the absence of endogenous OPN appears to increase sensitivity to ischemia, as OPN-knockout animals have greater kidney damage after renal ischemia (Noiri et al, 1999). Interestingly, in our studies reported here, the absence of OPN does not appear to predispose mice to greater injury in the CNS, as we found similar infarcts in wild-type and OPN-knockout mice after MCAO (Figure 5).
Previous reports suggest that OPN may activate multiple prosurvival mechanisms, including decreased induction of nitric oxide synthase (Denhardt et al, 1995), increased NF-κB activity (Das et al, 2003), and increased PI3K activation (Zheng et al, 2000). Here we show that the neuroprotective action of OPN depends, in part, on PI3K activation as neuroprotection is lost in the presence of LY294002. Blocking the activation of p42/p44 MAPK also reduced the neuroprotective effect of OPN. Subsequent phosphorylation and activation of Akt by PI3K or p42/p44 MAPK by Mek appears to be involved in mediating the neuroprotection seen in vitro. This finding is consistent with previous studies wherein activation of Akt and p42/p44 MAPK leads to increased phosphorylation of Bad, a protein whose pro-cell death functions are blocked by such phosphorylation events (Bonni et al, 1999). Furthermore, the transcription factor CREB, which regulates the expression of a number of pro-cell survival proteins, is also a substrate for these protein kinases (Bonni et al, 1999; Pugazhenthi et al, 2000). Given that OPN's protective effects are also blocked by a protein synthesis inhibitor, it would appear that the activation of multiple intracellular neuroprotective protein kinase cascades may result in the synthesis of new proteins resulting in reduced cell death after ischemia. Changes in gene expression after administration of OPN into the CNS are currently under further investigation.
Exogenous administration of OPN directly into the brain has a marked neuroprotective effect in ischemia. These results offer promise in the identification and development of neuroprotectants in stroke. Therapeutic use can be limited for proteins that must be delivered directly into the brain. Efficient delivery to the brain in animal models and humans has been achieved with a number of proteins using intranasal administration (Liu et al, 2001). This approach has already been used successfully with a limited number of proteins and peptides in the treatment of CNS diseases or injury, including stroke (Liu et al, 2001), and may allow the rapid administration of OPN into the brain.
Further drug development is needed in the case of OPN. Pursuit of an OPN-mimetic with the similar biological properties of OPN, but improved potency and pharmacokinetics, would be a viable therapeutic agent for further development. Interestingly, the biological effects of OPN have been mimicked by a 20-amino-acid peptide containing the RGD integrin receptor-interacting motif of OPN (Hwang et al, 1994). Hence, OPN or short OPN analogues may lead the way to the design of a drug as an effective therapy for use in stroke. As such, further investigation of the therapeutic potential of OPN or an OPN mimetic is warranted.
Footnotes
Acknowledgements
This work was supported by the Medical Research Foundation of Oregon (RM) and by NIH grants NS24728 (RPS), NS35965 (RPS, MS-P) and NS046827 (MS-P). The work reported in this manuscript was also supported in part by Virogenomics, Inc., a company that may have a commercial interest in the results of this research. OHSU and investigators, MS-P and RPS, have a financial interest in Virogenomics. This potential conflict of interest has been reviewed and managed by the OHSU Conflict of Interest in Research Committee.