
Abstract
The transcription factor NF-κB is a key regulator of inflammation and cell survival. NF-κB is activated by cerebral ischemia in neurons and glia, but its function is controversial. To inhibit NF-κB selectively in neurons and glial cells, we have generated transgenic mice that express the IκBα superrepressor (IκBα mutated at serine-32 and serine-36, IκBα-SR) under transcriptional control of the neuron-specific enolase (NSE) and the glial fibrillary acidic protein (GFAP) promoter, respectively. In primary cortical neurons of NSE-IκBα-SR mice, NF-κB activity was partially inhibited. To assess NF-κB activity in vivo after permanent middle cerebral artery occlusion (MCAO), we measured the expression of NF-κB target genes by real-time polymerase chain reaction (PCR). The induction of c-myc and transforming growth factor-β2 by cerebral ischemia was inhibited by neuronal expression of IκBα-SR, whereas induction of GFAP by MCAO was reduced by astrocytic expression of IκBα-SR. Neuronal, but not astrocytic, expression of the NF-κB inhibitor reduced both infarct size and cell death 48 hours after permanent MCAO. In summary, the data show that NF-κB is activated in neurons and astrocytes during cerebral ischemia and that NF-κB activation in neurons contributes to the ischemic damage.
Introduction
The central nervous system reacts to noxious stimuli with a complex genomic response. Recent advances in expression profiling have identified hundreds of genes that are upregulated on damage to brain tissue. In an attempt to investigate transcriptional programs rather than individual genes, the functional role of transcriptional activators has been studied in various paradigms of brain damage. In the case of cerebral ischemia, there is clear evidence for the activation of transcription factors such as hypoxia inducible factor, interferon regulatory factor 1, and NF-κB (Bergeron et al, 1999; Iadecola et al, 1999; Schneider et al, 1999; Stephenson et al, 2000).
Originally, NF-κB was discovered in an immunologic context but it is also present in the brain (O'Neill and Kaltschmidt, 1997). Five subunits, p50, p52, p65 (RelA), RelB, and c-Rel, form homo- and heterodimers. In an inactive state, NF-κB dimers are sequestered in the cytoplasm by the specific inhibitors IκBα, IκBβ, and IκBɛ. On stimulation, IκB is phosphorylated by the IκB kinase (IKK) complex, ubiquitinated, and then degraded by the 26S proteasome. However, there are also pathways of NF-κB activation that are independent of IKK or the proteasome (Bui et al, 2001; Schölzke et al, 2003). In cerebral ischemia, most investigators found activation of NF-κB in neurons (Huang et al, 2001; Schneider et al, 1999; Stephenson et al, 2000). In addition, there is also evidence for the activation of NF-κB in endothelial and glial cells during cerebral ischemia (Carroll et al, 1998; Gabriel et al, 1999; Terai et al, 1996). NF-κB activity in the brain consists mainly of the subunits p50 and p65 (RelA). To show the functional significance of NF-κB activation, we previously used p50-deficient mice. A reduced infarct size in the absence of p50 suggested that NF-κB is involved in ischemic brain damage (Nurmi et al, 2004; Schneider et al, 1999). However, further interpretation of this result is complicated because p50 both represses and transactivates gene transcription, depending on the dimerization partner, the promoter, and cell type that are studied (Franzoso et al, 1992; Kang et al, 1992; Kurland et al, 2001). Moreover, the absence of p50 in all cells and tissues of the body causes slight defects in immune responses (Sha et al, 1995) and potentially also other as yet unknown systemic effects (e.g., changed levels of proinflammatory cytokines released from endothelial cells during ischemia), which might influence ischemic brain damage indirectly. To obtain more definite evidence for the functional significance of NF-κB activation in cerebral ischemia, we have now generated transgenic mice that express a specific dominant inhibitor of NF-κB activation under transcriptional control of neuron- or astrocyte-specific promoters. As a dominant inhibitor of NF-κB, we used IκBα mutated at the phosphorylation sites for IKK (serine-32 and serine-36, IκBα superrepressor, IκBα-SR), which has been successfully used as a transgene before (Hettmann et al, 1999; Vallabhapurapu et al, 2001). Our results show that selective inhibition of NF-κB in neurons reduces ischemic cerebral damage.
MATERIALS AND METHODS
Generation of Transgenic Mice
The IκBα-SR transgene is preceded by the β-globin initiation signal to maximize translation efficiency and by a T7 tag to facilitate detection (Vallabhapurapu et al, 2001). A 2.1-kb fragment of the human growth hormone gene downstream of the cDNA provides intron and polyadenylation sequences. A 1.8-kb fragment of the rat neuron-specific enolase (NSE) promoter, which has been shown to direct gene transcription into neurons (Forss-Petter et al, 1990), was inserted 5′ to the IκBα-SR cDNA (pNSE-IκBα-SR). To construct pGFAP-IκBα-SR (GFAP: glial fibrillary acidic protein), the NSE promoter was replaced by a 2.2-kb DNA fragment of the human GFAP promoter that was derived from the plasmid pEGFP-GFAP (Nolte et al, 2001). This promoter directs transgene expression into astrocytes (Brenner et al, 1994; Nolte et al, 2001). Transgenic mice were obtained by pronuclear injection into oocytes that were derived from female F1 hybrids (C57Bl/6 × DBA2/Crl) and male C57Bl/6 mice. Founders were backcrossed for four to six generations on a C57Bl/6 background. Genomic DNA obtained from tail biopsies was tested by polymerase chain reaction (PCR) using the following primers resulting in a PCR product of 233bp: nm-map1, 5′-cct gtg ttc act agc aac ctc aaa cag aca cc; nm-map2, 5′-gta atc ctc gtc ctt cat ggc gtc caa gcc ggc-3′.
Middle Cerebral Artery Occlusion
At an age of 4 to 8 months, male mice were anesthetized by intraperitoneal injection of 150 μL 2.5% avertin per 10 g body weight. A skin incision was made between the ear and the orbit on the left side. The parotid gland and the temporal muscle were removed by electrical coagulation. The stem of the middle cerebral artery (MCA) was exposed through a burr hole and was occluded by microbipolar coagulation (Erbe, Tübingen, Germany). Surgery was performed under a microscope (Hund, Wetzlar, Germany). Mice were kept at a body temperature of 37°C on a heating pad. After 48 hours they were deeply reanesthetized with avertin and perfused intracardially with Ringer's solution. Brains were removed and immediately frozen on dry ice. Coronal cryosections (20 μm) were cut every 400 μm, starting rostrally. Sections were stained with a silver technique to determine the infarct size (Herrmann et al, 2003). Stained sections were scanned at 600 dpi and the infarct area was measured (ScnImage, Scion, Frederick, MD, USA). The total infarct volume was obtained from integrating infarcted areas corrected for brain edema (Swanson et al, 1990). Surgery and infarct measurement were obtained without a knowledge of the genotype. For the measurement of physiologic parameters in a subgroup of mice, the right femoral artery was cannulated. Samples (100 μL per mouse) were collected for analysis of arterial blood gas, hemoglobin, and glucose. For laser Doppler measurements, the probe (P415–205; Perimed, Järfälla, Sweden) was placed 3 mm lateral and 6 mm posterior to the bregma. Relative perfusion units were determined (Periflux 4001; Perimed, Järfälla, Sweden).
Immunohistochemistry
Coronal cryosections (10 μm) were prepared and air-dried for 1 hour. They were fixed in acetone (–20°C) for 2 mins, air-dried again, and blocked with biotin-blocking solution for 20 mins and with peroxidase-blocking reagent for 5 mins (DAKO, Hamburg, Germany). For staining of the T7 epitope, a monoclonal mouse anti-T7 antibody (Novagen, Madison, WI, USA) was prelabeled with biotin using DAKO ARK (DAKO, Hamburg, Germany). Sections were incubated overnight at 4°C with biotin-labeled anti-T7 (2.5 μg/mL) in phosphate-buffered saline (PBS). For detection of the anti-T7 antibody, a streptavidin-peroxidase complex (ARK, DAKO, Hamburg, Germany) was used. Peroxidase was visualized with 3,3′-diaminobenzidine (DAB). For immunohistochemistry of neurofilament-200 kDa, a rabbit anti-neurofilament-200kDa antibody (1:500, Sigma, Munich, Germany) was used. After washing with PBS, sections were incubated with an alkaline phosphatase-conjugated goat anti-rabbit antibody (1:80, DAKO, Hamburg, Germany) for 30 mins at room temperature. Alkaline phosphatase was visualized with 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium (BCIP/NBT).
Real-time RT-PCR
Mice were reanesthetized and perfused with Ringer's solution 24 hours after middle cerebral artery occlusion (MCAO); the ischemic and the corresponding contralateral cortices were quickly dissected and frozen on dry ice. Tissues were stored at –80°C. RNA was extracted with peqGOLD RNAPure (peqLAB, Erlangen, Germany), according to the manufacturer's instructions. In total 10 μg RNA was transcribed with MMLV reverse transcriptase and random hexamers. Primers for the quantitative real-time PCR are listed in Table 1. Polymerase chain reaction was performed according to the following protocol: 10 mins at 95°C, 15 secs at 95°C, and 1 minute at 60°C (40 cycles). Amplification was quantified with the Gene Amp 5700 sequence detector and the SYBR Green kit (PE Diagnostik, Weiterstadt, Germany). A linear concentration-response curve was established by diluting pooled samples. Quantified results for individual cDNAs were normalized to mean levels for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), cyclophilin, and β-actin. The purity of the amplified products was checked by the dissociation curve.
Primers for real-time PCR
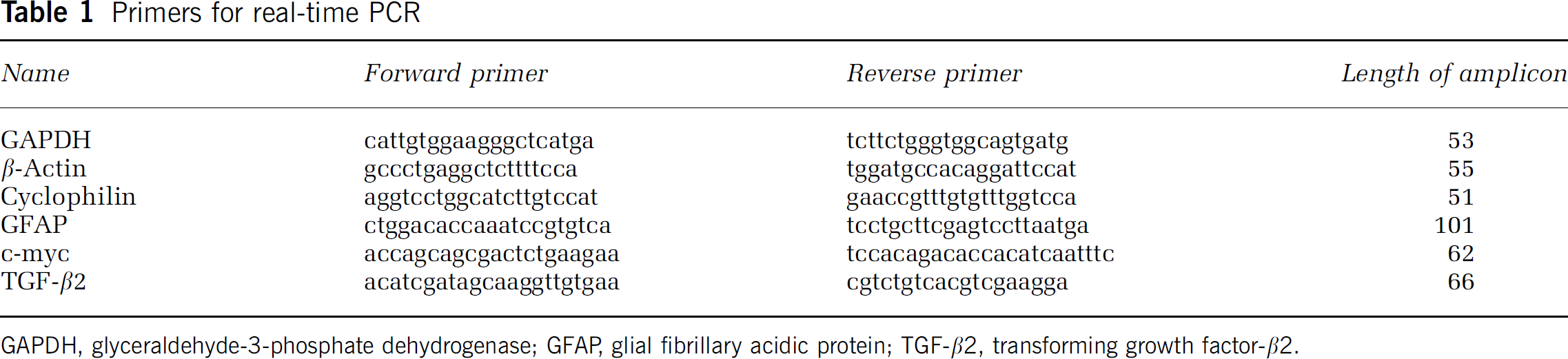
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFAP, glial fibrillary acidic protein; TGF-β2, transforming growth factor-β2.
TUNEL Staining
For terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick end labeling (TUNEL) staining, sections were fixed in 4% paraformaldehyde at room temperature for 30 mins. Then sections were washed twice in PBS for 5 mins and permeabilized for 2 mins with 200 μL permeabilization solution (0.1% Triton X-100 and 0.1% sodium citrate in PBS) at 4°C. After washing, sections were incubated with 50 μL TUNEL Reaction Mix (enzyme solution diluted 1:6 in labeling solution; In Situ Cell Detection Kit, Fluorescein, Roche, Mannheim, Germany) for 1 hour at 37°C in the dark. Then sections were mounted with medium containing 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, Vectashield). TUNEL-and DAPI-positive cells were detected with a fluorescence microscope. In three fields of 0.04 mm2 at the dorsal, ventral, and middle border of the infarct at the level of the anterior commissure, TUNEL-positive cells and DAPI-positive nuclei were counted. TUNEL-positive cells were expressed as percent of total cell count. Cells were quantified without a knowledge of the genotype.
Immunoblot
Tissue lysates (in 50 m mol/L Tris-HCl, pH 7.0, 1% sodium dodecyl sulfate (SDS), 2% 2-mercaptoethanol) were resolved by 10% SDS-polyacrylamide gel electrophoresis and transferred to Hybond nitrocellulose (Amersham, Freiburg, Germany). Membranes were rinsed in TBST (20 m mol/L Tris-HCl, pH 7.6, 150 m mol/L NaCl, 0.1% Tween 20), incubated in 5% nonfat dry milk dissolved in TBST for 2 hours, and then incubated with rabbit anti-IκBα antibodies (0.4 μg/mL, Santa Cruz, Heidelberg, Germany) or mouse anti-T7 (0.3 ng/mL, Novagen, Madison, WI, USA) in TBST. Antibody-antigen complexes were detected with ECL reagents (Amersham, Freiburg, Germany).
Cell Culture and Transfection
Cortical neurons were derived from embryonic day 16 (E16) mice. The cells from individual brains were dissociated and cultured in 24-well plates precoated with poly-d-lysine (50 μg/mL) at a density of 200,000 cells/well. These cells were then incubated in neurobasal medium (Invitrogen, Karlsruhe, Germany) supplemented with B27 (Invitrogen), 0.5 μ mol/L l-glutamine, penicillin (100 IU/mL), and streptomycin (100 μg/mL). After 10 days in vitro, cells were transfected using Lipofectamine 2000 (Invitrogen) and 1 μg/well of the NF-κB reporter plasmid pNF-κB-Luc comprising five tandem repeats of NF-κB binding sites (Stratagene, Amsterdam, Netherlands), according to the manufacturer's protocol. After 24 hours, cells were stimulated as indicated and harvested. To control for cell viability after stimulation with 10 μM camptothecin, cells were cotransfected with 0.1 μg/well of the reference plasmid phRL-TK (Promega, Mannheim, Germany). Renilla luciferase was measured with the Dual-Luciferase Reporter Assay, according to the manufacturer's protocol (Promega). Firefly luciferase activity was measured as described (Sallmann et al, 2000). Cell death was quantified after 16 hours of exposure to 10 μ mol/L camptothecin by staining neurons with Vectashield and counting the cells with condensed and normal nuclei.
The preparation and culture of primary mouse astrocytes has been described before (Schwaninger et al, 2000). Cells were transfected with pNF-κB-Luc (2 μg/6-cm well) and GFAP-IκBα-SR using Transfast (Promega, Mannheim, Germany), according to the manufacturer's instructions. Cotransfections were performed with a constant DNA concentration, which was maintained by adding pBluescript (Stratagene, La Jolla, CA, USA). At 42 hours after transfection, cells were stimulated for 6 hours and harvested.
Statistical Analysis
Data are illustrated as mean ± s.d. Statistical comparisons of three or more groups were made by analysis of variance followed post hoc by Tukey's honestly significant difference (Tukey-HSD) or Fisher's protected least-squares difference (LSD) as indicated. Two groups were compared by a two-sided t-test. Values were considered to be significant at P<0.05.
RESULTS
The DNA constructs that were used for the generation of transgenic mice are schematically depicted in Figure 1. For selective inhibition of NF-κB in neurons or astrocytes, the superrepressor of NF-κB IκBα-SR was put under transcriptional control of the rat 1.8-kb, neuron-specific NSE or the human 2.2-kb, astrocyte-specific GFAP promoter, respectively. To verify that the T7-tagged, mutated form of IκBα (IκBα-SR) acts as an inhibitor of NF-κB, we transfected the construct pGFAP-IκBα-SR together with an NF-κB reporter gene into primary mouse astrocytes. NF-κB activity stimulated by phorbol 12-myristate 13-acetate (TPA) plus thapsigargin was inhibited by expression of IκBα-SR in a dose-dependent manner (Figure 2).
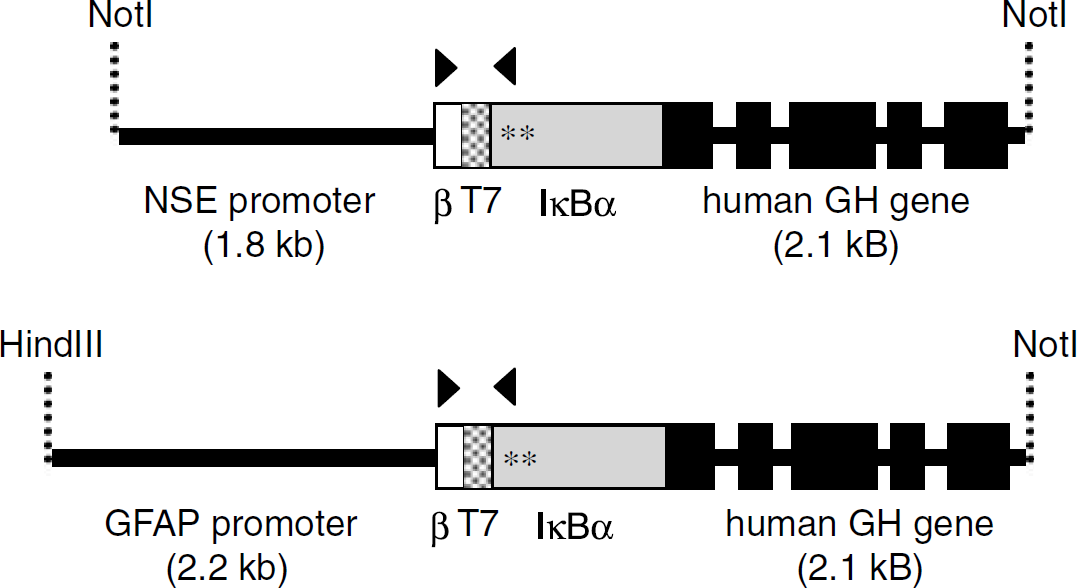
Schematic drawing of the DNA fragments for the generation of NSE-IκBα-SR (upper panel) and GFAP-IκBα-SR transgenic mice (lower panel). β, β-globin translational initiation site; T7, T7-antibody tag; arrowheads, primers for genotyping; *, mutation of serine to alanine; GH, growth hormone sequences.
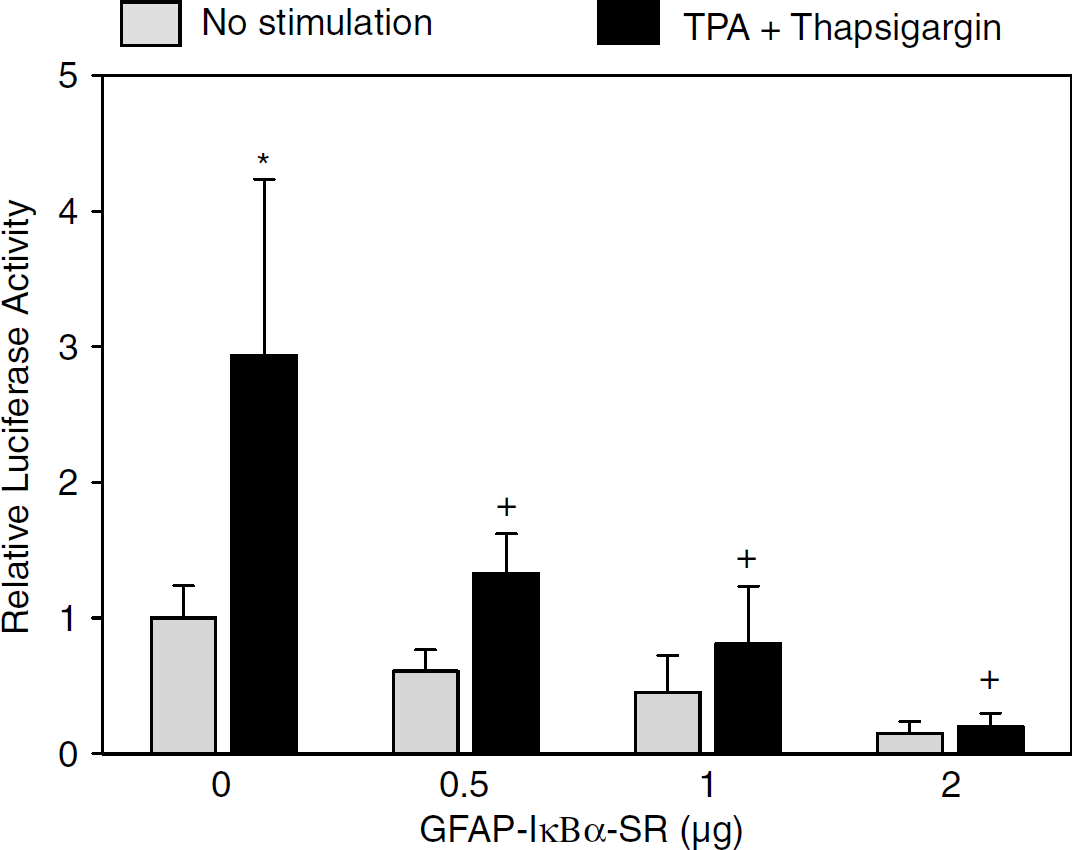
Expression of IκBα-SR in astrocytes inhibits NF-κB activation. Primary astrocytes were transiently transfected with the construct pGFAP-IκBα-SR and stimulated with 300 n mol/L TPA plus 1 μ mol/L thapsigargin as indicated. Luciferase activity is expressed relative to the unstimulated control group of the experiment. Values are means ± s.d. of two independent experiments each performed in triplicate. P<0.0001 (one-way ANOVA). *P<0.0001 compared with the unstimulated control group. +P<0.0001 compared with the stimulated group without pGFAP-IκBα-SR (Tukey-HSD post hoc test).
We obtained three founders for the NSE-IκBα-SR construct and four founders for the GFAP-IκBα-SR construct. Two founder lines for each construct were further investigated (NSE-IκBα-SR211, NSE-IκBα-SR241, GFAP-IκBα-SR211, GFAP-IκBα-SR192). The transgenes did not have any obvious effect on behavior or breeding nor was gross brain morphology altered by the transgene (data not shown).
The transgenic IκBα-SR has a higher molecular weight than the endogenous form because of the T7 tag. This allowed a direct comparison of the expression of transgenic and endogenous IκBα by immunoblotting. Transgenic IκBα-SR levels in brain extracts were lower than endogenous IκBα in all transgenic lines (Figure 3A). The specificity of the band in immunoblots was confirmed by detection of the transgene with an antibody against T7 (Figure 3A). Transgene expression was restricted to the brain and could not be detected in heart, kidney, liver, or spleen (data not shown).
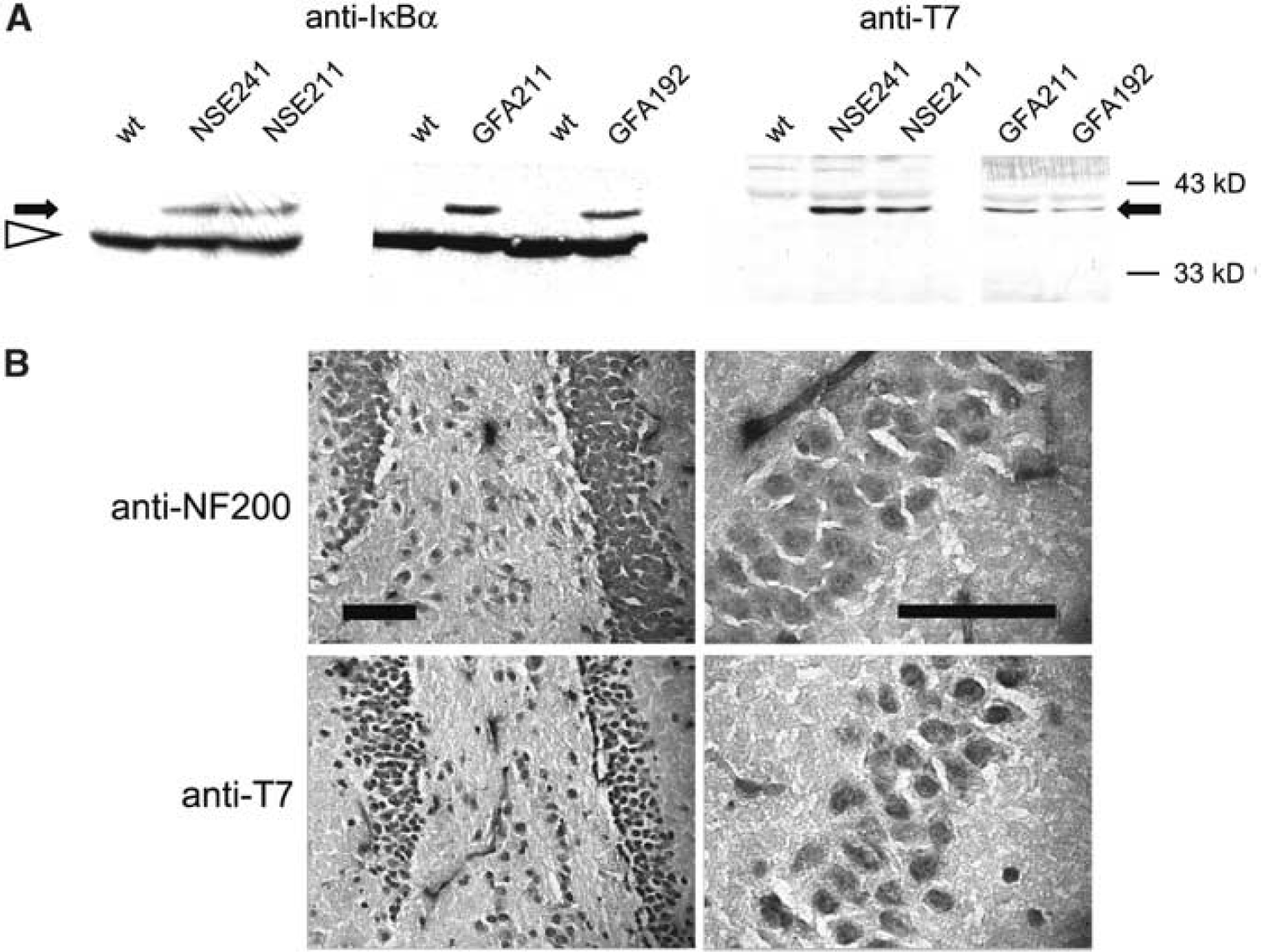
Characterization of the expression of the IκBα-SR transgene. (
The cell specificity of the transgene expression in NSE-IκBα-SR mice was further verified by immunohistochemistry. The neuronal cell layer of the dentate gyrus of the hippocampus that expresses the neuronal marker neurofilament-200 was also stained by an antibody against the transgene epitope T7 (Figure 3B). In addition, neurons in the granular layer of the cerebellum and in the cortex were positive for the T7 marker (not shown). Primary cortical neurons in vitro expressed the transgene as shown by immunoblots of cell extracts (Figure 4A). To test whether transgene expression interferes with NF-κB activation, we used a reporter gene assay in primary cortical neurons from mice with the NSE-IκBα-SR transgene and from wild-type littermates. NF-κB-driven luciferase expression was stimulated by 10 ng/mL TNF-α, a classical NF-κB inducer. In cortical neurons from transgenic mice, NF-κB activation was preserved, but was significantly lower (Figure 4B). To investigate the effect of NF-κB inhibition on neuronal cell survival, we used camptothecin, a DNA-damaging agent (Park et al, 1997). DNA damage is an important component of the pathophysiology of cerebral ischemia, well in advance of DNA fragmentation caused by the apoptotic process (Chen et al, 1997; Cui et al, 2000; Tobita et al, 1995). Camptothecin (10 μ mol/L for 16 h) stimulated NF-κB-driven luciferase expression 4.0 ± 1.0-fold over controls (n = 6, P<0.0001). Camptothecin exposure for 16h also induced neuronal apoptosis that was quantified by counting cells with condensed nuclei. In prior experiments nuclear condensation corresponded closely to TUNEL staining. The percentage of apoptotic neurons was significantly smaller in primary cortical cultures expressing the transgene IκBα-SR (Figure 4C). Thus, in primary neurons of NSE-IκBα-SR mice, the transgene is expressed and inhibits NF-κB activation and apoptosis.
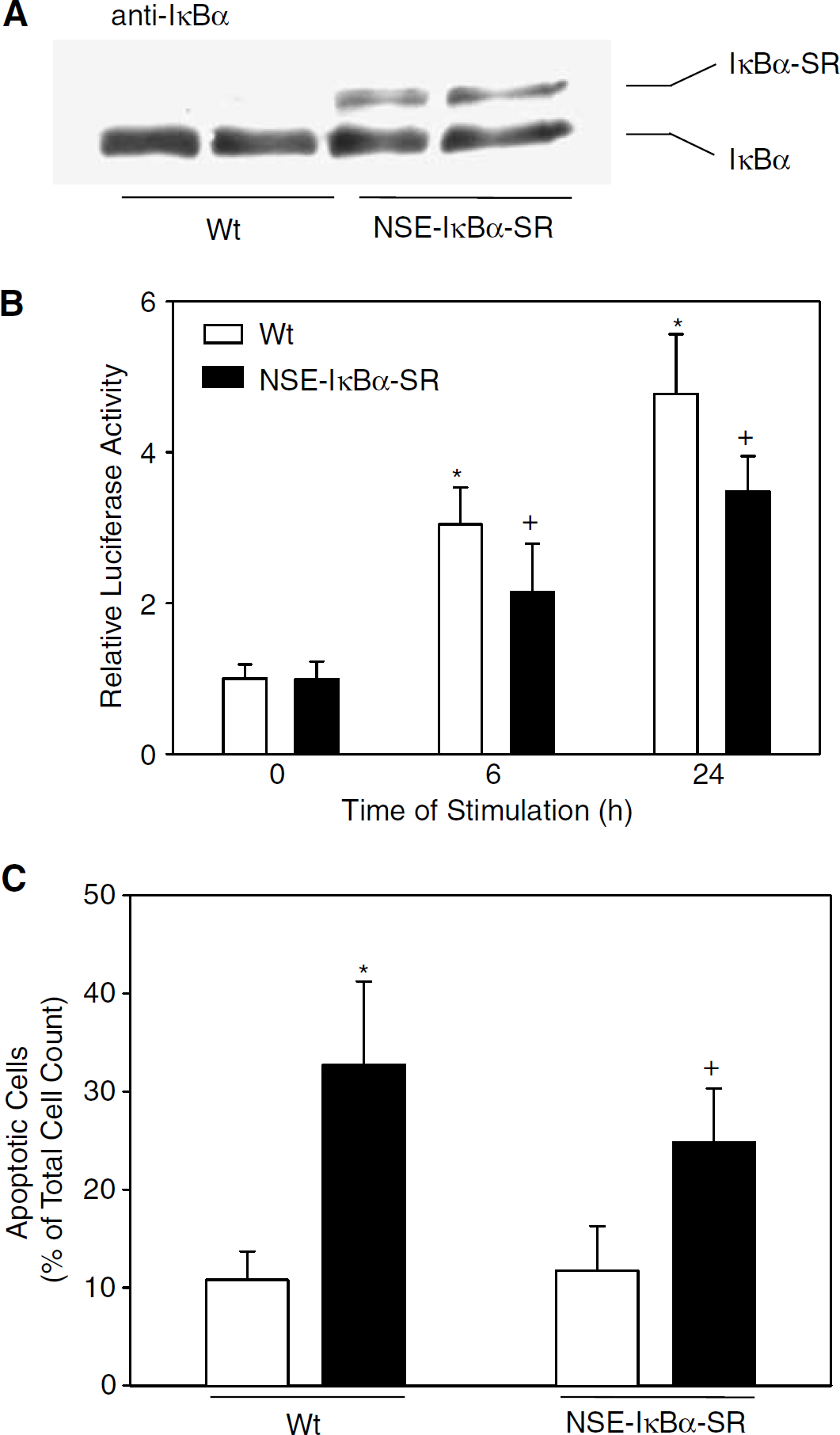
Expression and functions of IκBα-SR in primary cortical neurons of NSE-IκBα-SR211 mice. (
To evaluate the effect of the IκBα-SR transgene on NF-κB activation in vivo, we measured mRNA accumulation of specific NF-κB target genes in the cortex by real-time RT-PCR. The promoter of the c-myc gene contains two functional NF-κB sites and the transforming growth factor-β2 (TGF-β2) promoter contains a putative NF-κB site (Duyao et al, 1990; La Rosa et al, 1994; Malipiero et al, 1990). Both c-myc and TGF-β2 are known to be induced in neurons by cerebral ischemia (Ata et al, 1999; Huang et al, 2001). Middle cerebral artery occlusion for 24 hours induced a significant increase in the mRNA concentration of c-myc and TGF-β2 in wild-type mice (Figures 5A, B). However, in NSE-IκBα-SR mice, MCAO did not stimulate mRNA levels of c-myc and TGF-β2, supporting the notion that the NSE-IκBα-SR transgene inhibits the transcriptional activity of NF-κB in neurons after MCAO. Glial fibrillary acidic protein is a cell-specific marker for astrocytes and contains a promoter element that binds the NF-κB subunit p50 (Chen and Swanson, 2003; Krohn et al, 1999). In MCAO, GFAP expression was upregulated in both wild-type and NSE-IκBα-SR mice, indicating astrocytic activation (Figure 5C). In GFAP-IκBα-SR mice, however, the induction of GFAP was significantly reduced (Figure 5C). Immunohistochemical study of GFAP after 48 hours of MCAO supported a reduced induction of GFAP in GFAP-IκBα-SR mice compared with wild-type littermates and NSE-IκBα-SR mice (data not shown). These data indicate a partial but significant inhibition of NF-κB activity in astrocytes and neurons in the respective transgenic line.
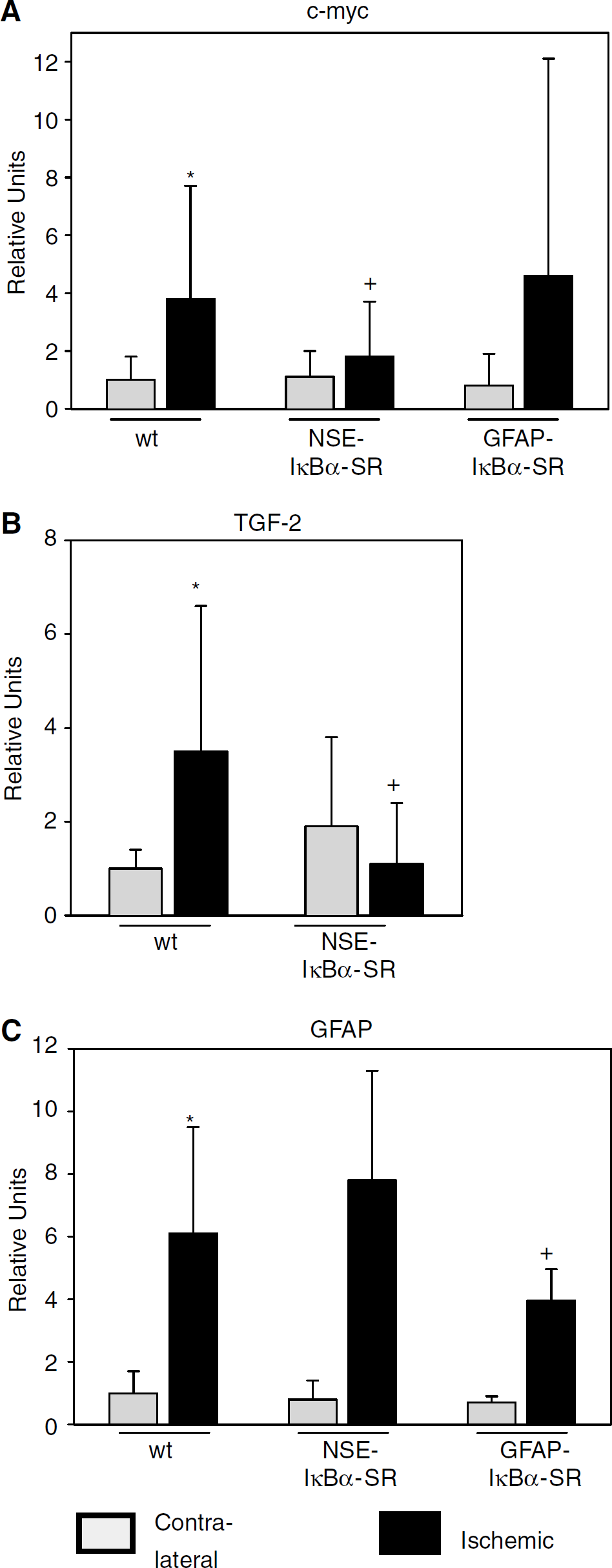
Cell-type-specific expression of IκBα-SR reduces the induction of NF-κB target genes by cerebral ischemia. mRNA accumulation was quantified by reverse transcription and real-time polymerase chain reaction (PCR) 24 hours after onset of middle cerebral artery occlusion (MCAO). (
To investigate the consequences of cell type-specific inhibition of NF-κB on ischemic damage, we compared the infarct size in IκBα transgenic lines and wild-type littermates after 48-hour MCAO. In NSE-IκBα-SR211 mice, the infarct size was significantly smaller (26 %) than in controls (Figure 6, left panel; 18.8 ± 2.4 mm3 (n = 15) in NSE-IκBα-SR211 versus 25.4 ± 2.1 mm3 (n = 12) in controls; P<0.05). To exclude an insertional effect of the transgene, the experiment was repeated with another founder line (NSE-IκBα-SR241) expressing the same transgene. In this line, infarct size was also significantly smaller than in wild-type littermates (reduction by 43%, n = 10; P<0.05). Physiologic parameters during the surgery did not differ between NSE-IκBα-SR mice and wild-type littermates. In addition, the reduction in cerebral blood flow between groups was nearly identical (Table 2). In contrast to NSE-IκBα-SR lines, the infarct size in two GFAP-IκBα-SR mouse lines did not differ from controls (GFAP-IκBα-SR192, Figure 6, right panel; GFAP-IκBα-SR211, data not shown). These data demonstrate that inhibition of NF-κB in neurons, but not in astrocytes, reduces the infarct size after permanent MCAO.
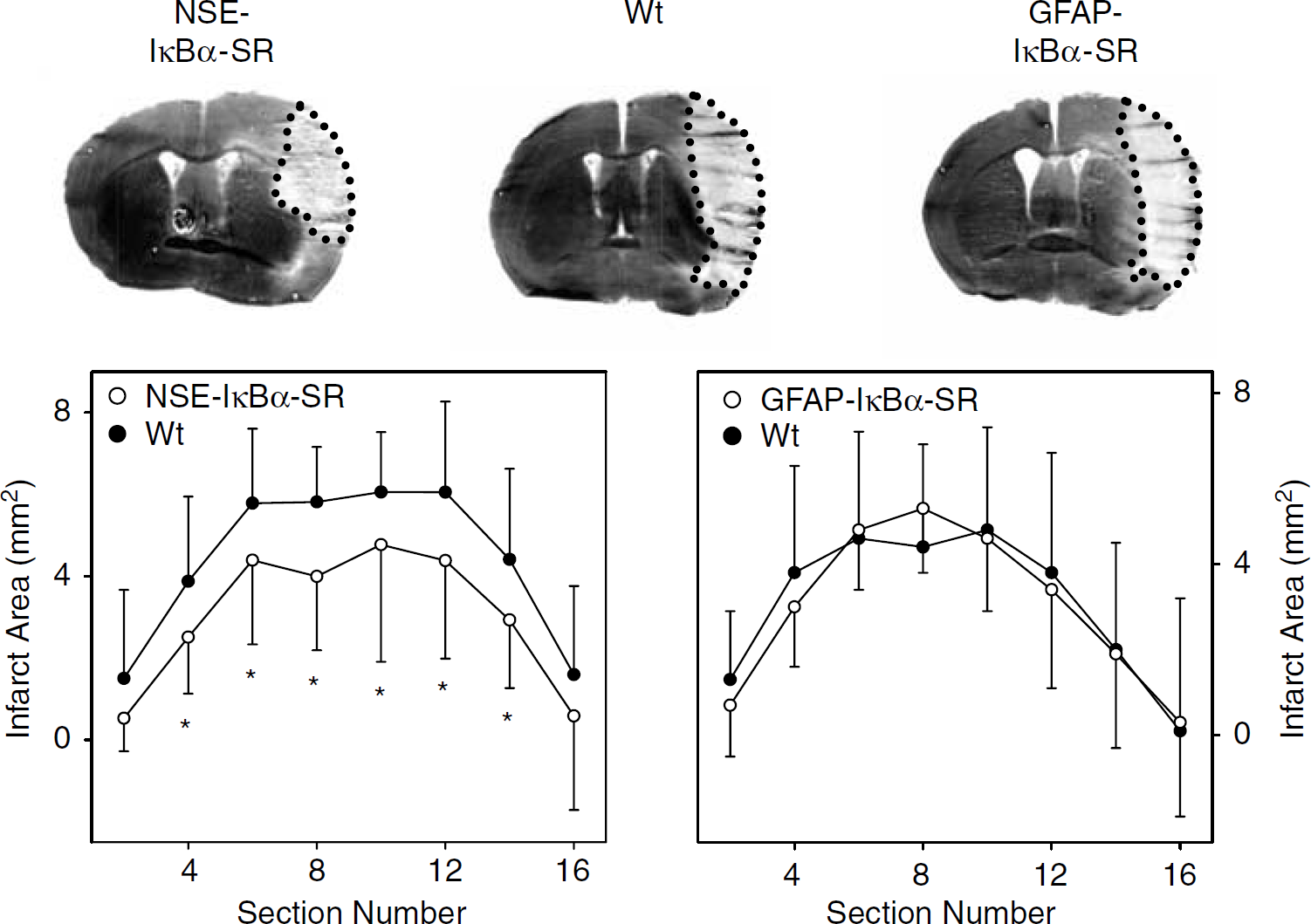
Neuronal, but not astrocytic, expression of the NF-κB inhibitor IκBα-SR reduces the infarct size. After 48 hours of permanent middle cerebral artery occlusion (MCAO), infarct areas were determined on consecutive coronal brain sections using silver staining. Typical sections at the level of the anterior commissure are shown at the top. Infarct areas of NSE-IκBα-SR211 mice, GFAP-IκBα-SR192 mice, or the respective wild-type littermates (Wt) are plotted from rostral to caudal. Values are means ± s.d. (n = 12 to 15) of the average infarct area on two consecutive coronal brain sections. The anterior commissure is seen on section 10. *P<0.05 compared with wt (ANOVA, LSD post hoc test).
One possible explanation for the observed reduction in infarct size is protection of neurons against ischemic cell death. Staining with the fluorescent dye fluoro-jade B, which specifically marks dying neurons but does not distinguish between necrotic and apoptotic neurons (Schmued and Hopkins, 2000), showed slightly reduced neuronal cell death in the outer zone of the infarct in NSE-IκBα-SR mice, but this did not reach statistical significance (data not shown). TUNEL staining is considered to be more specific for apoptotic cell death than fluoro-jade B. In accordance with this notion, only about 40% of fluoro-jade B-positive cells were TUNEL positive. The number of TUNEL-positive cells was significantly reduced in infarcts of NSE-IκBα-SR mice. In GFAP-IκBα-SR mice, there was a slight reduction in the number of TUNEL-positive cells that did not reach statistical significance (Figure 7, P = 0.075). Previous work has shown that, after MCAO, most TUNEL-positive cells in the brain are neurons (Schneider, 1999). These data support the concept that NF-κB in neurons contributes to the induction of apoptotic neuronal cell death.
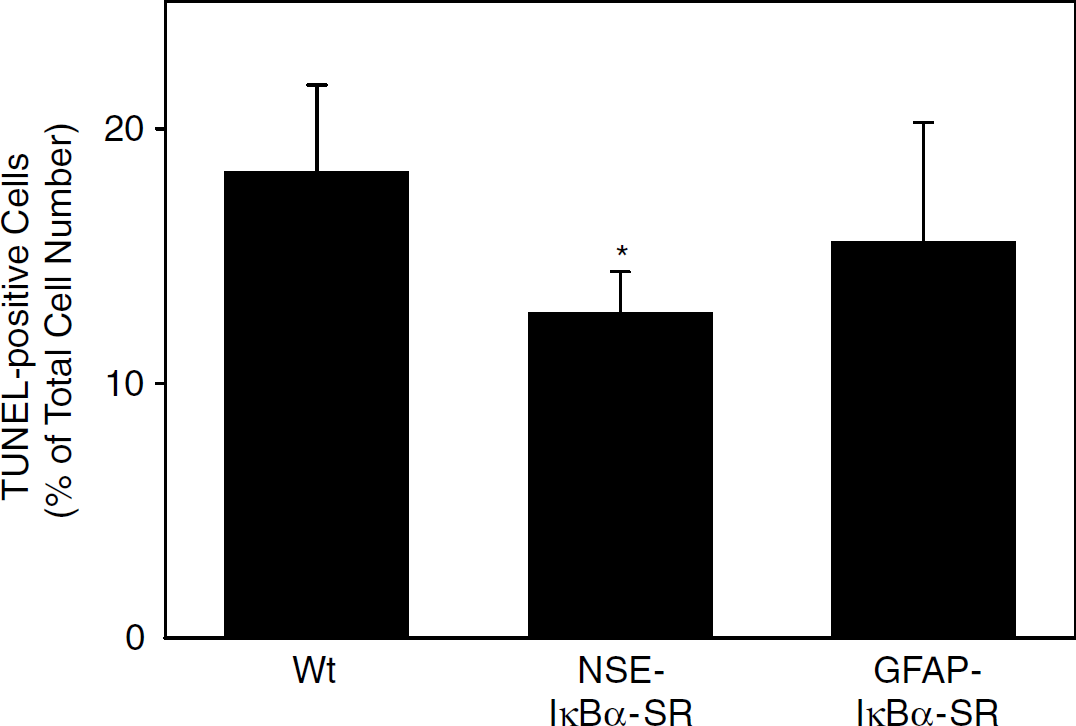
Neuronal, but not astrocytic, expression of the NF-κB inhibitor IκBα-SR reduces ischemic cell death. TUNEL-positive cells were counted at the border of the infarct at the level of the anterior commissure and expressed as percent of DAPI-positive cells. Values are means ± s.d. (n = 5 for NSE-IκBα-SR and GFAP-IκBα-SR; n = 10 for wild-type, Wt). *P = 0.05 (ANOVA, Tukey-HSD post hoc test).
DISCUSSION
To investigate the contribution of the transcription factor NF-κB to brain pathology, we have generated mice that express the NF-κB superrepressor IκBα-SR under transcriptional control of an NSE and an astrocyte-specific promoter (GFAP). Mice with neuronal and astrocytic expression of the superrepressor have an apparently normal phenotype, although formal behavioral tests have not yet been performed. In addition, body weight was the same as in littermates. Transgene expression effectively reduced the induction of neuron- or astrocyte-specific target genes of NF-κB after MCAO. However, only neuronal expression of IκBα-SR interfered with infarct volume after 48 hours of permanent MCAO. The neuroprotective effect of neuronal IκBα-SR was reproduced in another founder line. Our data provide definitive evidence for the detrimental role of NF-κB during the pathogenesis of stroke. Previous transgenic approaches could not clarify this point because deficiency of p65, the main transactivating subunit of NF-κB in the brain, causes embryonic death (Beg et al, 1995) and the use of p50 –/– mice is obscured by reports that p50 can both activate and repress NF-κB activity. Future work will have to investigate whether inhibition of NF-κB also leads to a long-term reduction of the infarct size.
Physiologic parameters 15 mins before and 15 mins after MCAO in control and NSE-IκBα-SR241 mice
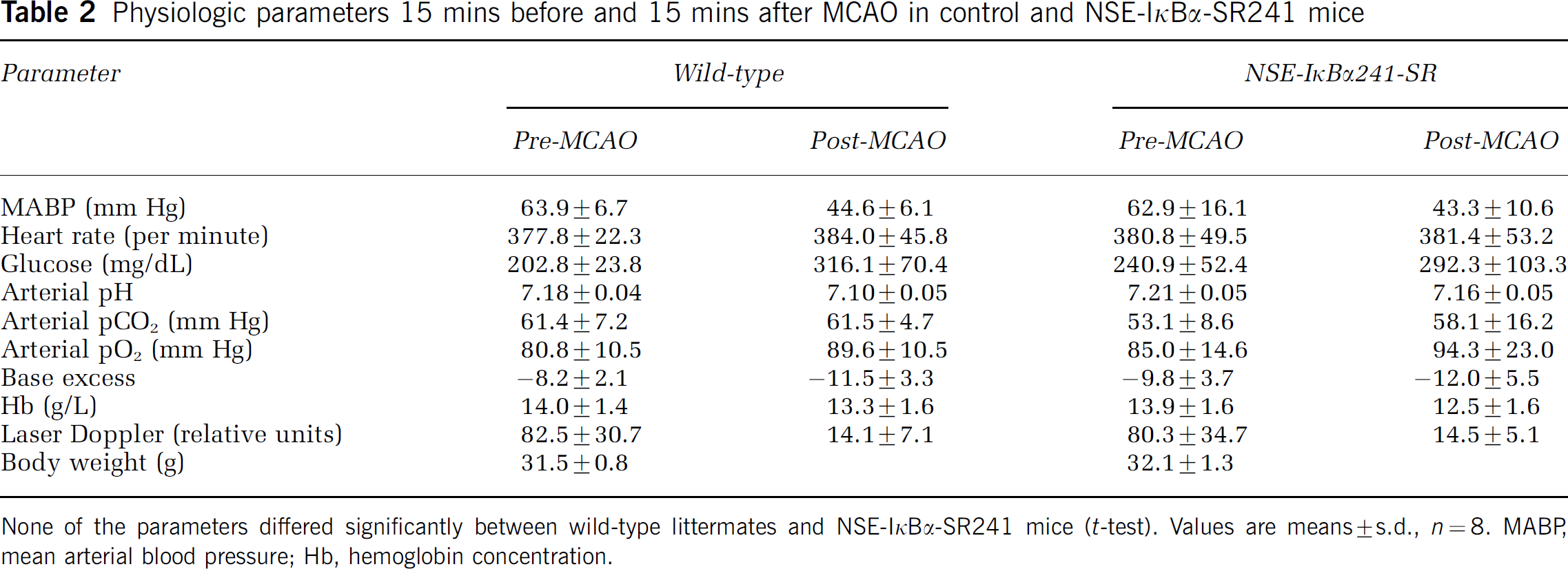
None of the parameters differed significantly between wild-type littermates and NSE-IκBα-SR241 mice (t-test). Values are means ± s.d., n = 8. MABP, mean arterial blood pressure; Hb, hemoglobin concentration.
Several investigators have found that NF-κB is activated in neurons after cerebral ischemia (Huang et al, 2001; Schneider et al, 1999; Stephenson et al, 2000). However, previous in vitro work of the role of NF-κB in neuronal cell death only provided ambiguous results in contrast to nonneuronal cell types. Several studies have found an antiapoptotic effect of NF-κB in neurons in vitro (reviewed in Mattson and Camandola, 2001). However, NF-κB can also promote neuronal cell death in vitro (de Erausquin et al, 2003; Pizzi et al, 2002; Qin et al, 1999). Indeed, cortical neurons expressing the NF-κB superrepressor were partially protected against camptothecin-induced apoptosis (Figure 4C). Using the same superrepressor of NF-κB, Fridmacher et al (2003) recently observed that inhibition of neuronal NF-κB reduced spontaneous cell death but increased FeSO4-induced cell death in vitro. Whether the effect is pro- or anti-apoptotic depends on the stimulus, the cell type, the activated subunits, and the duration of NF-κB activation (Kaltschmidt et al, 2002; Pizzi et al, 2002; Ryan et al, 2000). In the ischemic brain, multiple potential stimuli of NF-κB are released, which makes it difficult to predict the net effect on NF-κB activation from in vitro data. Our results show in vivo that neuronal activation of NF-κB in cerebral ischemia contributes to ischemic brain damage. Possibly, the neuroprotective effect of the super-repressor is due to the moderate expression level in our mouse lines because there is evidence that a critical NF-κB dosage is required for cell survival and either too much or too little activation is detrimental (Goudeau et al, 2003).
Numerous target genes of NF-κB are upregulated in cerebral ischemia. Candidates, which may mediate the toxic effect of NF-κB in neurons, are c-myc and TGF-β2 (Ata et al, 1999; Huang et al, 2001). c-myc functions as a proapoptotic regulator in cells of various types and under a variety of conditions (Packham and Cleveland, 1995). It has been linked to N-methyl-d-aspartate (NMDA) receptor- and NF-κB-mediated apoptosis in neurons (Qin et al, 1999). Administration of TGF-β1 has been reported to protect against focal cerebral ischemia (Prehn et al, 1993). However, endogenous TGF-β is required in vivo for apoptosis of axotomized motoneurons as shown by the application of neutralizing antibodies (Krieglstein et al, 2000).
In GFAP-IκBα-SR mice, expression of the transgene IκBα-SR in astrocytes apparently inhibited NF-κB activity because the upregulation of GFAP was reduced (Figure 3). Glial fibrillary acidic protein is a marker of astroglia activation and a target gene of NF-κB (Krohn et al, 1999), implying a role of NF-κB in glial activation in vivo. However, inhibition of astrocytic NF-κB had no effect on infarct size. A neuroprotective function of GFAP has been reported in cerebral ischemia (Nawashiro et al, 2000). Nevertheless, the moderate inhibition of GFAP induction in our mice had no detrimental effect or was overridden by the effect on other target genes. Regulation of the endogenous GFAP gene suggests that IκBα-SR, which is directed by the human GFAP promoter, also exerts negative feedback on its own expression but is still upregulated in ischemic cortex compared to the nonischemic side.
Pharmacological studies support our concept of the neurodegenerative function of NF-κB in cerebral ischemia although the inherent unspecificity of drugs only provides correlative evidence. The antioxidant LY341122 (Stephenson et al, 2000), the salicylate triflusal (Acarin et al, 2001), the proteasome inhibitor MLN519 (Williams et al, 2003), the ubiquitin ligase inhibitor pyrrolidine dithiocarbamate (Hayakawa et al, 2003; Nurmi et al, 2004), and the cannabinoid dexanabinol (Jüttler et al, 2004) all inhibit NF-κB activation and exert neuroprotective effects in cerebral ischemia. The most specific step in the NF-κB signaling is the activation of the IKK complex (Li and Verma, 2002). Because the mutation of IKK phosphorylation sites in IκBα prevents degradation of IκBα and inhibits NF-κB, our results from NSE-IκBα-SR transgenic mice suggest that activation of NF-κB through IKK is a critical step in cerebral ischemia. Pharmacological IKK inhibitors might, therefore, represent a specific and effective treatment of stroke.
Footnotes
Acknowledgements
Dr F Kirchhoff, Berlin, kindly provided pEGFP-GFAP. The authors thank Anja Buhl for technical assistance.