
Abstract
The expression profile of the protease-activated receptor-2 (PAR-2) and effects of PAR-2 gene knockout (PAR-2 KO) on the infarct size were investigated after 60 minutes of transient middle cerebral artery occlusion (tMCAO) in mice in relation to phosphorylated extracellular signal-regulated kinase (p-ERK) and astrocyte activation. PAR-2 was normally distributed mainly in neurons of the central nervous system (CNS), and strongly upregulated at 8–24 hours after tMCAO. Deficiency of PAR-2 gene significantly increased the infarct volume and the number of TUNEL-positive cells at 24 hours of reperfusion. The strong neuronal expression of p-ERK was induced at 5 minutes as a peak after reperfusion in wild-type mice, but the signal change was significantly reduced in PAR-2 KO mice. Astroglial activation was also greatly inhibited at 24 hours after tMCAO in PAR-2 KO mice. These results show that the deficiency of PAR-2 gene increases the acute ischemic cerebral injury associating with suppression of neuronal ERK activation and reactive astroglial activation.
Introduction
Protease-activated receptor-2 (PAR-2), a member of seven transmembrane G protein-coupled receptors, is widely distributed in the central nervous system (CNS) (Striggow et al, 2001), and is known to play important roles in response to injury, inflammation, neurodegeneration, ischemia, neuronal signaling and pain pathway (Smith-Swintosky et al, 1997; Steinhoff et al, 2000; Striggow et al, 2001; Macfarlane et al, 2001; Vergnolle et al, 2001a, 2001b; Fiorucci and Distrutti, 2002; Oliver and Hill, 2002).
PAR-2 is first activated within proteolytic cleavage of the extracellular N-terminal domain by proteases such as trypin and tryptase (Nystedt et al, 1994; Macfarlane et al, 2001), couples to Gαq/11, and then results in activation of intracellular signals, such as mobilization of Ca2+, protein kinase C (PKC) and mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) (Macfarlane et al, 2001; Cottrell et al, 2003). The activated MAPK/ERK plays critical roles in regulating cell growth, differentiation, inflammation and promoting survival under normal and pathological conditions (Bonni et al, 1999; Lee et al, 2000; Han and Holtzman, 2000; Wang et al, 2000; Gu et al, 2001; von Gise et al, 2001; Morita et al, 2003). A number of recent reports suggest that activation of PAR-2 induces activating of MAPK/ERK signaling pathway (DeFea et al, 2000b; Macfarlane et al, 2001; Koo et al, 2002; Cottrell et al, 2003).
Astrocytes are the most numerous cell type in the CNS. They provide structural, trophic and metabolic support to neurons and thus modulate synaptic activity under normal conditions. On brain injury, astrocytes are transformed into the activated form, and critically influence neuronal survival by affecting glutamate reuptake, free radical scavenging, water transport, and the production of cytokines and nitric oxide (Stoll et al, 1998; Chen and Swanson, 2003). Neurons cannot survive in the brain without the close interaction with astrocytes, both under normal and pathological conditions. PAR-2 is also involved in activation of astroglia in vitro (Ubl et al, 1998; Wang et al, 2002), and glial fibrillary acidic protein (GFAP) expression is strikingly enhanced in the trypsinogen IV transgenic mice (Minn et al, 1998). However, the exact relationship between PAR-2 and astroglial activation is unclear.
In recent years, there has been increasing interest in functions of PAR-2 as a mediator of neurogenic inflammation and nociception. However, many studies performed to date have shown only the effect of PAR-2 by administrating PAR-2 agonists or performing in vitro study, while the intracerebral targets and pathological roles of PAR-2 in vivo are largely unknown. In this study, therefore, we examined an expression profile of the PAR-2 after transient focal brain ischemia, and the effect of PAR-2 gene knockout on the infarct volume in relation to ERK and astrocyte activations.
Materials and methods
Generation of Protease-Activated Receptor-2-Deficient Mouse
The PAR-2-deficient mice with C57BL/6 background, which replaced almost the entire exon 2 coding sequence with a cassette TAG3/IRES/LacZpA/MC1neopA (Figure 1A), were generated as described previously (Ferrell et al, 2003). The PAR-2-deficient strain was maintained by backcrossing a heterozygous (PAR-2+/−) male with C57BL/6 females at each generation at Kowa Tokyo New Drug Research Laboratories. The genotype of the mice was confirmed by Southern blot analysis (Figure 1B). Homozygous (PAR-2−/−) and wild-type (WT; PAR-2+/+) female mice generated from male and female PAR-2+/− mice at backcross generation eight were used at 8 weeks of age for the experiments. Saliva-secreting response to the PAR-2-activating peptide SLIGRL-NH2 was measured and compared with control mice to confirm the PAR-2−/− phenotype (Figure 1C, PAR-2 knockout (PAR-2 KO)). The PAR-2−/− mice were of the same body size and vitality when compared with PAR- 2+/− and PAR-2+/+ littermates, and, as previously reported, they exhibited no evidence of macroscopic abnormalities (Lindner et al, 2000).
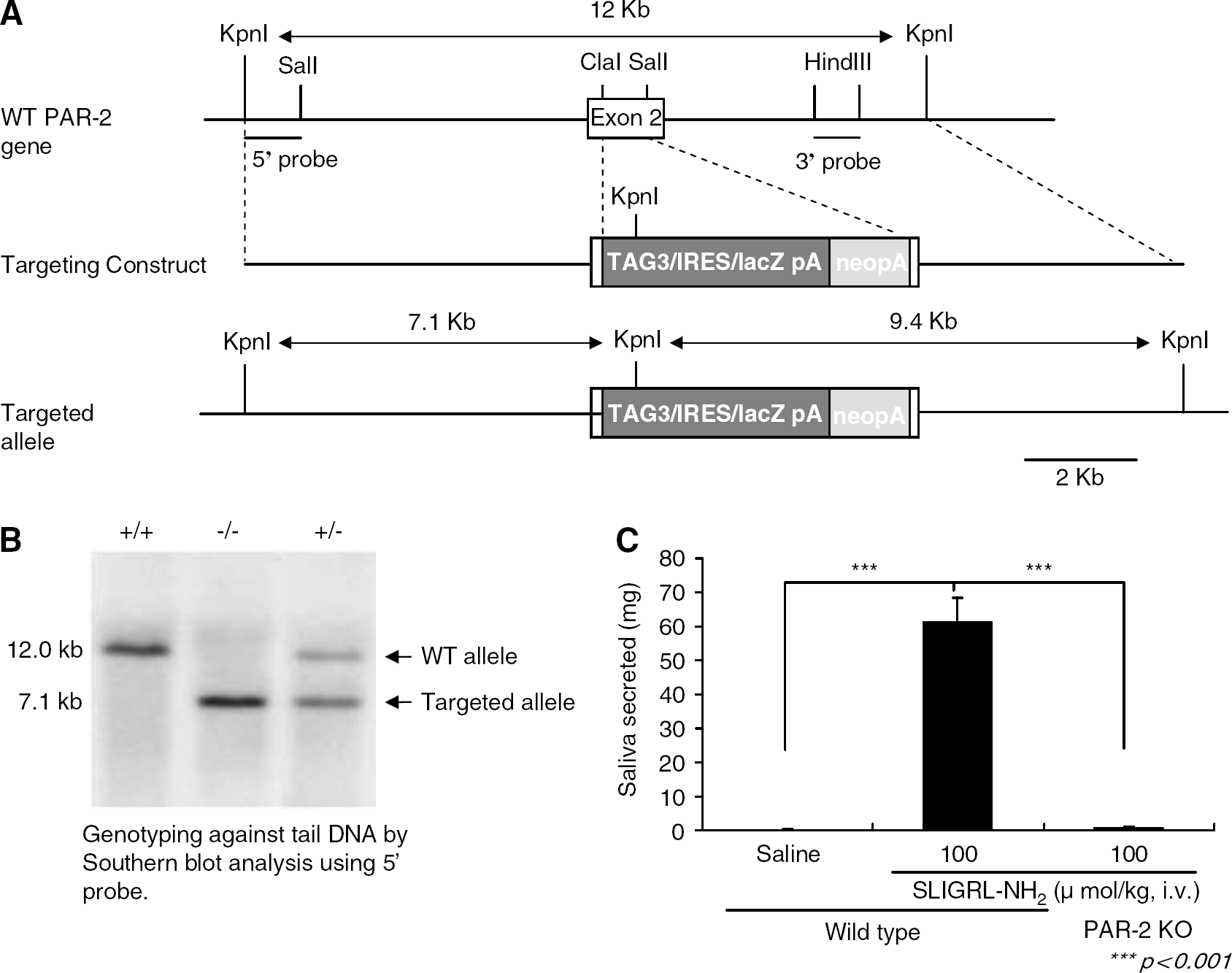
Generation of PAR-2-deficient mice. (
Focal Cerebral Ischemia
The PAR-2 KO and WT male mice (20–22 g) were subjected to transient focal cerebral ischemia by intraluminal middle cerebral artery (MCA) blockade with a nylon suture, as described previously (Connolly, Jr et al, 1996). Anesthesia was induced with 2% halothane in a mixture of 70% nitrous oxide and 30% oxygen delivered by face mask. The rectal temperature was controlled at 37°C, and the regional CBF (rCBF) of the right front parietal cortex was continually monitored during surgical procedure. Under an operating microscope, the right common carotid artery (CCA) and external carotid artery (ECA) were exposed through a midline neck incision and its branches were electrocoagulated. An 11-mm 6–0 monofilament nylon suture, blunted at the end, was inserted via the proximal ECA into the internal carotid artery (ICA), occluding MCA. After 60 minutes of MCA occlusion (MCAO), blood flow was restored by withdrawal of the nylon suture. The CBF was confirmed down to 10±5% of basal level after MCA occlusion, and returned to 90% of normal CBF level after reperfusion. The experimental protocol and procedures were approved by the Animal Committee of the Okayama University Medical School.
Calculation of Infarction Volume
The animals (n=6) were killed and the brains were removed 24 hours after MCAO reperfusion. Fourteen serial 10 μm-thick slice coronal sections from each brain were cut at 600 μm intervals beginning at 1 mm from the front using a cryostat. Each slice was stained with cresyl violet, and the infarction area was measured with the use of computer-assisted image analysis software SigmaScan pro 5 (SPSS Inc.). Total infarct volume was calculated by adding the area of infarction of each slice and multiplying by the distance between each slice.
Single Immunohistochemistry
Mice brains were removed at 5 minutes, 8 hours and 24 hours after MCAO reperfusion in both PAR-2+/+ and PAR-2−/− groups (each n=4), and quickly frozen in liquid nitrogen. Coronal sections at the caudate level were cut on a cryostat at −20°C to 10 μm thickness, and collected on glass slides. With these sections, immunohistochemistry for PAR-2, phosphorylated ERK (p-ERK), and histochemistry for TUNEL were performed. The fresh-frozen sections were fixed by 4% paraformaldehyde (PFA) and rinsed three times in PBS (pH 7.4). After blocking with 3% to 10% bovine serum albumin (BSA) for 1 hour, the slides were incubated with primary antibodies overnight at 4°C. The primary antibodies used and the dilutions for each were as follows: goat polyclonal anti-PAR-2 antibody at 1:100 (Santa Cruz Biotechnology), rabbit polyclonal anti-p-ERK antibody at 1:100 (Cell Signaling, CA, USA) and goat polyclonal anti-GFAP antibody at 1:200 (Santa Cruz Biotechnology). The sections were then washed and incubated for 1 hour with relevant biotinylated anti-goat or anti-rabbit IgG at 1:400 (Vector Laboratories, CA, USA), followed by incubation with avidin–biotin–peroxidase complex (Vectastatin ABC kit, Vector Laboratories) for 30 minutes. Diaminobenzidine tetrahydrochloride (DAB) was used as a color substrate. A set of sections was stained in a similar way without the primary antibody.
Double Fluorescent Immunohistochemistry of Protease-Activated Receptor-2 with NeuN or GFAP
We performed double staining for PAR-2 with NeuN or GFAP to investigate the expression of PAR-2 on the neuron and astrocyte. For the immunofluorescent detection, the coronal brain sections fixed by 4% PFA as above were first immunostained with the PAR-2 antibody, followed by fluorescein isothiocyanate (FITC)-conjugated rabbit anti-goat IgG antibody (Vector) at a dilution of 1:500; then, the sections were treated with an MOM kit (BMK-2202, Vector Laboratories, Bulingme, CA, USA) to prevent nonspecific binding, and finally reacted with mouse monoclonal anti-NeuN antibody (Chemicon, Temecula, CA, USA) at a dilution of 1:200 or mouse monoclonal anti-GFAP antibody (61011, PROGEN Biotechnik GmbH) at a dilution of 1:10, followed by Rhodamine-conjugated goat anti-mouse IgG antibody (Chemicon) at a dilution of 1:500. Subsequently, the slides were covered with VECTASHIELD mounting medium with 4′,6′-diamidino-2-phenylindole (DAPI) (H-1200; Vector Laboratories). Signals were examined using an Olympus microscope (BX51; Olympus, Japan) equipped with FITC and rhodamine filter set.
Double Fluorescent Immunohistochemistry of p-ERK with NeuN or GFAP
We performed double staining for p-ERK with NeuN or GFAP to investigate the expression of p-ERK on the neuron and astrocyte. For double staining of p-ERK and NeuN, the sections were immunostained with p-ERK antibody, followed by FITC-conjugated donkey anti-rabbit IgG (Chemicon) at a dilution of 1:500. Subsequently, the sections treated with the MOM kit, and reacted with anti-NeuN antibody and secondary antibody, as described above. For double staining of p-ERK and GFAP, the sections were incubated together with p-ERK antibody and goat polyclonal anti-GFAP antibody, followed by FITC-conjugated donkey anti-rabbit IgG (Chemicon) and Alexa fluor 546-conjugated donkey anti-goat IgG (Molecular Probes) antibody at a dilution of 1:500. After mounting, fluorescence was observed, as mentioned above.
Western Blotting Analysis
Whole-cell protein extraction was performed. Samples were obtained from both the cortex and caudate on the sham controls and ischemic sides after 5 mins, 8 hours, 24 hours (n=4 each) of reperfusion. The brain tissues were homogenized in 10 vol of cold protein extraction buffer (complete tablet; 1697498, Roche Molecular Biochemicals, Mannheim, Germany). The homogenate was centrifuged at 10,000g for 20 mins at 4°C, and the supernatant was used for the analysis. After adding the 2 vol of Laemmli sample buffer (Bio-Rad Laboratories) to the supernatant, equal amounts of the samples were loaded per lane (4 μg protein). The primary antibodies were a 1:1000 dilution of rabbit polyclonal antibody (Cell Signaling) against p-ERK1/2 (Thr202/Tyr204), and a 1:1000 dilution of monoclonal PAR-2 antibody (Santa Cruz). Western blots were performed with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Amersham, Buckinghamshire, UK) or anti-mouse IgG (PI-2000, Sigma, Buringame, USA) using a chemiluminescence Western blotting detection system kit (ECLTM-RPN 2106, Amersham Pharmacia Biotech, Uppsala, Sweden). The results were scanned with a LAS-1000 image analyzer (LAS-1000-mini, FUJIFILM, Tokyo), and quantified using Scion Image software (NIH).
Apoptosis Assay
For detection of DNA fragmentation, terminal deoxynucleotidyl dUTP nick-end labeling (TUNEL) was performed using an Apoptag In Situ Apoptosis Detection kit (TACS in situ apoptosis detection kit #4810–30k, Trevigen, Gaithers-burg, MD, USA) at 24 hours after the reperfusion. Briefly, the sections fixed by 4% paraformaldehyde were incubated with dioxigenin-labeled dUTP in the presence of terminal deoxynucleotidyl transferase (TdT) at 37°C for 1 hour, then incubated with HRP-conjugated anti-digoxigenin antibody for 1 hour at room temperature and stained with DAB. The positive cell stainings were counted in the three 1 × 1 mm2 regions of the inner boundary zone of subsequent infarction.
Data analyses and Statistics
The data are expressed as mean±s.d., except the extra notation. Statistical analysis was performed using the Student's t-test as P<0.05 statistically significant.
Results
Expression of Protease-Activated Receptor-2 in the Normal WT Mice
In all sham control brain areas examined, PAR-2 immunoreactivity was detected mainly in the cells with the morphological appearance of neurons, where the reaction product was mainly localized in neuronal perikarya (Figure 2). The staining of PAR-2 was slightly stronger in the cerebral cortex (Figure 2A) than that in the caudate putamen (Figure 2B). No immunoreactivity was found in the whole brain sections without the primary antibody (data not shown).
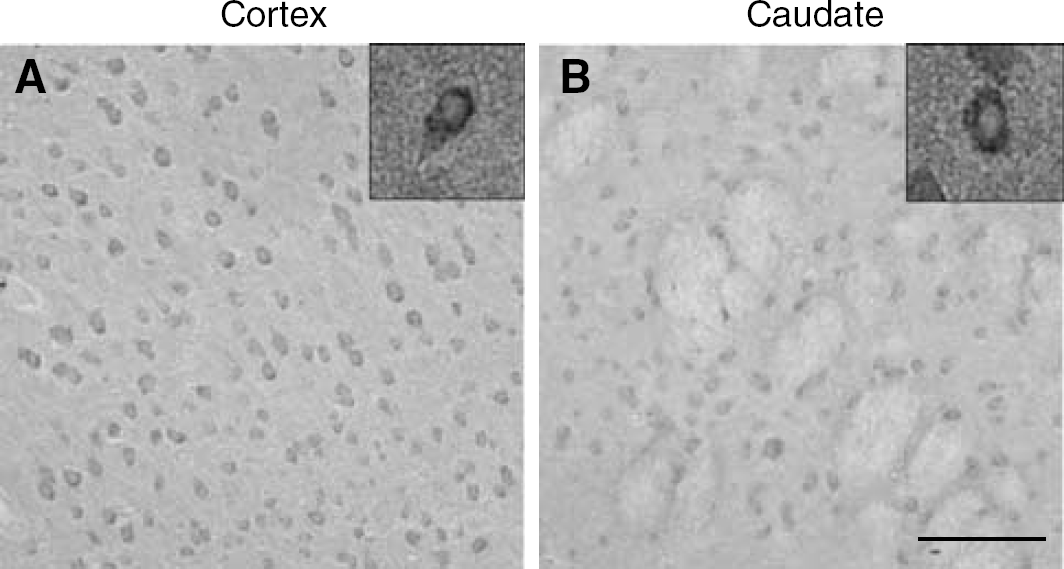
Immunoreactivity for PAR-2 on the cerebral cortex (
Protease-Activated Receptor-2 Upregulation in the WT Mice after Transient MCAO
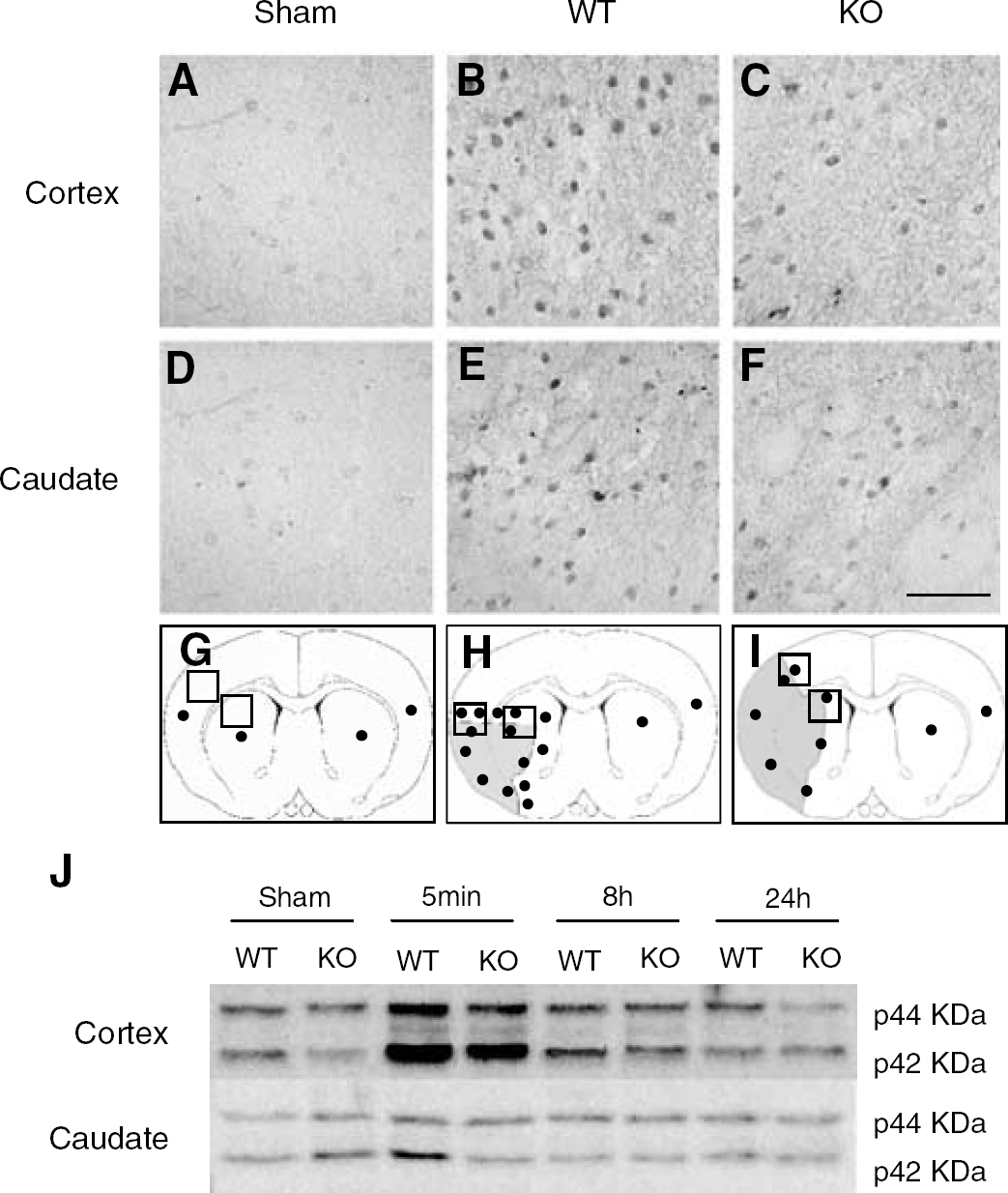
At 5 mins after transient MCAO (tMCAO), no significant difference was observed for the expression of PAR-2 compared with the sham control (data not shown). However, the expression of PAR-2 became increased significantly in the ischemic territory at 8 hours after reperfusion (Figure 3), and stronger staining was observed mostly in the larger cells of the peri-ischemic area of the cerebral cortex (Figure 3B), then the caudate (Figure 2E). The strong immunoreactivity of PAR-2 remained until 24 hours after reperfusion in the penumbra of the ischemic cortex (Figure 3C), where the number of PAR-2-positive cells also remained. In contrast, the number of PAR-2-positive cells was significantly reduced at 24 hours after reperfusion in the ischemic caudate putamen, where the dense stained cells were still detectable (Figure 3F, arrow).
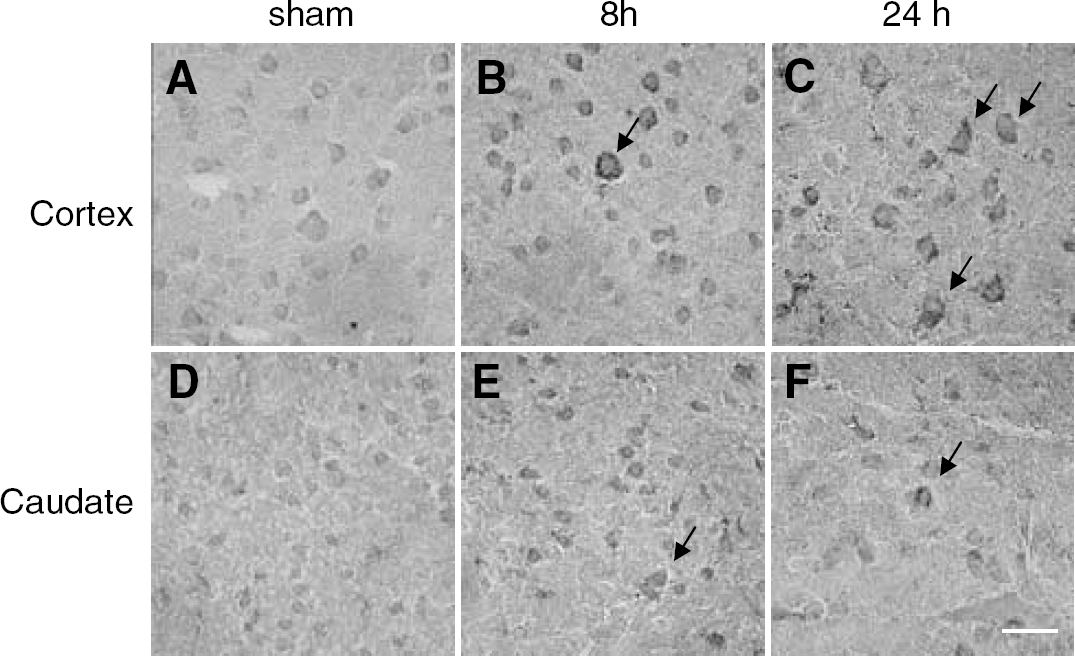
Immunoreactivity for PAR-2 on the WT mice cortex (
With Western blot analysis, PAR-2 immunoreactivity was evident as a band with a molecular mass of 55 kDa (Figure 4), and no bands were detected without primary antibody. After tMACO, the level of PAR-2 increased in the cerebral cortex (Figure 4A) at 8 and 24 hours, while that in the caudate putamen showed a transient increase at 8 hours (Figure 4B).
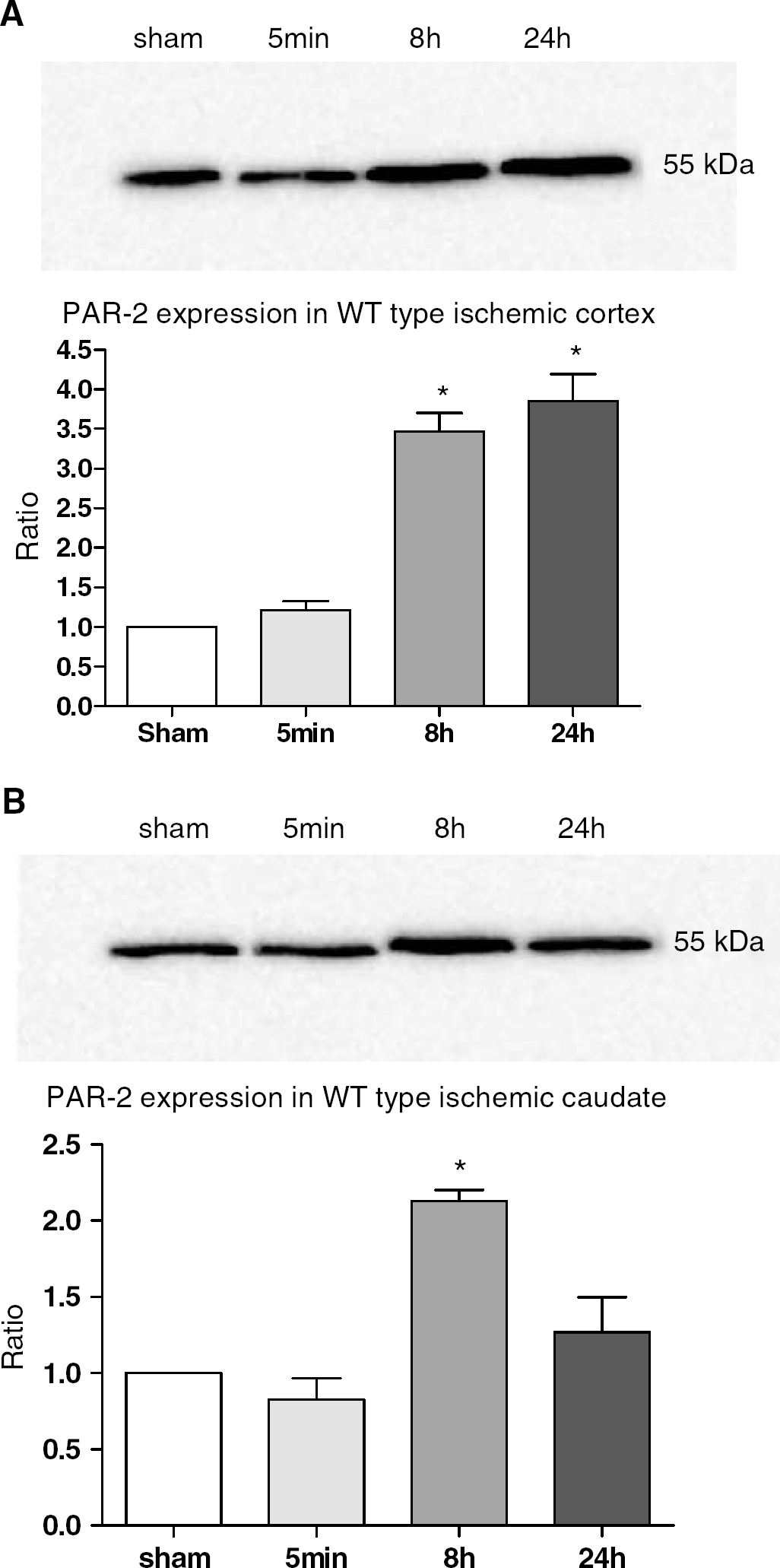
Western bolt analysis of PAR-2 in the cerebral cortex (
Double immunofluorescence for PAR-2 (green) (Figures 5A, 5D, 5G and 5J) and NeuN (red) (Figures 5B, 5E and 5H) showed that the expression of PAR-2 colocalized with some neurons in the sham control brain (Figure 5C). The PAR-2 + NeuN double-positive neurons are the cells with stronger NeuN stainings (Figure 5B). After tMCAO, strong induction of PAR-2 (Figures 5D and 5G) shows double positive cells with NeuN (Figures 5F and 5I), where double-positive cells are the cells with weaker NeuN stainings (Figures 5E and 5H). Although GFAP (Figure 5K, red) is not generally colocalized (Figure 5L) with PAR-2 (Figure 5J) in the control and postischemic brains, a few cells which appeared typical reactive astrocytes were double stained in the peri-ischemic area (Figures 5M, 5N and 5O).
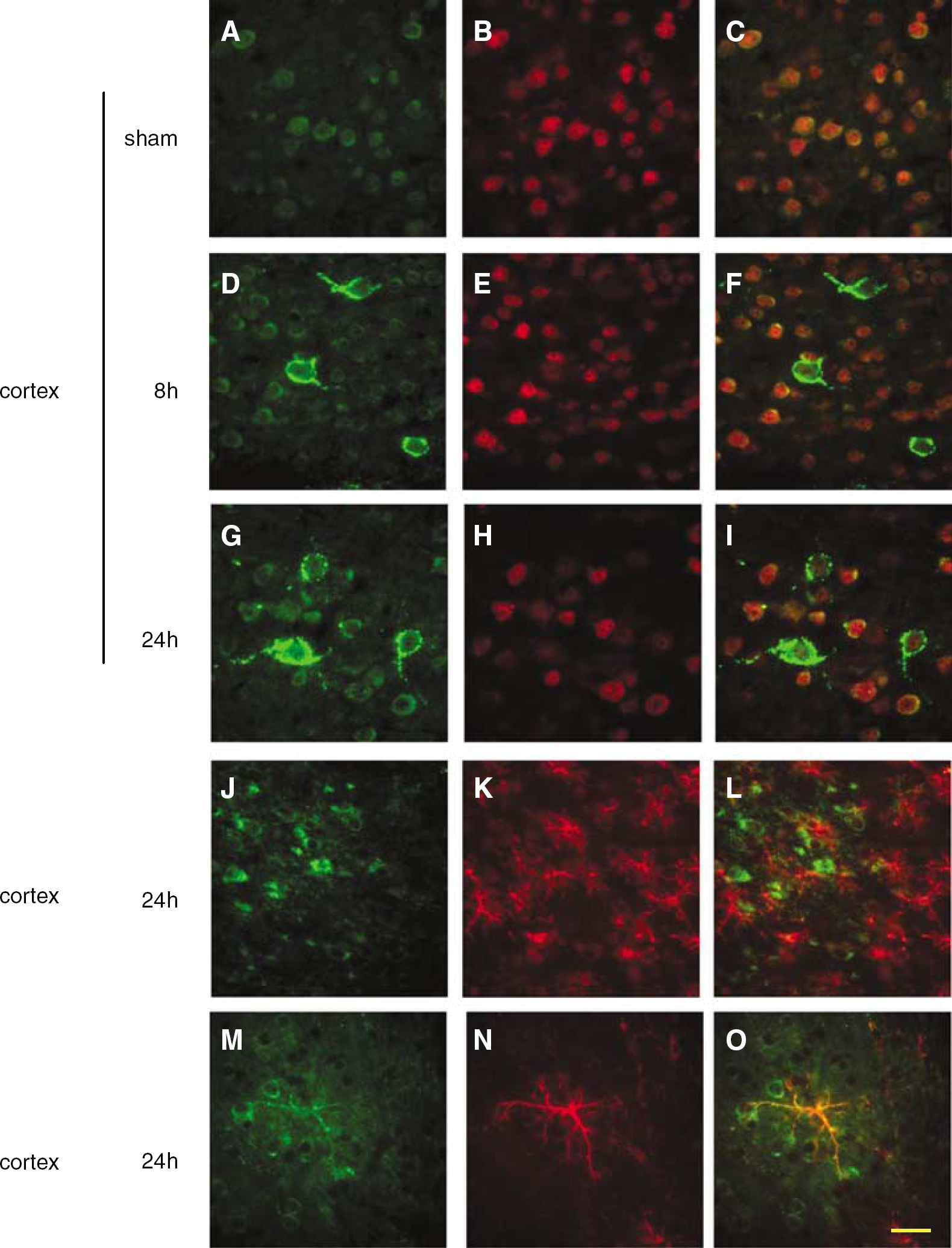
Double immunostaining for PAR-2 (green,
Evaluation of Infarct Volume in Protease-Activated Receptor-2 KO Mice
As for physiological parameters such as pH, PO2, PCO2, body temperature, and CBF, there was no significant difference between PAR-2 KO and WT groups before and after MCAO. The infarct volumes of the PAR-2 KO group (38.9±14.4 mm3) (Figures 6B and 6C) were significantly increased at 24 hours of reperfusion compared with the WT group (23.8±7.7 mm3) (Figures 6A and 6C) (n=6, *P<0.05).
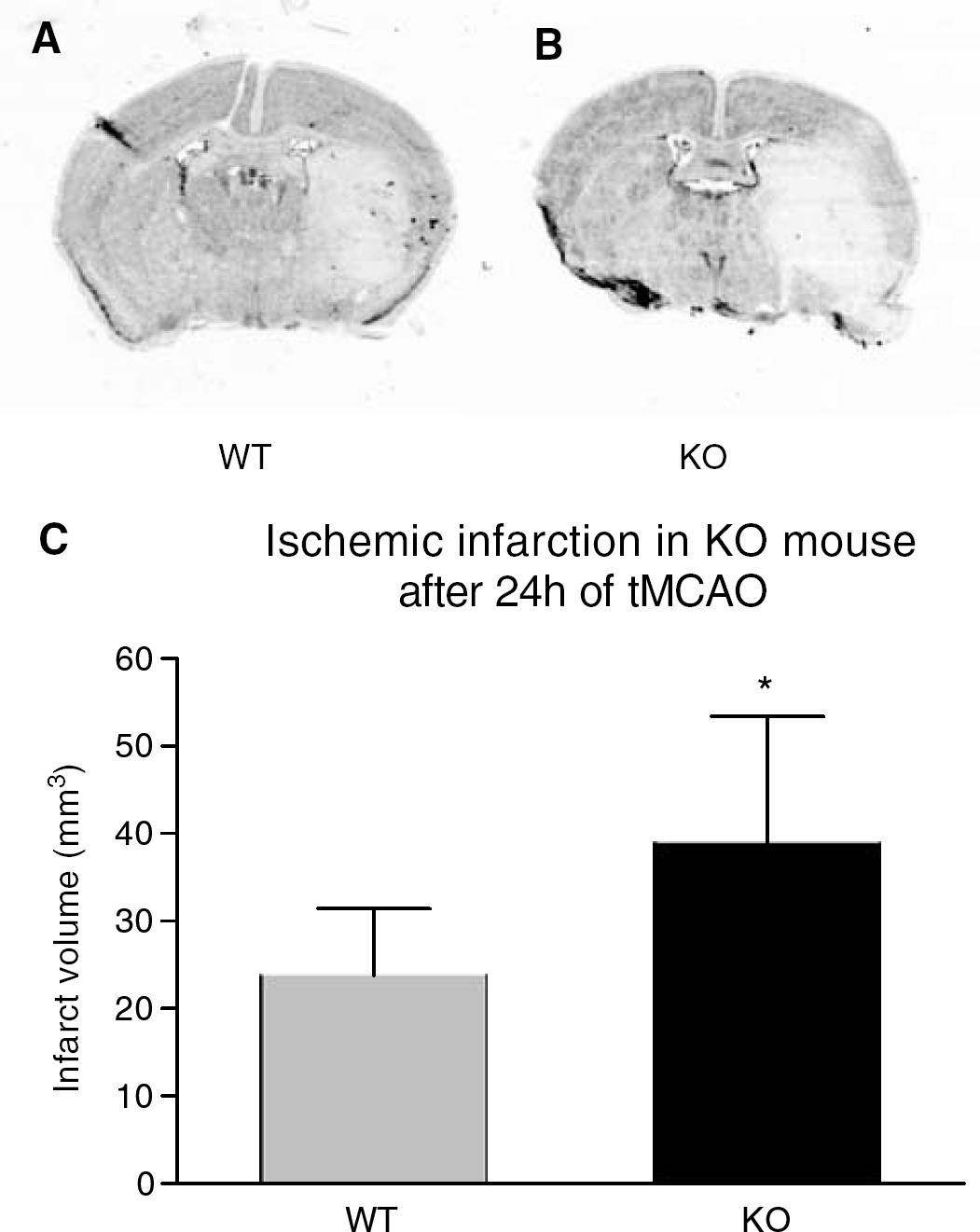
Effects of PAR-2 deficiency on the infarct volume at 24 hours after 60 minutes of tMCAO. The infarct volume of PAR-2 KO mice (
Neuronal p-ERK1/2 Expression in WT and Protease-Activated Receptor-2 KO Mice
p-ERK was only minimally detectable in the neuronal cells of cerebral cortex in the sham control brain (Figures 7A and 7D). At 5 mins after tMCAO, p-ERK became strongly positive in the ischemic cortex of the MCA territory of WT mice (Figures 7B and 8A) with immunoreactivity in the neuronal nuclei. Comparatively, p-ERK moderately increased in the caudate putamen (Figure 7E). In the PAR-2 KO mice, the staining of p-ERK in the cortex and caudate putamen was significantly reduced compared with that in the WT mice at the same time points (Figures 7C, 7F and 8D). With Western blot analysis, the bands of p-ERK were observed at 44 kDa (p-ERK1) and 42 kDa (p-ERK2) (Figure 7J) in the sham control brains of WT and KO mice. With reperfusion after tMCAO, protein level of p-ERK1/2 was significantly increased at 5 mins and 8 hours in the ischemic cortex with a peak at 5 mins, while that in the caudate putamen only showed a slight increase at 5 mins in WT mice. In the PAR-2 KO mice, the expression of p-ERK in the ischemic cortex was reduced compared with that in the WT mice, and did not change in the caudate putamen compared with the sham control. Schematic illustration (Figures 7G–7I) showed spatial distribution of p-ERK positive cells in the sham, WT, and PAR-2 KO mice, respectively. The expression of p-ERK in the contralateral hemisphere after reperfusion was not different from that in the normal brain (data not shown).
Immunoreactivity for p-ERK on the cerebral cortex (
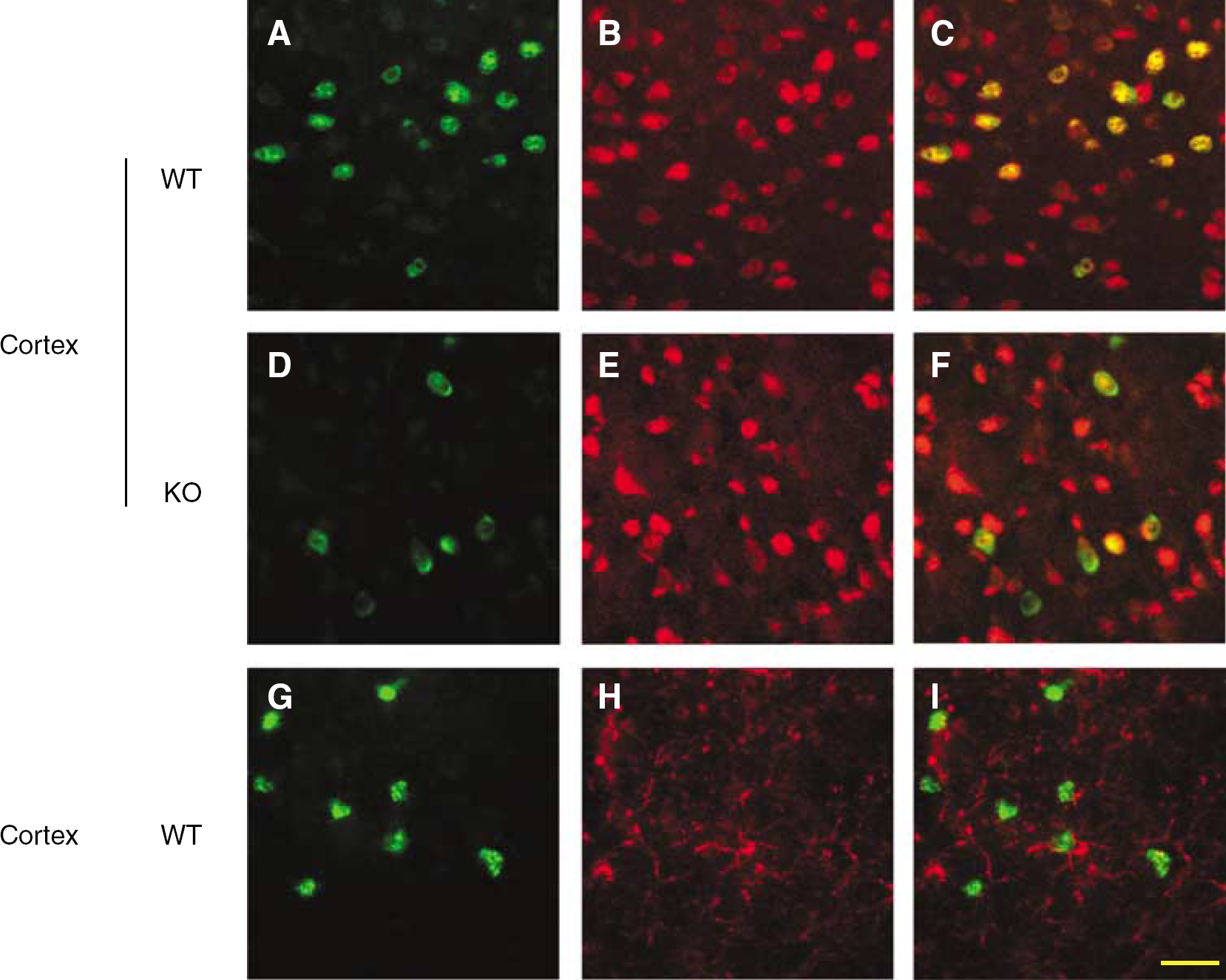
Double immunostaining for p-ERK (green,
With double immunofluorescence, the staining of p-ERK (green) was colocalized (Figures 8C and 8F) with the neuronal marker NeuN (red) (Figures 8B and 8E) at 5 mins of reperfusion, but did not coexpress with glial marker GFAP (red) (Figures 8G–8I), showing that p-ERK is expressed primarily in neurons after tMCAO.
Expression of GFAP in the Protease-Activated Receptor-2 KO Mice
In the sham control brain, GFAP immunoreactivity was principally located in the hippocampus and corpus callosum. At 24 hours after tMCAO, GFAP immunoreactivitity was strongly found in the peri-ischemic area, with thick numerous processes as typical reactive astrocytes (Figure 9A). In contrast, the expression of GFAP was greatly reduced in PAR-2 KO mice at 24 hours of reperfusion with reduced numbers of positive cells and processes (Figure 9B). The integral densitometric analysis for GFAP staining (intensity plus number) was significantly decreased in PAR-2 KO mice quantified using SigmaScan pro 5 software (Figure 9C) (*P<0.05).
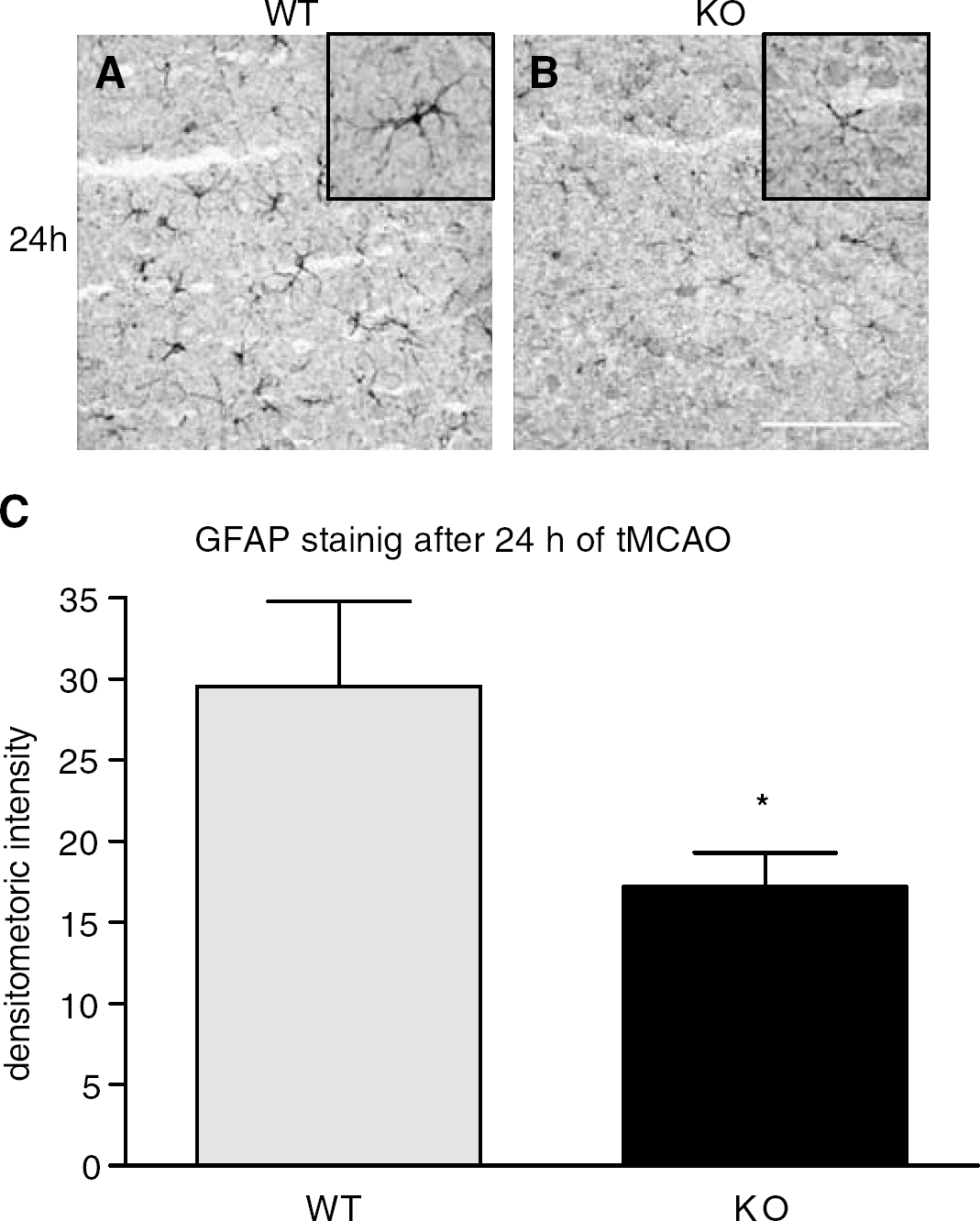
Immunoreactivity for GFAP on the peri-ischemic area in WT (
TUNEL Staining after Transient Middle Cerebral Artery Occlusion in Protease-Activated Receptor-2 KO Mice
TUNEL staining at 24 hours of reperfusion showed that positive cells were mainly distributed in the border of the occluded MCA area in both WT and PAR-2 KO mice. TUNEL labeling was essentially found in the nuclei of cells (Figures 10A and 10B). As compared with WT mice (261.2±41.3, n=6) (Figures 10A and 10C), the number of TUNEL-positive cells in PAR-2 KO mice was significantly increased (356.7±71.8, n=6; *P<0.05) (Figures 10B and 10C). TUNEL staining was negative in the sham control (data not shown).
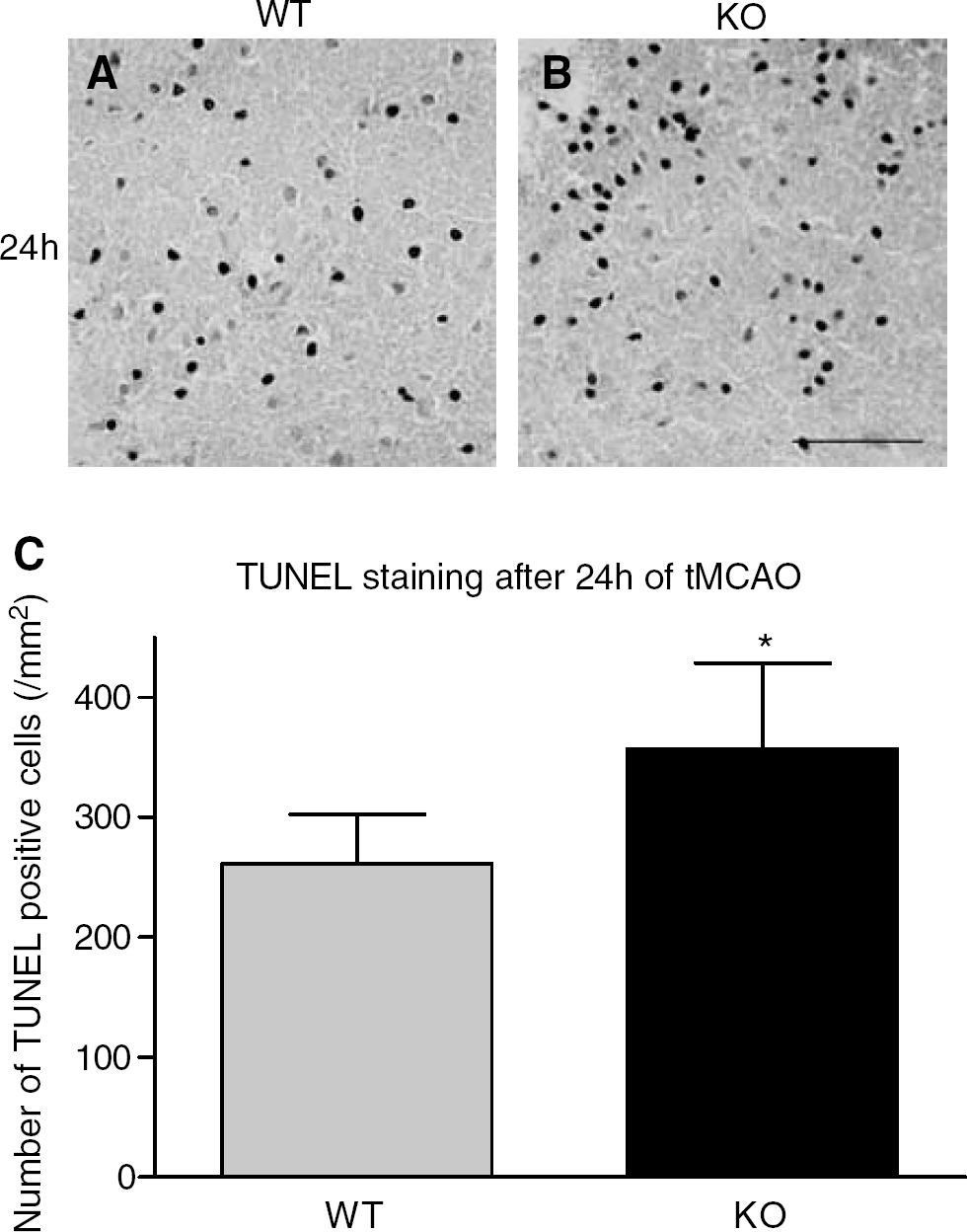
Staining of TUNEL on the border of the occluded MCA area in WT (
Discussion
The current study shows the important roles of PAR-2 in the transient focal cerebral ischemia with the PAR-2 KO mice. We observed that, first, PAR-2 was widely distributed in the sham control brains and significantly upregulated after 8 and 24 hours of tMCAO in the WT mice. Second, the infarct volume in the PAR-2 KO mice was increased after 24 hours of ischemia compared with WT mice. Third, the neuronal expression of p-ERK was induced after 5 minutes of tMCAO in both WT and PAR-2 KO mice, but the signals were significantly reduced in the PAR-2 KO mice. Fourth, the astrocyte activation was greatly reduced in the PAR-2 KO mice. Fifth, the number of TUNEL positive cells was increased in the PAR-2 KO mice.
The PAR-2 was widely distributed in the CNS and had important effects on distinct cerebral processes under physiological conditions such as learning and memory (Striggow et al, 2001). The expression of PAR-2 was enhanced by ischemic stimulation (Smith-Swintosky et al, 1997; Ubl et al, 1998; Striggow et al, 2001). In the current study, the basal neuronal expression of PAR-2 was widely distributed in the WT sham controls (Figure 2), reflecting the physiological activity of PAR-2 in the normal brain. Upregulation of PAR-2 at 8 and 24 hours after tMCAO (Figures 3 and 4) indicates the role of PAR-2 under ischemic conditions. Of interest is the fact that the strong expression of PAR-2 was found mainly in the large neurons of the peri-ischemic area (Figures 5D and 5G), and those cells were double positive with the weak NeuN positive cell (Figures 5E, 5F, 5H and 5I). These large neurons are known to have a high metabolic rate and the weak expression of NeuN was considered to be induced due to axonal injury (McPhail et al, 2004), suggesting that PAR-2 might be closely involved in the repair or cell-protective mechanism in these cells.
Activation of PAR-2 mobilizes intracellular Ca2+, and induces the assembly of a MAPK/ERK signaling module by a mechanism that depends on β-arrestins (Dery et al, 1998; DeFea et al, 2000a, 2000b; Macfarlane et al, 2001). Previous studies indicate that PAR-2 is involved in the activation of ERK subfamily of MAPK relatively selectively by forming a multiprotein signaling complex that contains PAR-2, β-arrestin, Raf-1 and p-ERK1/2 (Belham et al, 1996; Defea et al, 2000a, 2000b; Cottrell et al, 2003). Extracellular signal-regulated kinase is abundant in the CNS, and is activated by phosphorylation, which requires elevation of intracellular Ca2+ during various physiological and pathological events such as brain ischemia and epilepsy (Fukunaga and Miyamoto, 1998). P-ERK plays a critical role in mediating intracellular signal transduction in response to a variety of stimulation, and morphological differentiation and inflammatory response, and in promoting survival in neurons (Fukunaga and Miyamoto, 1998; Erhardt et al, 1999; Irving et al, 2000; Han and Holtzman, 2000; Fahlman et al, 2002; Hasegawa et al, 2003; Obata and Noguchi, 2004; Tian et al, 2004). Our previous reports also showed that the total ERK and p-ERK were induced after tMCAO and promoted neuronal survival in the ischemic brain (Kitagawa et al, 1999; Li et al, 2002). In the present study, the strong expression of p-ERK was observed predominantly in the peri-ischemic neurons from the very early period of reperfusion (Figure 7), suggesting that the neural cells in the peri-ischemic area were more supported by p-ERK than those in the ischemic core. In contrast, the activation of p-ERK was significantly reduced in the PAR-2 KO mice compared with the WT mice (Figures 7 and 8). These results indicate that PAR-2 was involved in activation of the p-ERK pathway after tMCAO, so that the activation of p-ERK by PAR-2 was inhibited, and the neuronal supporting induced by p-ERK was reduced in the PAR-2 KO mice. Although both our results and accumulating reports show p-ERK benefits to cell survival, some papers show that p-ERK does not have neuronal protective effects (Alessandrini et al, 1999). The functional role of p-ERK may differ depending on types of cells or kinds of insults. In the present study, the strong expression of p-ERK showed earlier than PAR-2 protein induction. This must be the reason why these signaling events occurred rapidly without inducing additional PAR-2 protein expression, which usually needs time after ischemic stimulation.
A prominent response of glia to many types of injuries or insults in the CNS is the activation of astrocytes, which is typically characterized by an upregulation of GFAP expression (Norton et al, 1992). Reactive astrogliosis is a key component of the cellular response to CNS injury (Ridet et al, 1997). During acute brain injury, astrocytes support neuronal survival by glutamate reuptake, release of metabolic intermediates, scavenging of oxygen-free radical, buffering of extracellular K+ and H+, and secreting neurotrophic factor and cytokines (Goss et al, 1998; Hailer et al, 2001; Chen and Swanson, 2003; Dhandapani et al, 2003). Early astroglial dysfunction plays a critical role in determining the outcome of acute hypoxic–ischemic injury by compromising neuronal–glial interactions (Liu et al, 1999). Wang et al (2002) reported a PAR-2-induced proliferation of astrocytes in rat cultured cells. Minn et al (1998) reported that the expression of GFAP was strikingly enhanced in the trypsinogen IV transgenic mice. In the present study, the expression of GFAP was greatly enhanced at 24 hours after tMCAO in the WT mice (Figure 9A), and this was significantly inhibited in the PAR-2 KO mice (Figure 9B). Consequently, the number of TUNEL-positive cells and the infarction volume increased in the PAR-2 KO mice (Figures 6 and 10), suggesting that the increase of brain injury was also partly attributable to astroglial dysfunction induced by deficient PAR-2.
One mechanism for astrocyte activation by PAR-2 was considered via intracellular signalings such as Ca2+, PKC and ERK in astrocytes (Yu et al, 1997; Cazaubon et al, 1997; Abe and Saito, 2000; Wang et al, 2002; Fiorucci et al, 2003). However, in the present study, the PAR-2 was expressed mainly in the neurons (Figures 5C, 5F and 5I), whereas only a few activated large astrocytes became positive with PAR-2 at 24 hours after tMCAO (Figures 5M and 5O). A recent report also showed that PAR-2 was expressed in limited astrocytes after the brain injury by the trimethyltin (Pompili et al, 2004). These results suggest a possible involvement of other indirect mechanisms in astroglial activation. Microglia is known to mediate astrocyte activation by the secretion of inflammation cytokines after ischemic injury. In fact, we also investigated the activation of microglia and macrophage in the WT and KO mice by immunostaining, but we did not find significant difference in the two groups (data not shown), similar to previous reports (Minn et al, 1998; Pompili et al, 2004). However, the accumulating evidence indicates that neuronal regulation of astrocyte function by neurotransmitters, cytokines and calcium oscillations was an important mechanism of astroglial activation (Hu and Levin, 1994; Matsutani and Yamamoto, 1997; Acarin et al, 2000; Zonta and Carmignoto, 2002; Zonta et al, 2003). In our study, the neuronal response for ischemic injury was found in the very early period of reperfusion as p-ERK induction (Figures 7 and 8). The early neuronal activation probably plays an important role in remodeling astrocyte processes. The exact mechanism on the relationship between PAR-2 and astrocyte activation remains to be determined further.
Some studies report that PAR-2 is associated with neurodegeneration or mediation of neurotoxic factors such as tumor necrosis factor alpha (TNF-α) (Smith-Swintosky et al, 1997; Kim et al, 2002). The neuroprotective or neurodegenerative effects may be concomitant in the pathological processes, leading to the final outcome. The current study showed that the deficiency of PAR-2 leads to the increase of the infarction volume after acute transient cerebral focal ischemia, and that neuronal p-ERK activation and astroglial activation were involved in the mechanism. Further investigations of cellular signal transduction mechanisms and their long-term effect are necessary for understanding the role of PAR-2 further.
Footnotes
Acknowledgements
This work was partly supported by Grant-in-Aid for Scientific Research (B) 15390273 and (Hoga) 15659338 and National Project on Protein Structural and Functional Analyses from the Ministry of Education, Science, Culture and Sports of Japan, by grants (Itoyama Y, Kimura I and Kuzuhara S) from the Ministry of Health and Welfare of Japan, and by grants from Kanae Foundation for Life and Socio-Medical Science.