
Abstract
The serine-threonine kinase, Akt, plays an important role in the cell survival signaling pathway. A proline-rich Akt substrate, PRAS40, has been characterized, and an increase in phospho-PRAS40 (pPRAS40) is neuroprotective after transient focal cerebral ischemia. However, the involvement of PRAS40 in the cell death/survival pathway after spinal cord injury (SCI) is unclear. Liposome-mediated PRAS40 transfection was performed to study whether overexpression of pPRAS40 is neuroprotective. We further examined the expression of pPRAS40 after SCI by immunohistochemistry and Western blot using copper/zinc-superoxide dismutase (SOD1) transgenic (Tg) rats and wild-type (Wt) littermates. We then examined the relationship between PRAS40 and Akt by injection of LY294002, a phosphatidylinositol 3-kinase (PI3K) pathway inhibitor, or Akt inhibitor IV, a compound that inhibits Akt activation after SCI. Our data demonstrated that increased pPRAS40 resulted in survival of more motor neurons compared with control complementary DNA transfection. Phosphorylated PRAS40 increased in the Wt rats after SCI, whereas there was a greater and prolonged increase in the SOD1 Tg rats. Coimmunoprecipitation showed that binding of pPRAS40 with 14-3-3 increased 1 day after SCI in the Wt rats, whereas there was a significant increase in the Tg rats. The inhibitor studies showed that phospho-Akt and pPRAS40 were decreased after injection of LY294002 or Akt inhibitor IV. We conclude that an increase in pPRAS40 by transfection after SCI results in survival of motor neurons, and overexpression of SOD1 in the Tg rats results in an increase in endogenous pPRAS40 and a decrease in motor neuron death through the PI3K/Akt pathway.
Introduction
The serine-threonine kinase, Akt, plays an important role in the cell death/survival pathway (Franke et al, 1997). Akt is activated through phosphatidylinositol 3-kinase (PI3K) and inhibits apoptosis by phosphorylating several different substrates: Bad (Datta et al, 1997), glycogen synthase kinase-3 (Cross et al, 1995), forkhead transcription factors (Brunet et al, 2001), and caspase-9 (Cardone et al, 1998). A temporal increase in Akt phosphorylation was proven to be neuroprotective after cerebral ischemia (Noshita et al, 2001; Ouyang et al, 1999), traumatic brain injury (Noshita et al, 2002), and spinal cord injury (SCI) (Yu et al, 2005). The proline-rich Akt substrate, PRAS40, has been characterized and found to be phosphorylated by activated Akt (Kovacina et al, 2003). Increased expression of phospho-PRAS40 (pPRAS40) by liposome-mediated gene transfer of PRAS40 complementary DNA (cDNA) after focal cerebral ischemia resulted in the inhibition of apoptotic neuronal cell death (Saito et al, 2004). Increased pPRAS40 has also been seen in premalignant and malignant breast and lung cancer cell lines, suggesting its contribution to cell survival during cancer progression (Huang and Porter, 2005). However, the relationship between PRAS40 and the PI3K/Akt pathway and their roles in the cell death/survival pathway after SCI are not clear.
Cationic liposome-mediated gene transfer is non-immunogenic and nontoxic at therapeutic doses and has been proven to be safe and efficient (Cao et al, 2002; Zou et al, 1999). Liposomes condense DNA and carry it into cells by membrane fusion and endocytosis (Cao et al, 2002; Zou et al, 1999). The development of a more efficient liposome formulation makes liposome-mediated gene transfer promising in treating central nervous system injury (Yang et al, 1997). Dimethylaminoethane-carbamoylcholesterol liposome has been approved by the FDA for a clinical trial for cancer immunotherapy (Zou et al, 1999). N,N,N-Trimethyl-2-3-bis ((1-oxo-9-octa-decenyl)oxy)-(Z,Z)-1-propanaminium methyl sulfate) (DOTAP) liposomes have been used to deliver neurotrophic genes into the brain by a single intrathecal injection in several in vivo studies and have proven to be effective (Cao et al, 2002; Saito et al, 2004). In this study, we used liposome-mediated gene transfer to examine whether increased phosphorylation of PRAS40 is neuroprotective after SCI.
Reactive oxygen species play important roles in the pathogenesis of central nervous system injury. Human copper/zinc-superoxide dismutase (SOD1) is a crucial endogenous enzyme present in both the cytosol and the mitochondria (Higgins et al, 2002; Yu et al, 2006) and is responsible for eliminating superoxide. Overexpression of SOD1 in rodents has been shown to protect neurons from death after focal cerebral ischemia (Noshita et al, 2001), global ischemia (Chan et al, 1998), and SCI (Sugawara et al, 2002). Apoptosis plays an important role in neuronal loss after central nervous system injury (Crowe et al, 1997; Liu et al, 1997; Yakovlev et al, 2001). Oligodendrocytes, neurons, and glia undergo apoptosis in the spinal cord after SCI (Crowe et al, 1997; Liu et al, 1997; Yong et al, 1998). Previous studies have provided evidence that caspase cascades such as caspase-3 and caspase-9, and cytochrome c and Bad are involved in apoptosis after SCI (Springer et al, 1999; Sugawara et al, 2002). We have developed a SCI model with compression of the spinal cord at T13 using a vascular clip; it produces selective and delayed motor neuron death without astrocyte damage, which is milder than other compression or contusion models (Sugawara et al, 2002). Using this model, we have shown that overexpression of SOD1 in transgenic (Tg) rats prevents neuronal apoptosis after SCI by increasing Akt/Bad signaling (Yu et al, 2005). Overexpression of SOD1 in Tg mice increases PRAS phosphorylation after stroke, compared with wild-type (Wt) mice (Saito et al, 2006). In this study, we further examined the expression of endogenous pPRAS40 after SCI using SOD1 Tg rats.
Materials and methods
Copper/Zinc-Superoxide Dismutase Transgenic Rats
Heterozygous SOD1 Tg rats with a Sprague-Dawley background, carrying human SOD1 genes, were derived from the founder stock and further bred with Wt Sprague-Dawley rats to generate heterozygous rats as described previously by our group (Chan et al, 1998). The phenotype of the SOD1 Tg rats was identified by isoelectric focusing gel electrophoresis as described (Chan et al, 1998). There were no observable phenotypic differences in brain vasculature between the Tg and Wt rats (Chan et al, 1998).
Surgery
All animals were treated in accordance with Stanford University guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Adult female SOD1 Tg rats (250 to 300 g) and their Wt littermates were used in this experiment. The animals were anesthetized with 2.0% isoflurane in 70% N2O and 30% O2 using a face mask. Lumbar enlargement was exposed by a partial laminectomy of the T13 vertebra, and a vascular clip (closing force 15 g; Ohwa Tsusho, Tokyo, Japan) was applied extradurally for 5 secs according to our previous study (Sugawara et al, 2002). The rectal temperature was controlled at 37.0±0.5°C during surgery with a home-othermic blanket.
Drug Injection
To investigate the role of the PI3K pathway after SCI, we administered a PI3K inhibitor, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002; Cell Signaling Technology, Beverly, MA, USA), as described previously (Noshita et al, 2001). LY294002 was dissolved in dimethyl sulfoxide (DMSO) to make a stock solution of 100 mmol/L, and further diluted to 25 mmol/L in filtered phosphate-buffered saline (PBS, pH 7.4) before use. A partial laminectomy was performed and lumbar enlargement was exposed. Two microliters of 25 mmol/L LY294002 and the same amount of the vehicle (25% DMSO in PBS) were injected intrathecally 2 h before SCI. Animals were killed 1, 4, and 24 h after surgery and the spinal cords were collected for Western-blot analysis. Another two groups of animals were killed 3 days after SCI for an apoptotic cell death assay.
To investigate the role of Akt phosphorylation on PRAS40 activation, we administered Akt inhibitor IV (Calbiochem, San Diego, CA, USA), a cell-permeable benzimidazole compound that inhibits Akt phosphorylation by targeting the adenosine triphosphate binding site of a kinase upstream of Akt, but downstream of PI3K (Kau et al, 2003). Akt inhibitor IV was dissolved in DMSO to make a stock solution of 10 mmol/L, and further diluted to 100 μmol/L in filtered PBS (pH 7.4) before use. A partial laminectomy was performed and lumbar enlargement was exposed. Four microliters of 100 μmol/L Akt inhibitor IV and the same amount of the vehicle (10% DMSO in PBS) were injected intrathecally 2 h before SCI. Animals were killed 1, 4, and 24 h after surgery and the spinal cords were collected for Western-blot analysis.
Liposome-Mediated PRAS40 Complementary DNA Transfection
Myc-tagged plasmid of PRAS40 was a kind gift from Dr Richard A Roth and was generated by subcloning the entire coding region of human cDNA (GenBank accession number BC007416, IMAGE clone number 2988648) into a pcDNA3.1(+) vector (Invitrogen, Carlsbad, CA, USA) (Kovacina et al, 2003). Both the PRAS40 plasmid and plasmid pcDNA3.1(+) vector were grown overnight, and plasmid was extracted using a Qiagen kit (Valencia, CA, USA). Both the PRAS40 plasmid and pcDNA3.1(+) were mixed with cationic liposome DOTAP (Biontex, Germany) at the ratio of 1:2 (microgram of cDNA to microliter of liposome) and allowed to complex for 15 mins before injection, according to our previous study (Saito et al, 2004). We used an intracisternal injection because it has been shown to be acceptable up to 300 μL solution (Pulford et al, 1999; Solomon et al, 1985). One-hundred microliters of the complex were injected intracisternally 1 day after SCI. Three days after SCI, the spinal cord was collected and embedded in paraffin. Immunostaining of c-Myc and pPRAS40 was used to examine the efficacy of transfection and pPRAS40 expression. Neuron-specific nuclear protein (NeuN) staining was performed to examine the number of surviving neurons. We used a method established by Grossman et al (2001) to determine motor neuron survival. If the cells were in the lower ventral horn and their profiles were larger than half of the sampling square (20 × 20 μm), they were determined to be ventral horn motor neurons. The cells above the line, 150 μm ventral from the central canal, were excluded (Grossman et al, 2001) (n =4 in each group).
Immunohistochemistry
Anesthetized animals were perfused with 10 U/ml heparin saline and subsequently with 3.7% formaldehyde in PBS 1, 3, and 7 days after SCI. Spinal cords at the lumbar enlargement were collected and postfixed with the same fixative for 24 h. The tissue was embedded in paraffin and sectioned 6-μm thick with a microtome. We focused on the area peripheral to the lesion according to our previous study (Yu et al, 2005). For immunohistochemistry, the sections were deparaffinized and incubated with 3% H2O2 in PBS. We used a 5% blocking serum, and then incubated the sections with rabbit polyclonal anti-pPRAS40 (1:50; Biosource International, Camarillo, CA, USA), mouse anti-Myc (1:100; Covance Inc., Berkeley, CA, USA), or mouse anti-NeuN (1:100; Chemicon International, Temecula, CA, USA) at 4°C overnight. The sections were then reacted with biotin-conjugated goat anti-rabbit or horse anti-mouse immunoglobulin G (1:100; Vector Laboratories, Burlingame, CA, USA) at room temperature for 1 h, then incubated with avidin-biotin horse radish peroxidase solution (ABC kit; Vector Laboratories) for 30 mins at room temperature and visualized using 0.025% 3,3′-diaminobenzidine hydrochloride (Vector Laboratories). Finally, the sections were counterstained with methyl green and mounted with permount (Fisher Scientific, Pittsburgh, PA, USA).
Western Blot Analysis
To obtain the whole-cell fraction, spinal cord from the area peripheral to the lesion was collected 1, 3, and 7 days after SCI and gently homogenized by douncing 30 times in a glass tissue grinder (Wheaton, Millville, NJ, USA) in 5 volumes of cold suspension buffer (20 mmol/L HEPES-KOH, pH 7.5, 250 mmol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, one tablet of cocktail inhibitor (Roche Diagnostics, Mannheim, Germany) in 25 ml buffer). Approximately 30 μg of total protein per lane were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis on Tris-glycine gel (Invitrogen) and transferred to a polyvinylidene difluoride membrane (Invitrogen). The membrane was incubated in polyclonal rabbit anti-pPRAS40 (1:1000; Biosource International) and then incubated in the peroxidase-conjugated secondary antibody. The signals were detected with a chemiluminescence kit (Amersham Biosciences, Uppsala, Sweden) and exposed on X-ray film. To confirm equal loading, the membranes were then stained with β-actin. After the film was scanned with a GS-700 imaging densitometer (Bio-Rad Laboratories, Hercules, CA, USA), a quantitative analysis was performed using Multi-Analyst software (Bio-Rad). Optical density of each band was measured on the same membrane and the results are presented as ±s.e.m.
Coimmunoprecipitation
To obtain cytosolic and mitochondrial fractions, spinal cord from the area peripheral to the lesion was collected 1, 3, and 7 days after SCI and gently homogenized by douncing 30 times in a glass tissue grinder (Wheaton) in 7 volumes of cold suspension buffer (same as above, for whole-cell fraction). The homogenates were centrifuged at 750 × g for 10 mins at 4°C and then at 8000 × g for 20 mins at 4°C. The supernatant was further centrifuged at 100,000 × g for 60 mins at 4°C. The supernatant was then collected and protein concentrations were determined by the Bradford method (Bio-Rad).
Precipitation was performed as described previously (Yu et al, 2005). Approximately 300 μg of protein from the cytosolic fraction were used for coimmunoprecipitation. Whole spinal cord extract was included as a positive control. The protein sample was mixed with 50% slurry of protein G-Sepharose (Amersham Biosciences) and incubated for 1 h at 4°C, then was centrifuged at 14,000 × g for 1 min. The supernatant was incubated with 2 μg of polyclonal mouse anti-14-3-3 antibody (Cell Signaling Technology) and 15 μL of protein G-Sepharose (50% slurry) for 1 h at 4°C. The negative control was prepared with protein G-Sepharose without an antibody. After centrifugation at 14,000 × g, the pellets were washed three times and used as samples bound to each antibody. The same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen) was added to each sample and the samples were boiled to remove the Sepharose beads. After centrifugation at 14,000 × g for 1 min, the supernatant was subjected to Western blot analysis with the anti-pPRAS40 antibody (1:1000; Biosource International) as described above.
Apoptotic Cell Death Assay
To quantify apoptosis-related DNA fragmentation, we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Diagnostics). This assay detects apoptotic but not necrotic cell death (Bonfoco et al, 1995). Samples were obtained from the area peripheral to the lesion of the LY294002-treated rats and control rats 3 days after SCI. Fresh tissue was taken and homogenized with a Teflon homogenizer in 5 volumes of ice-cold buffer (50 mmol/L KH2PO4 and 0.1 mmol/L EDTA, pH 7.8) and centrifuged for 10 mins at 750 × g at 4°C. The supernatant was collected and centrifuged for 20 mins at 10,000 × g at 4°C. The resulting supernatant was collected, and the protein concentration was determined. A cytosolic volume containing 20 μg of protein was used for the enzyme-linked immunosorbent assay, following the manufacturer's protocol. A statistical study was carried out in the same manner as mentioned under Western-blot analysis.
Quantification and Statistical Study
Significance was determined by analysis of variance followed by Fisher's protected least significant difference test and accepted when P <0.05.
Results
Liposome-Mediated PRAS40 Complementary DNA Transfection
Liposome-mediated PRAS40 transfection was performed to examine whether the increase in pPRAS40 was neuroprotective. Immunostaining with c-Myc, pPRAS40, and NeuN was performed after PRAS40 cDNA and pcDNA3.1(+) transfection to examine transfection efficacy, pPRAS40 expression, and motor neuron survival, respectively. Positive c-Myc staining was observed in the cytosol of motor neurons in both the PRAS40-transfected group and the pcDNA3.1(+)-transfected group, whereas only a slight staining was observed in the nontransfected group, indicating that the transfection was effective in the first two groups. Phospho-PRAS40 staining was much stronger in the PRAS40-transfected group than in the nontransfected group or the pcDNA3.1(+)-transfected group. There were more NeuN-positive motor neurons in the ventral horn of the PRAS40-transfected group than in the other two groups (Figure 1). At the epicenter (0 mm), almost no motor neurons survived in any groups. The numbers of surviving motor neurons 2 mm rostral to the epicenter were 20.0 ± 0.4, 13.6 ± 1.7, and 11.3 ± 2.3 in the PRAS40-transfected group, pcDNA3.1(+)-transfected group, and nontransfected group, respectively. More motor neurons were observed in the PRAS40-transfected group compared with the other groups (n = 4; P <0.05). The numbers 2 mm caudal to the epicenter were 20.8 ± 1.0, 15.7 ± 0.8, and 13.6 ± 3.8 in the PRAS40-transfected group, pcDNA3.1(+)-transfected group, and nontransfected group, respectively. More motor neurons survived in the PRAS40-transfected group than in the other groups (n = 4; P <0.05). Four millimeters rostral to the epicenter, the numbers were 24.5 ± 0.8, 21.0 ± 1.6, and 19.3 ± 2.9 in the PRAS40-transfected group, pcDNA3.1(+)-transfected group, and nontransfected group, respectively. No significant difference was seen among these groups (n = 4). Four millimeters caudal to the epicenter, the numbers were 24.4 ± 0.7, 21.3 ± 1.5, and 22.0 ± 2.8 in the PRAS40-transfected group, pcDNA3.1(+)-transfected group, and nontransfected group, respectively. No significant difference was seen among these groups (n = 4) (Figure 2).
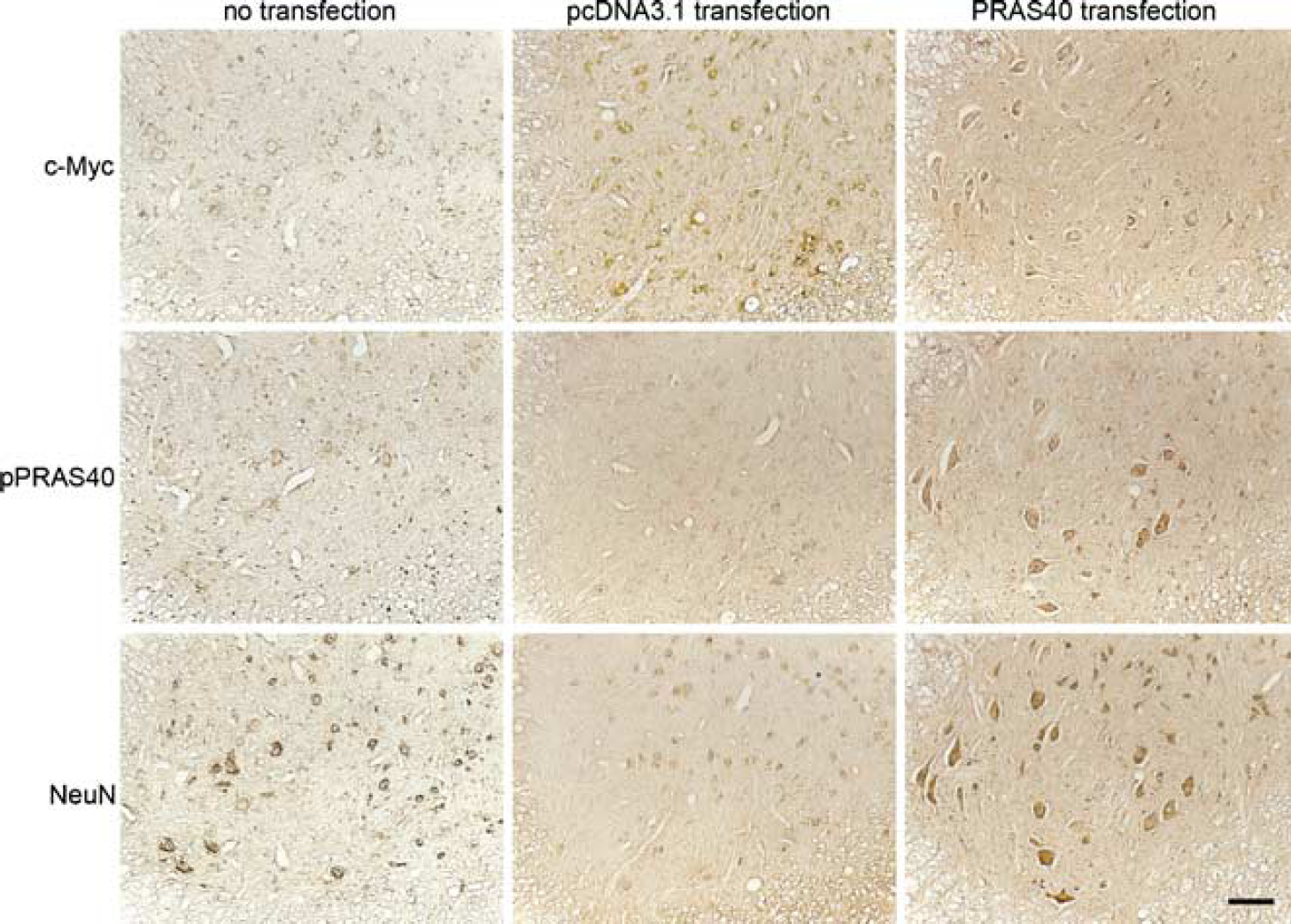
Representative photomicrographs of c-Myc, pPRAS40, and NeuN staining after no transfection and PRAS40 cDNA and pcDNA3.1(+) transfection 2 mm from the epicenter 3 days after SCI. There was c-Myc staining in the cytosol of motor neurons in both the PRAS40-transfected group and pcDNA3.1(+)-transfected group, whereas there was slight staining in the nontransfected group. The pPRAS40 staining was much stronger in the PRAS40-transfected group than in the pcDNA3.1(+ Hransfected group or the nontransfected group. There were more NeuN-positive large cells in the ventral horn of the PRAS40-transfected group than in the pcDNA3.1(+)-transfected group or the nontransfected group. Scale bar= 100 μm.
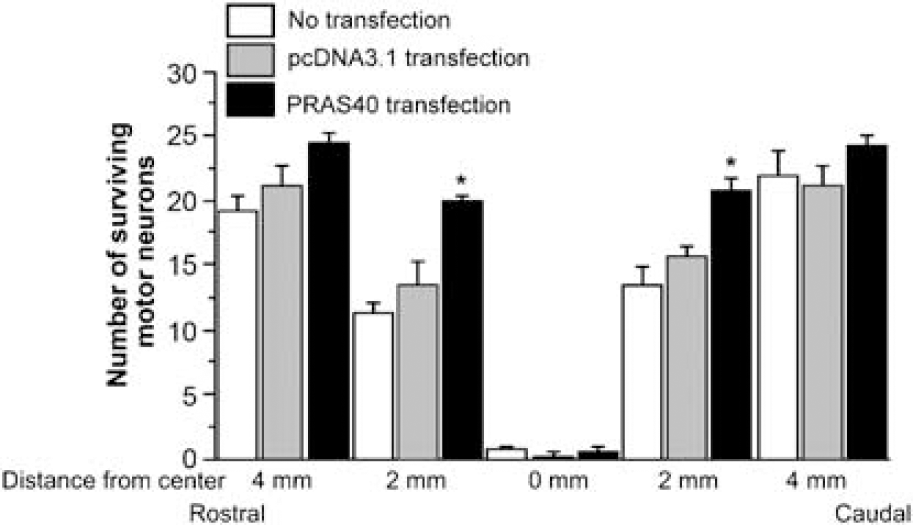
Cell count of surviving motor neurons after transfection study. At the epicenter (0 mm), almost no motor neurons survived in any group. The number of surviving motor neurons 2 mm rostral to the epicenter was greater in the PRAS40-transfected group than in the pcDNA3.1(+ Hransfected group or the nontransfected group (n = 4; P <0.05). The number 2 mm caudal to the epicenter was greater in the PRAS40-transfected group than in the other two groups (n = 4; P <0.05). Four millimeters rostral to the epicenter, there was no significant difference in the number of surviving motor neurons among the three groups (n = 4; P > 0.05). Four millimeters caudal to the epicenter, there was no difference in the number among the three groups (n = 4; P > 0.05). *P <0.05.
Phospho-PRAS40 Expression After Spinal Cord Injury
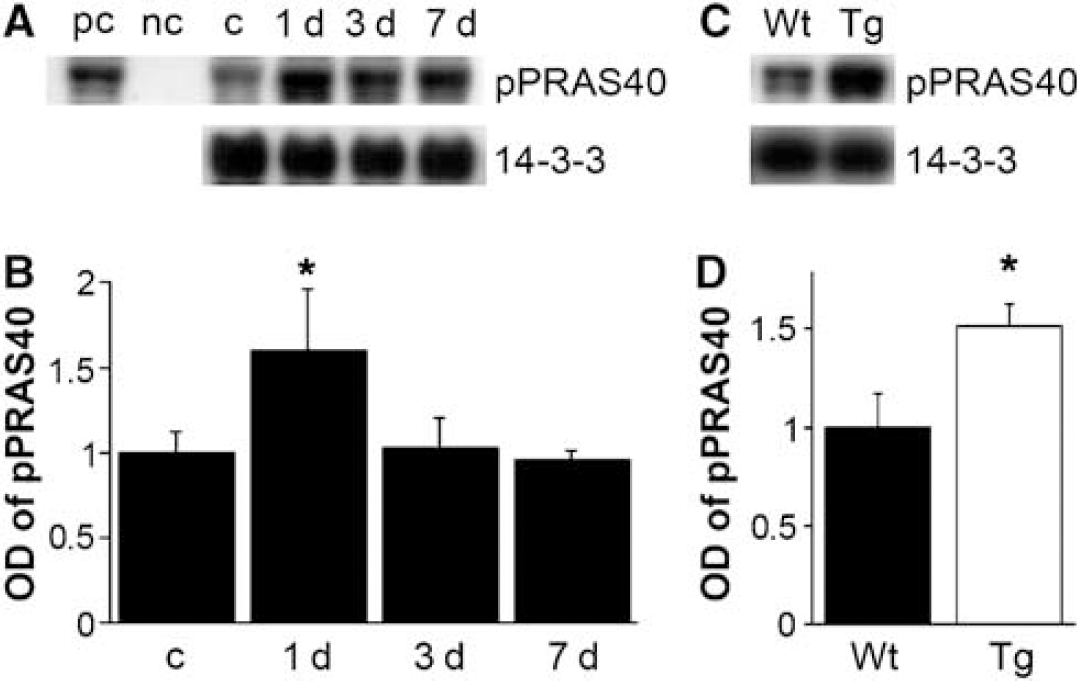
An immunohistochemistry study showed that pPRAS40 was expressed in the cytosol of ventral horn motor neurons of spinal cord in the control Wt and SOD1 Tg rats (Figure 3). In the Wt rats, pPRAS40 immunostaining was increased in the area peripheral to the lesion 1 day after SCI and then returned to the basal expression 3 and 7 days after SCI (Figure 3). In the SOD1 Tg rats, pPRAS40 was increased 1 day after SCI, was sustained until 3 days, and then returned to the baseline 7 days after SCI (Figure 3). Phospho-PRAS40 was visualized as a 40-kDa band in the Western-blot study. Expression of pPRAS40 was significantly increased 1 day (n = 5, P <0.05) after SCI in the Wt rats and 1 day (n = 5, P <0.01) and 3 days (n = 5, P <0.05) in the Tg rats. There was a significant difference in pPRAS40 expression between the Wt and Tg rats 1 day (n = 5, P <0.05) and 3 days (n = 5, P <0.05) after SCI (Figure 4).
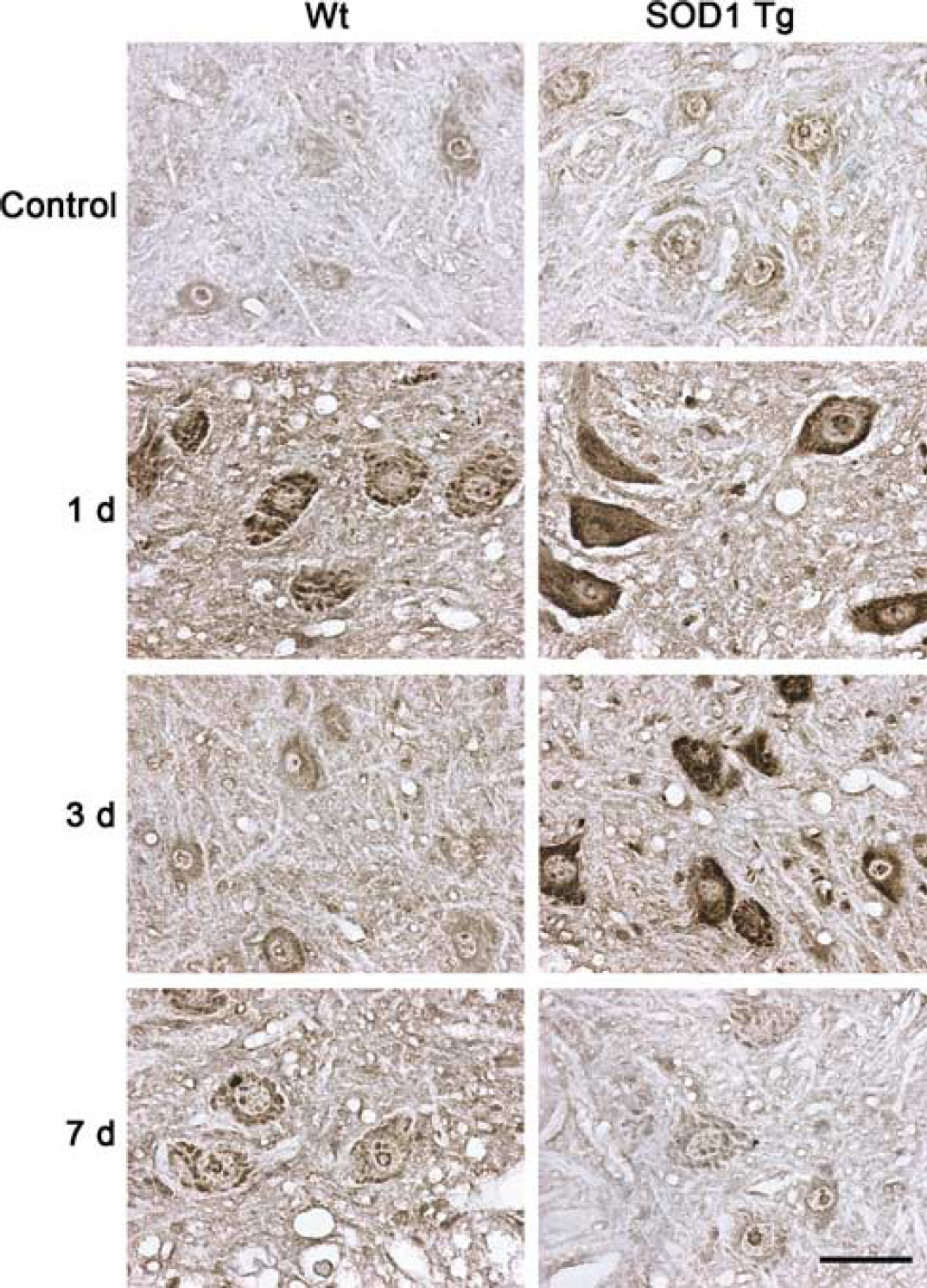
Representative photomicrographs of pPRAS40 in the area peripheral to the lesion after SCI. Phospho-PRAS40 was constitutively expressed in the cytosol of motor neurons in both the Wt and SOD1 Tg rats. There was increased expression 1 day after SCI in both the Wt and SOD1 Tg rats, whereas there was stronger staining in the Tg rats than in the Wt rats. Three days after SCI, expression of pPRAS40 returned to the baseline in the Wt rats, whereas the increase was sustained in the SOD1 Tg rats. Seven days after SCI, pPRAS40 expression was the same in both the Wt and the Tg rats. Scale bar = 50 μm.
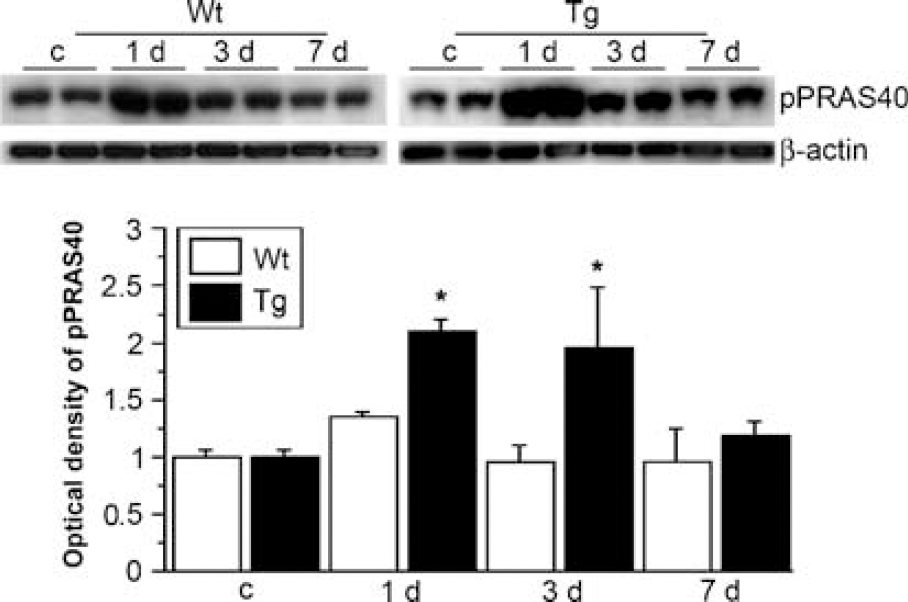
Western-blot analysis of pPRAS40 in the area peripheral to the lesion after SCI. Phospho-PRAS40 was seen as a 40-kDa band. It was significantly increased 1 day after SCI in the Wt rats (n = 5, P < 0.05) and at 1 day (n = 5, P < 0.01) and 3 days (n = 5, P < 0.05) in the SOD1 Tg rats. There was a significant difference in pPRAS40 expression between the Wt and SOD1 Tg rats 1 day (n = 5, P < 0.05) and 3 days (n = 5, P < 0.05) after SCI. β-Actin was used as an internal control and no difference in loading was observed between the samples. *P <0.05.
Coimmunoprecipitation of Phospho-PRAS40/14-3-3
Coimmunoprecipitation of pPRAS40 and 14-3-3 was performed using the cytosolic fraction from the area peripheral to the lesion of the spinal cords 1, 3, and 7 days after SCI. The binding of pPRAS40/14-3-3 was significantly increased in the Wt rats 1 day after SCI (Figures 5A and B) (n = 6, P <0.05). There was a significant increase in pPRAS40/14-3-3 binding in the Tg rats compared with the Wt rats 1 day after SCI (Figures 5C and D) (n = 6, P <0.05).
Coimmunoprecipitation of pPRAS40/14-3-3. (
The Phosphatidylinositol 3-Kinase Pathway and Akt Activation are Involved in the Phosphorylation of PRAS40
Involvement of the PI3K pathway was investigated with a study using the PI3K inhibitor, LY294002. In a group of rats injected with 50 nmol of LY294002, there was no difference in phospho-Akt (pAkt) or pPRAS40 expression 1 or 4 h after SCI, and this expression decreased in the area peripheral to the lesion 1 day after SCI compared with the vehicle-injected group (Figures 6A-C) (n = 6, P <0.05). We further examined whether inhibition by LY294002 can affect apoptotic cell death. Our data show that apoptotic cell death was increased 3 days after SCI in the LY294002-treated group compared with the vehicle-injected group (Figure 6D) (n = 6, P <0.05). Involvement of Akt activation was investigated with a study using Akt inhibitor IV. In the groups of rats injected with Akt inhibitor IV, pAkt and pPRAS40 decreased in the area peripheral to the lesion 1 h after SCI compared with the vehicle-injected group (Figures 7A-C) (n = 4, P <0.05), whereas there was no significant difference in pAkt and pPRAS40 expression 4 or 24 h after SCI (Figures 7A-C) (n = 4, P >0.05).
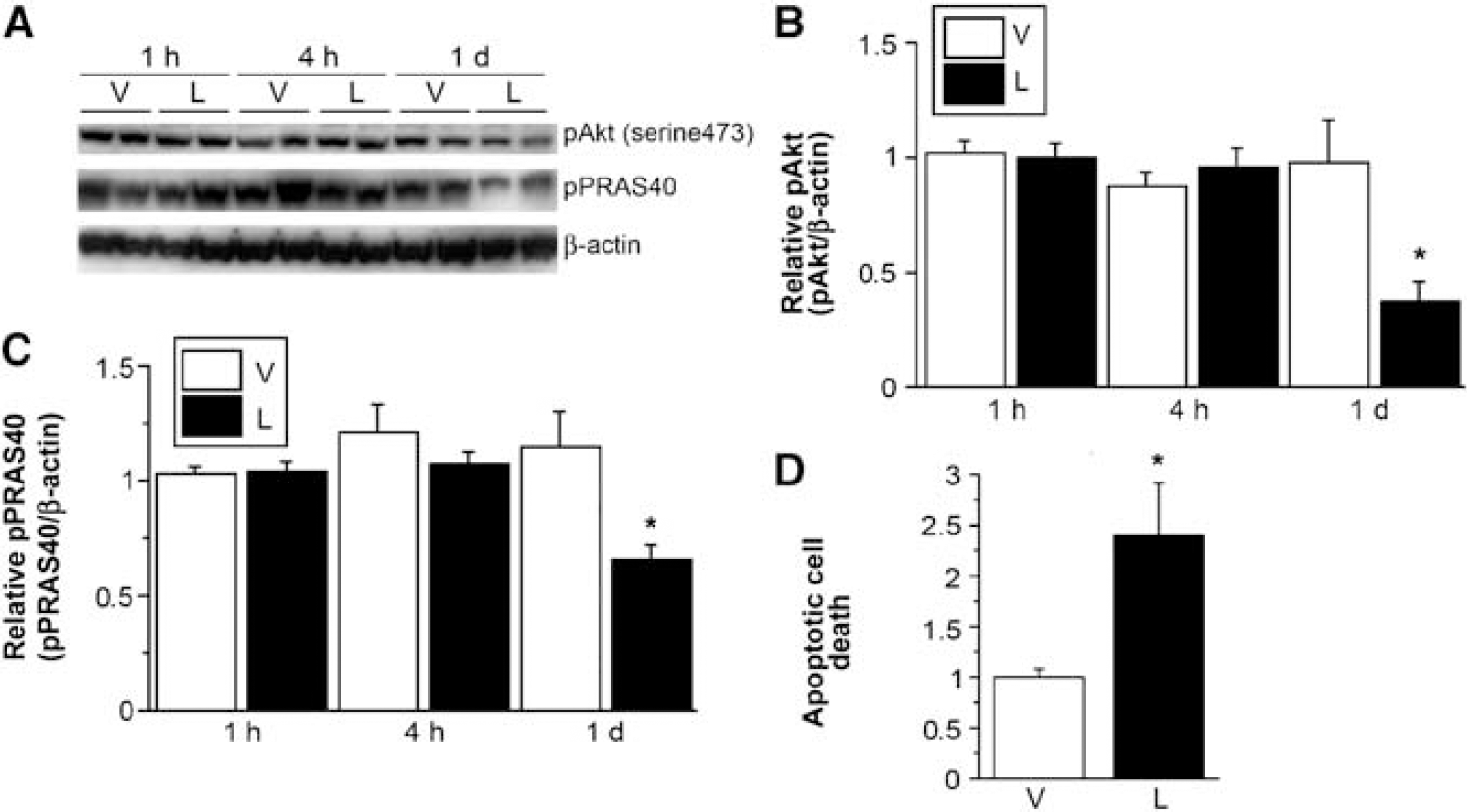
Western-blot analysis of pAkt and pPRAS40 in spinal cord samples with intrathecal injection of the vehicle or LY294002, a PI3k inhibitor. (
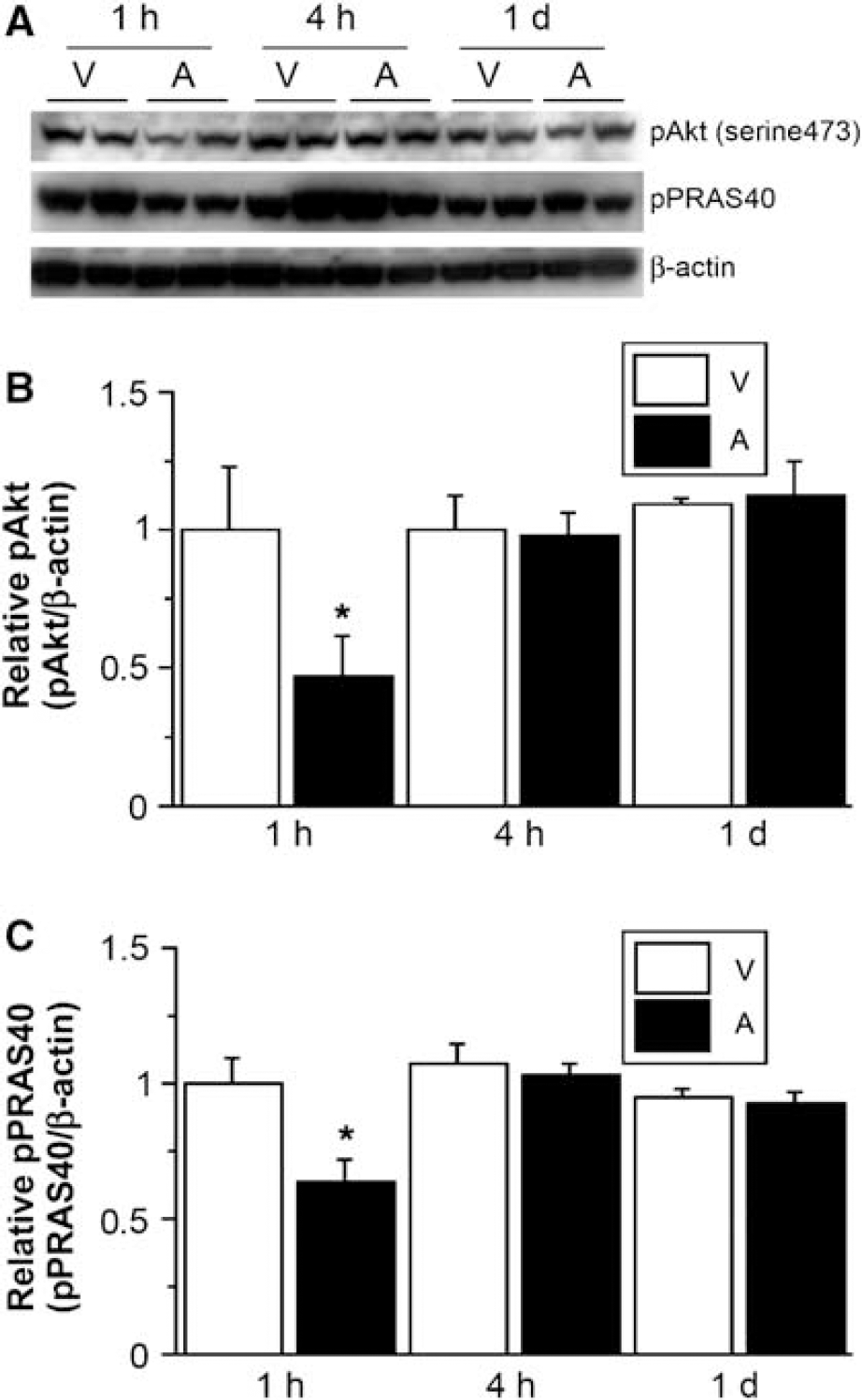
Western-blot analysis of pAkt and pPRAS40 in spinal cord samples with intrathecal injection of the vehicle or Akt inhibitor IV, a specific Akt inhibitor. (
Discussion
In this study, we demonstrated that liposome-mediated PRAS40 cDNA transfection in the spinal cord resulted in overexpression of pPRAS40 in the motor neurons compared with pcDNA3.1(+) transfection or no transfection and that the increase in pPRAS40 resulted in the survival of more motor neurons after SCI. Increased expression of endogenous pPRAS40 and its binding with 14-3-3 were observed in the area peripheral to the lesion after SCI, whereas overexpression of SOD1 in the Tg rats significantly increased the endogenous pPRAS40 level and the binding of pPRAS40 with 14-3-3 compared with the Wt rats. Finally, through the LY294002 and Akt inhibitor IV studies, we showed that PRAS40 phosphorylation is mediated by the PI3K/Akt pathway.
It is generally believed that traumatic SCI can result in primary and secondary injury involving necrosis and apoptosis. Studies have shown that different cell death/survival pathways contribute to apoptosis/cell survival. Cytochrome c release from mitochondria and caspase-3 cascade activation lead to apoptosis through a caspase-dependent pathway after SCI (Springer et al, 1999; Sugawara et al, 2002). c-Jun N-terminal kinase mediates apoptotic signals to the mitochondria and contributes to DP5 induction and subsequent apoptosis after SCI (Yin et al, 2005). Extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinases activate inducible nitric oxide synthase and induce nitric oxide-mediated neuronal degeneration after SCI (Xu et al, 2006). Nuclei translocation of apoptosis-inducing factor and endonuclease G induces neuronal apoptosis through caspase-independent pathways after SCI (Yu et al, 2006).
The PI3K/Akt pathway is one of the major cell survival pathways. Akt phosphorylation is increased in the cortex and contributes to neuronal survival, but is decreased in the ischemic core after cerebral ischemia (Noshita et al, 2001). In a traumatic brain injury model, Akt phosphorylation was decreased in the injured core but increased in the perifocal-damaged cortex and the CA1 subregion of the hippocampus (Noshita et al, 2002). Phospho-Akt and one of the major substrates, Bad, are upregulated in the area peripheral to the lesion and contribute to neuronal survival after SCI (Yu et al, 2005). A temporal increase in pAkt and phosphorylated Bad 3 h after reperfusion was reported in the peripheral area after stroke and contributed to neuronal survival (Kamada et al, 2007). PRAS40 was characterized to be another major substrate of Akt, and was phosphorylated at Thr246 after Akt activation (Kovacina et al, 2003). A recent study showed that pPRAS40 was increased in premalignant and malignant breast and lung cancer cell lines, which indicates that pPRAS40 may contribute to cell survival downstream of the PI3K/Akt pathway (Huang and Porter, 2005). An in vivo study showed that insulin mediated phosphorylation of PRAS40 in various tissues, such as skeletal, muscle, and liver, through the PI3K/Akt pathway (Nascimento et al, 2006). We demonstrated similar results showing that an increase in pPRAS40 decreased motor neuron death and that down-regulation of pPRAS40, using an inhibitor of the PI3K pathway, increased apoptotic cell death after SCI. PRAS40 is a major binding partner of 14-3-3 in binding studies (Kovacina et al, 2003). Another of our studies demonstrated that overexpression of PRAS40 results in an increase in pPRAS40 and its binding with 14-3-3, and protects neurons from apoptosis after cerebral ischemia (Saito et al, 2004). We also showed PRAS40/14-3-3 binding after SCI, yet the mechanism of pPRAS40 upregulation and how its binding with 14-3-3 increases cell survival remain to be clarified.
Oxidative stress plays an important role in the pathogenesis of central nervous system injury. Reactive oxygen species are generated and contribute to tissue damage after traumatic SCI (Azbill et al, 1997; Kamencic et al, 2001). An in vitro study showed that increased oxidative stress causes spinal cord neuron damage (Toborek et al, 1999). An in vivo study also showed that increased oxidative stress induces motor neuron death and neurological deficits after SCI (Bao and Liu, 2002). A decrease in oxidative stress caused by upregulation of glutathione or treatment with the mitochondrial uncoupling agent 2,4-dinitrophenol attenuates white matter damage and improves functional outcome (Jin et al, 2004; Kamencic et al, 2001). Overexpression of SOD1 in Tg rats also decreases oxidative stress and protects motor neurons from death after SCI (Sugawara et al, 2002). Other studies have demonstrated that decreased oxidative stress in SOD1 Tg mice results in an increase in Akt and PRAS40 phosphorylation after cerebral ischemia and a decrease in neuronal death (Kamada et al, 2007; Noshita et al, 2003; Saito et al, 2006).
Our spinal cord compression model is induced by milder injury (closing force/duration: 15 g/5 secs) compared with other compression models (27 to 95 g/30 to 60 secs) or severe injury models, and there is selective motor neuron loss with no cavity formation or astrocyte loss (Sugawara et al, 2002). We believe there is less mechanical damage but more ischemia/reperfusion injury compared with other SCI models. We continued with this model to study motor neuron injury and have shown that overexpression of SOD1 decreases neuronal death by increasing Akt/Bad signaling in the peripheral area after SCI (Yu et al, 2005). In this study, we further show that expression of endogenous pPRAS40 in SOD1 Tg rats is increased and contributes to neuronal protection after SCI.
Conclusion
We conclude that an increase in pPRAS40 by transfection after SCI increases survival of motor neurons, and that overexpression of SOD1 in Tg rats results in an increase in endogenous pPRAS40 and a decrease in motor neuron death by means of the PI3K/Akt pathway.
Footnotes
Acknowledgements
We thank Liza Reola and Bernard Calagui for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation. We also thank Dr Richard A Roth from Stanford University for the kind gift of PRAS40 cDNA.