
Abstract
Preconditioning with sublethal ischemia results in natural tolerance to ischemic stress, where multiple mediators of ischemic damage are simultaneously counteracted. Tumor necrosis factor alpha (TNF-α) has been implicated in development of ischemic tolerance. Using cellular models of ischemic tolerance, we have demonstrated that an effector of TNF-α– induced preconditioning is ceramide, a sphingolipid messenger in TNF-α signaling. TNF-α/ceramide-induced preconditioning protected cultured neurons against ischemic death and cultured astrocytes against proinflammatory effects of TNF-α. TNF-α activates a transcription factor NF-κB that binds promoters of multiple genes, thus ensuring pleiotropic effects of TNF-α. We describe here a mechanism that allows selective suppression of TNF-α/NF-κB–induced harmful genes in preconditioned cells while preserving cytoprotective responses. We demonstrate that in astrocytes activation of an adhesion molecule ICAM-1 by TNF-α is regulated through association of the phosphorylated p65 subunit of NF-κB with an adapter protein, p300, and that in preconditioned cells p65 remains unphosphorylated and ICAM-1 transcription is inhibited. However, TNF-α–activated transcription of a protective enzyme, MnSOD, does not depend on p300 and does not become inhibited in preconditioned cells. This new understanding of TNF-α–induced adaptation to ischemic stress and inflammation could suggest novel avenues for clinical intervention during ischemic and inflammatory diseases.
Keywords
There are multiple, interrelated mechanisms that cause progressive brain damage during the initial hours of ischemic stroke. They include dysfunction of ion homeostasis mechanisms, neurotransmitter release, failure of blood–brain barrier, compromised microcirculatory perfusion, oxidative stress, production of multiple cytokines, upregulation of adhesion molecules, and expression of apoptotic proteins. These reactions are thought to be counteracted by multiple neuroprotective mechanisms (Mattson et al., 2000a). Therapeutic interventions targeting single mechanisms in this complex network have produced only subtle effects on the outcome of stroke and overwhelmingly have led to negative clinical trials. An understanding of mechanisms by which the brain becomes tolerant to ischemic stress when preexposed to sublethal ischemia (Chen and Simon, 1997) could guide investigators to therapeutic targets that can simultaneously counteract multiple mediators of ischemic damage and produce more robust clinical effects.
Tumor necrosis factor alpha (TNF-α) has been implicated in both detrimental (Barone and Feuerstein, 1999; del Zoppo et al., 2000) and neuroprotective mechanisms of ischemic injury (Barger et al., 1995; Dawson et al., 1996). Recent studies in murine and rat models of brain ischemia demonstrate that TNF-α is a key mediator of ischemic preconditioning as well. It can substitute for ischemic preconditioning when applied intracisternally (Nawashiro et al., 1997) or elicited systemically by injection of lipopolysaccharide 48 to 72 hours before ischemic insult (Tasaki et al., 1997). Preexposure of cultured neurons to sublethal hypoxia or TNF-α caused an equal degree of protection against hypoxic injury, and inhibition of TNF-α in hypoxia-preconditioned cells abolished the tolerant state (Liu et al., 2000). We have demonstrated that a mediator of TNF-α–induced preconditioning is de novo synthesized ceramide, a sphingolipid messenger in TNF-α signaling (Liu et al., 2000). TNF-α/ceramide–induced preconditioning also protected astrocytes and brain endothelial cells against proinflammatory effects of TNF-α. (Ginis et al., 1999). In this model, TNF-α acted both as a stressor and a preconditioning effector. Preconditioning with TNF-α suppressed upregulation of ICAM-1 adhesion receptor, mRNA, and protein, in response to either TNF-α or hypoxia. The pathogenic role of endothelial ICAM-1 in ischemia/reperfusion injury has been demonstrated in multiple studies. One should not underestimate, however, the functional significance of ICAM-1 expression on astrocytes. Ionizing radiation (Kyrkanides et al., 1999) and stab wounds (Shibayama et al., 1996) activated ICAM-1 expression in astrocytes, probably through microglia. Monocyte–astrocyte interaction results in induction of cytokine and chemokine synthesis in monocytes (Hery et al., 1995; Andjelkovic et al., 2000). It is possible that ICAM-1 on astrocytes functions as a costimulatory molecule. It participates in antigen presentation by astrocytes, and cross-linking of ICAM-1 initiates signals for TNF-α production via ERK kinase and p38 kinase (Lee et al., 2000). We have previously reported that signaling via ICAM-1-LFA-1 interaction delays leukocyte apoptosis, thus promoting tissue injury (Ginis and Faller, 1997).
Because transcription factor NF-κB is solely responsible for ICAM-1 upregulation in astrocytes (Lee et al., 1998), these data suggested that cell tolerance results in alteration of mechanisms controlling activation of NF-κB. However, recent studies demonstrate that NF-κB activation is necessary for brain cell survival (Mattson et al., 2000b). This controversy was addressed in the investigation presented here. We show that initial steps of NF-κB activation (degradation of the inhibitor of NF-κB [IκB], nuclear translocation of NF-κB and its DNA binding activity) were not altered in tolerant cells. However, preconditioning with TNF-α or ceramide inhibited phosphorylation of the p65/RelA subunit of NF-κB, which was necessary for its interaction with p300 adapter protein and activation of ICAM-1 transcription. This mechanism of stress adaptation was specific for the ICAM-1 gene and did not affect expression of another NF-κB–dependent gene, MnSOD. These results demonstrate for the first time a molecular mechanism that allows cells to differentially control a potentially harmful stress response while preserving cytoprotective responses.
MATERIALS AND METHODS
Cortical astrocyte cultures were established from 2-day-old Sprague-Dawley rats as previously described (Ginis et al., 1999). Passages 1 to 3 were used.
TNF-α and ceramide preconditioning
Confluent astrocyte cultures were incubated for 4 hours with 50 ng/ml rat recombinant TNF-α (Chemicon International, Temecula, CA, U.S.A.). TNF-α–containing medium was replaced by fresh culture medium and cells were allowed to rest for 20 hours. They were then activated again with the same dose of TNF-α for indicated times. For ceramide preconditioning, N-acetylceramide (C-2 ceramide; Biomol, Plymouth Meeting, PA, U.S.A.) was added to the cultures at 10 μmol/L 1 hour before TNF-α addition.
RNA isolation and Northern blotting
Total RNA was extracted with Qiagen RNeasy Mini Kit (Qiagen Inc., Valencia, CA, U.S.A.) according to the manufacturer's instructions. RNA samples were electrophoresed, transferred to a nitrocellulose membrane, and baked in a vacuum oven for 2 hours at 80°C. Prehybridization was performed at 42°C with water solution of 6X SSC, 5X Denhardt's reagent, 0.5% sodium dodecyl sulfate (SDS), 100 μg/mL denatured salmon sperm DNA, and 50% formamide. Hybridization was carried out at 42°C for 24 hours by adding to the prehybridization solution (without Denhardt's reagent) 32P-DNA probes. Rat ICAM-1 cDNA was a gift from Dr. Donald Anderson (Pharmacia, Inc., Peapack, NJ, U.S.A.). Rat MnSOD cDNA was a gift from Dr. Andres Melendez (Albany Medical College, Albany, NY, U.S.A.). The blots were first washed with 2X SSC/0.1% SDS at room temperature and then with 1X SSC/0.1% SDS at 37°C, and then exposed to X-ray film (Kodak, Rochester, NY, U.S.A.).
Immunofluorescent staining for p65 subunit of NF-κB
Naïve and ceramide-pretreated astrocytes were incubated with 50 ng/mL TNF-α for 30 minutes. Cells were fixed with ethanol for 2 minutes and then with 3.7% formaldehyde for 5 minutes and immunostained with the anti-p65 subunit of NF-κB (Santa Cruz Biotechnology Inc., cat. no. sc109, Santa Cruz, CA, U.S.A.) at 1:50 dilution. Detection was performed with anti-rabbit biotinylated secondary antibody, followed by addition of streptavidin–fluorescein isothiocyanate. Digitized images of the fluorescent cells were generated using an Zeiss Axiovert 100 light microscope (20x) equipped with a digital CCD Camera C4742–95–12 (Hamamatsu Corporation, Bridge-water, NJ, U.S.A.).
Flow fluorocytometric analysis
Naïve and ceramide-pretreated astrocytes were activated with TNF-α for 24 hours, then fluorescently stained with anti-rat ICAM-1 monoclonal antibody as previously described (Ginis et al., 1999) and analyzed by means of a Becton-Dickinson (Franklin Lakes, NY, U.S.A.) FACScan flow cytometer.
Immunoprecipitation
To determine whether p65 associates with p300 on TNF-α activation, naïve and preconditioned astrocytes were activated with TNF-α for 30 minutes. At the end of incubation, nuclear extracts were prepared as has been described elsewhere (Ginis et al., 2000). Before immunoprecipitation, aliquots of nuclear extracts with equal amount of protein (usually 0.5–1 mg/sample) were diluted 1:4 with the buffer used for extraction (buffer B), but containing no NaCl (20 mmol/L HEPES, 50 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 10% (wt/vol) glycerol, pH = 7.9) to adjust salt concentration to 100 mmol/L, and then incubated with a mixture of two goat polyclonal anti-p65 antibody (Santa Cruz Biotechnology; cat. no. SC109 and SC-372) at 4 μg antibody per 1 mg protein overnight and then with 50 mL Protein G PLUS-Agarose (Santa Cruz Biotechnology) for 2 hours. The beads were washed 3 times with the diluted buffer B, boiled in 30 μl of sample-loading buffer/1 mmol/L dithiothreitol (DTT) for 3 minutes and electrophoresed on 4% to 12% Tris-glycine or on tris-acetate mini gels. p300 in precipitates was identified by Western blotting with p300-specific antibody (Upstate Biotechnology, cat. no. 05–267, Lake Placid, NY, U.S.A.) All electrophoresis buffers, minigels, nitrocellulose membranes, and electrophoresis equipment were from Novex (San Diego, CA, U.S.A.). For the studies of p65 phosphorylation, naïve and preconditioned astrocytes were activated with TNF-α for 15 minutes. At the end of incubation, the cells were washed twice with cold phosphate-buffered saline and immediately frozen on dry ice to inhibit phosphatase activity. Cells were scrapped into 600 mL of ice-cold lysis buffer [50 mmol/L Tris-HCl, (pH = 8.0), 1% Nonidet P-40, 150 mmol/L NaCl, 2 mmol/L EGTA, proteinase and phosphatase inhibitor cocktails (both from Sigma, St. Louis, MO, U.S.A.), 1 mmol/L DTT] and incubated on ice for 30 minutes, then microcentrifuged at 14,000g for 15 minutes and stored at −70°C. Samples equal in protein content (usually 2 mg protein) were diluted twofold with immunoprecipitation buffer [the aforementioned lysis buffer plus 0.1% SDS and 0.5% Na deoxycholate and immunoprecipitated with 10 (g anti-phosphoserine polyclonal antibody (Zymed Laboratories, San Francisco, CA, U.S.A.; cat. no. 61–8100) as described herein for p65 immunoprecipitation. Identification of p65 in immunoprecipitates was performed by Western blotting with anti-p65 antibody (Santa Cruz Biotechnology, cat. no. SC-372).
Western blots
For NF-κB activation studies, cytosolic or nuclear extracts were loaded on a gel at 10 μg protein/lane for determination of p65 concentrations in nuclear extracts, and at 15 μg protein/lane for IκB determination in cytosolic fractions. Anti-IκBα and anti-p65 rabbit polyclonal antibodies were from Santa Cruz Biotechnology (SC-203 and SC-372, respectively). For MnSOD blots, naíve and preconditioned cells were incubated with TNF-α for 24 hours; whole cell lysates were prepared as already described for p65 phosphorylation experiments; 15 μg protein/lane was loaded on a gel. Rabbit anti-MnSOD (a gift from Dr. Kato, Department of Biochemistry, Institute for Developmental Research, Aichi, Prefectual Colony, Kamiya, Kaugai, Aichi, 480–03, Japan) was used at 1:200 dilution followed by biotinylated goat-anti-rabbit secondary antibody (Jackson Immunoresearch Labs, West Grove, PA, U.S.A.) diluted 1:1,000. The bands were visualized by incubation with avidin–biotinperoxidase complex (ABC-Elite Kit, Vector Laboratories, Burlingame, CA, U.S.A.) and then with diaminobenzidine (DAB kit, Sigma).
Electrophoretic mobility shift assay was performed by using the Gel Shift Assay System (Promega Inc., Madison, WI, U.S.A.) according to the manufacturer's instructions, as previously described (Ginis et al., 2000).
PKA activity was determined using Promega PepTag Assay Kit (Promega Inc.), which uses fluorescent Kemptide peptide as a substrate for PKA. Whole cell lysates were immunoprecipitated with p65-specific antibodies as already described. Bead pellet was washed twice with immunoprecipitation buffer and resuspended in 30 μL of 350 mmol/L K3PO4/1 mmol/L DTT (pH 7.5). The bead suspension was used for the assay. Reaction was performed according to manufacturer's instructions, except no cAMP was added to the samples. Phosphorylated and nonphosphorylated peptide molecules were separated on an agarose gel. Phosphorylated bands were often distorted, probably because of the presence of large amount of antibodies in the samples. However, intensity of nonphosphorylated bands was inversely proportional to the fluorescence produced by 1, 2, and 4 ng of a standard PKA catalytic subunit (Promega Inc.). This standard curve remained linear for the assay conditions and was reproduced in each experiment and used to normalize the results of experimental samples. Nonphosphorylated bands were excised, melted, and transferred to a 96-well plate. Fluorescence was measured using CytoFluor 4000 fluorescent plate reader (PerSeptive Biosystems, Framingham, MA, U.S.A.) at excitation/emission wavelengths of 530/580 nm.
RESULTS
TNF-α and ceramide preconditioning have no effect on NF-κB activation
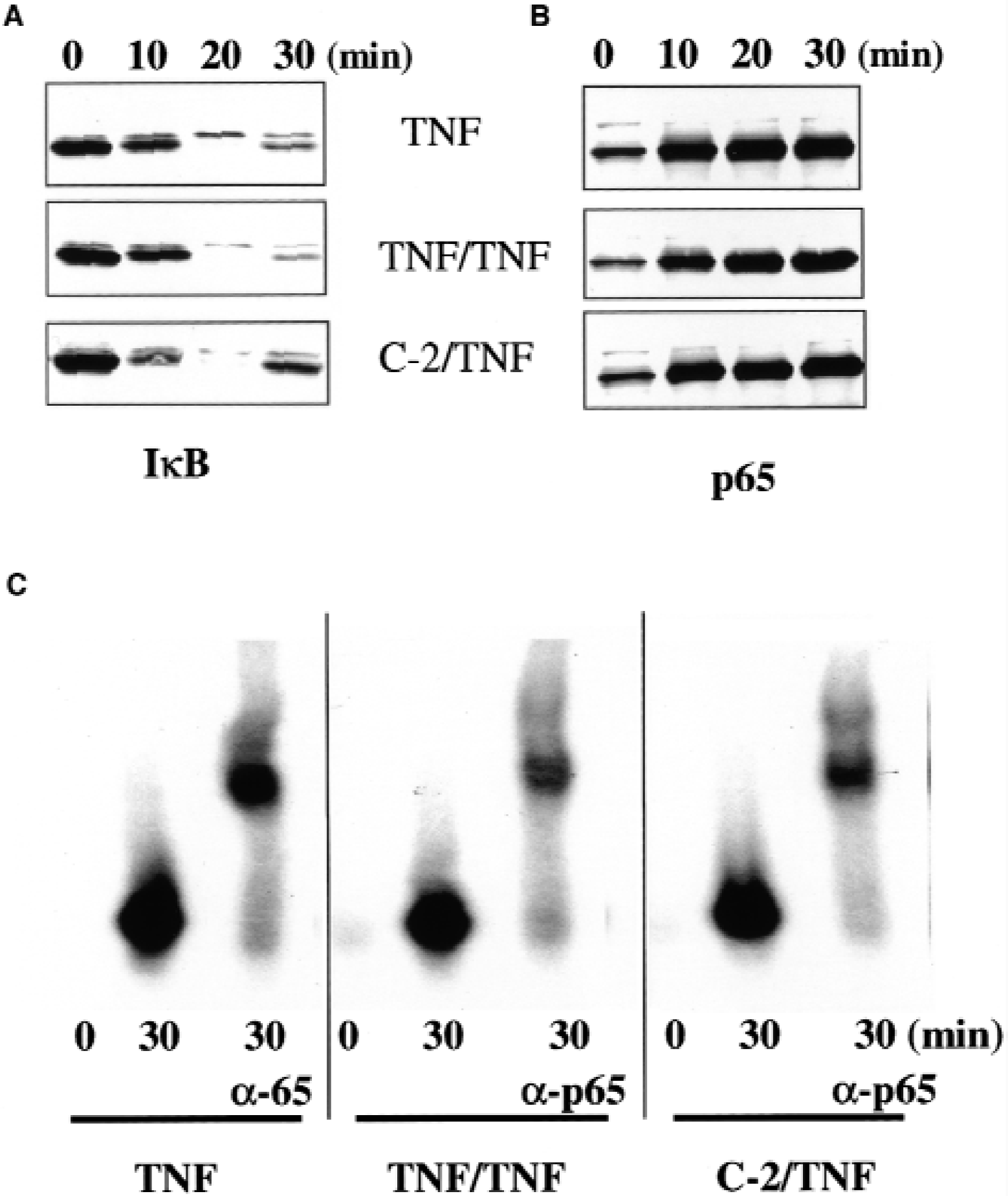
NF-κB is composed of two subunits, which belong to Rel family proteins. The most frequently found NF-κB heterodimer is p65/p50. In quiescent cells, most of the p65/p50 heterodimers are retained in cytoplasm as a complex with IκB. Activation with TNF-α causes degradation of IκB, allowing the complex to enter the nucleus and bind to DNA (Baeuerle, 1998). Based on these established mechanisms, we first investigated whether inhibition of ICAM-1 transcription in tolerant cells is caused by alterations in NF-κB activation. Naïve and ceramide-pretreated astrocytes were activated for 30 minutes with TNF-α, fixed, and immunostained with anti-p65 antibodies. Sister cultures were treated with TNF-α- and ICAM-1 mRNA and protein expression were investigated. In control untreated cells (Fig. 1A), p65 immunofluorescence was spread over cytoplasm and was not found in the nucleus. Treatment with TNF-α caused redistribution of fluorescence with the maximal signal coming from the nucleus (Fig. 1B). Ceramide-pretreated cells exhibited exactly the same pattern of distribution of p65 fluorescence (Fig. 1C). Despite the normal activation of NF-κB, TNF-α–induced ICAM-1 transcription was inhibited in ceramide-treated astrocytes (Fig. 1D). Ceramide also inhibited expression of ICAM-1 protein on the cell surface (Fig. 1E) in accordance with our previous findings, where TNF-α and C-2 ceramide preconditioning were shown to cause 74% and 75.5% inhibition of ICAM-1 surface expression and 31.6% and 49.3% of ICAM-1 transcription, respectively (Ginis et al., 1999). To further investigate NF-κB activation in tolerant cells, astrocytes were preconditioned either with TNF-α 24 hours before the second addition of TNF-α or with ceramide 1 hour before TNF-α addition, and the kinetics of IκB in cytosolic extracts and p65 in nuclear extracts were studied by Western blotting. In naïve cells without preconditioning, IκB almost completely disappeared from the cytosol, whereas p65 simultaneously accumulated in the nucleus at 20 minutes after addition of TNF-α (Figs. 2A and 2B, respectively, top panels). Exactly the same pattern of IκB degradation in the cytoplasm and p65 accumulation in the nucleus was observed in astrocytes preconditioned with TNF-α (Figs. 2A and 2B, middle panels) or with C-2 ceramide (Figs. 2A and 2B, bottom panels).
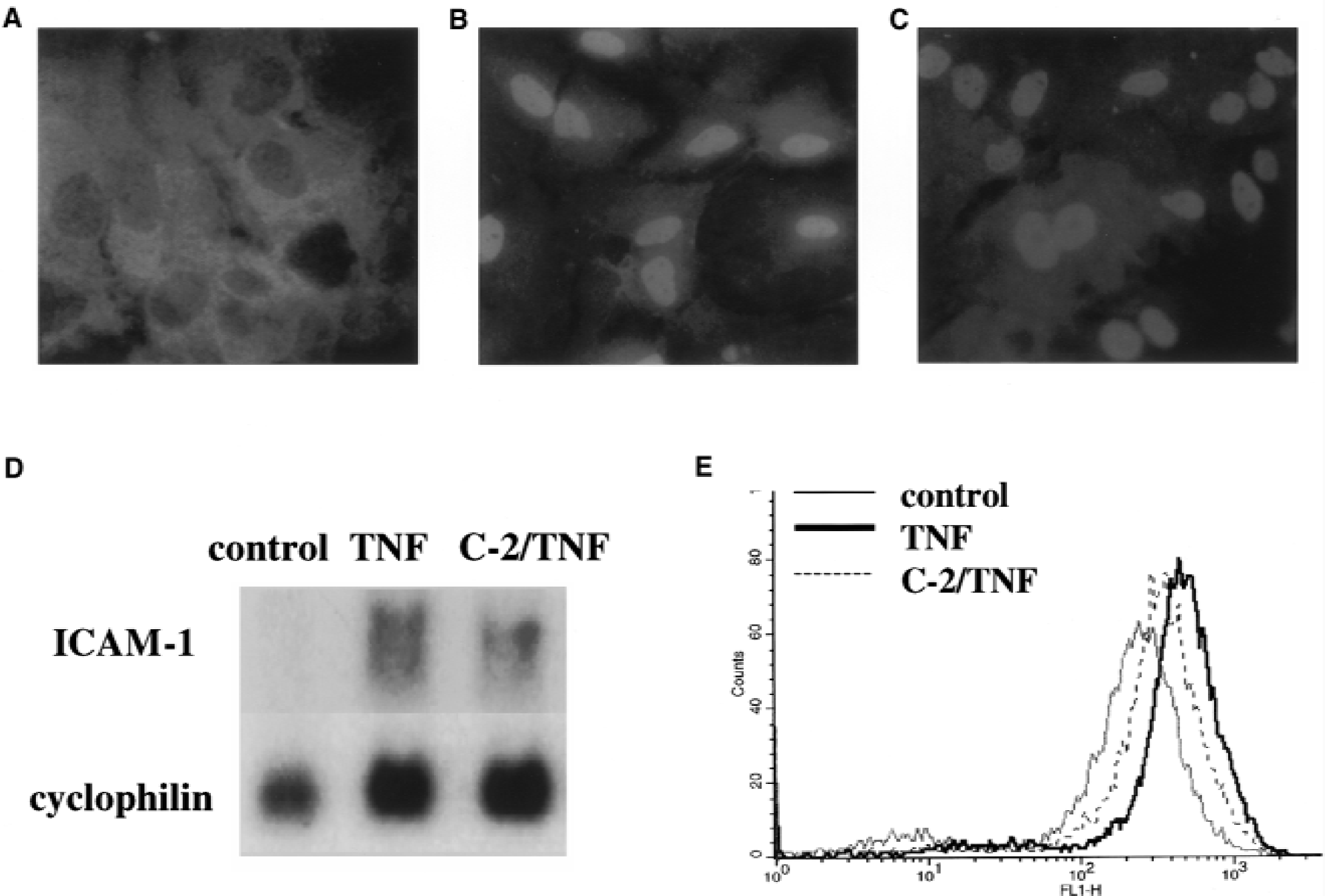
NF-κB translocation to the nucleus and ICAM-1 expression in ceramide-preconditioned cells. (A, B, C) Immunofluorescence with anti-p65 antibodies; x40 magnification.
Nuclear translocation and DNA binding of NF-κB in tolerant cells. Astrocytes were activated with tumor necrosis factor alpha (TNF-α) for indicated times.
TNF-α and ceramide preconditioning have no effect on p65 DNA-binding activity
NF-κB-dependent transcription in tolerant cells could be inhibited if p65 had lost its ability to bind DNA. To address this possibility, naïve or TNF-α– and ceramide-preconditioned astrocytes were incubated with TNF-α for 30 minutes, and DNA-binding activity in nuclear extracts was investigated in an electrophoretic mobility shift assay. No differences in DNA binding were found between nuclear extracts of naive and preconditioned cells (Fig. 2C). As was demonstrated by supershift experiments with anti-p65 antibody, the p65 subunit accounted for most of the DNA-binding complexes (Fig. 2C).
TNF-α and ceramide preconditioning prevent interaction of p65 with p300 adapter protein
It has been recently demonstrated that p65/RelA subunit of NF-κB is capable of association with a transcriptional coactivator p300, and this association regulates cell cycle progression (Perkins et al., 1997) or expression of the IL-6 gene by TNF-α (Vanden Berghe et al., 1999). Thus, we next tested a hypothesis that ICAM-1 gene expression is also dependent on association of p65 with p300 and that in preconditioned cells the formation p65/p300 complexes is interrupted, resulting in inhibition of transcription of the ICAM-1 gene. Indeed, antibody directed against p65 coprecipitated p300 from nuclear extracts of naive cells activated with TNF-α but not from nonactivated cells (“control”). Coprecipitated p300 was identified by Western blotting with p300-specific antibody (Fig. 3, “TNF”). In TNF-α or ceramide-preconditioned cells, however, little or no association between p65 and p300 was observed (Fig. 3, “TNF/TNF” and “C-2/TNF,” respectively).
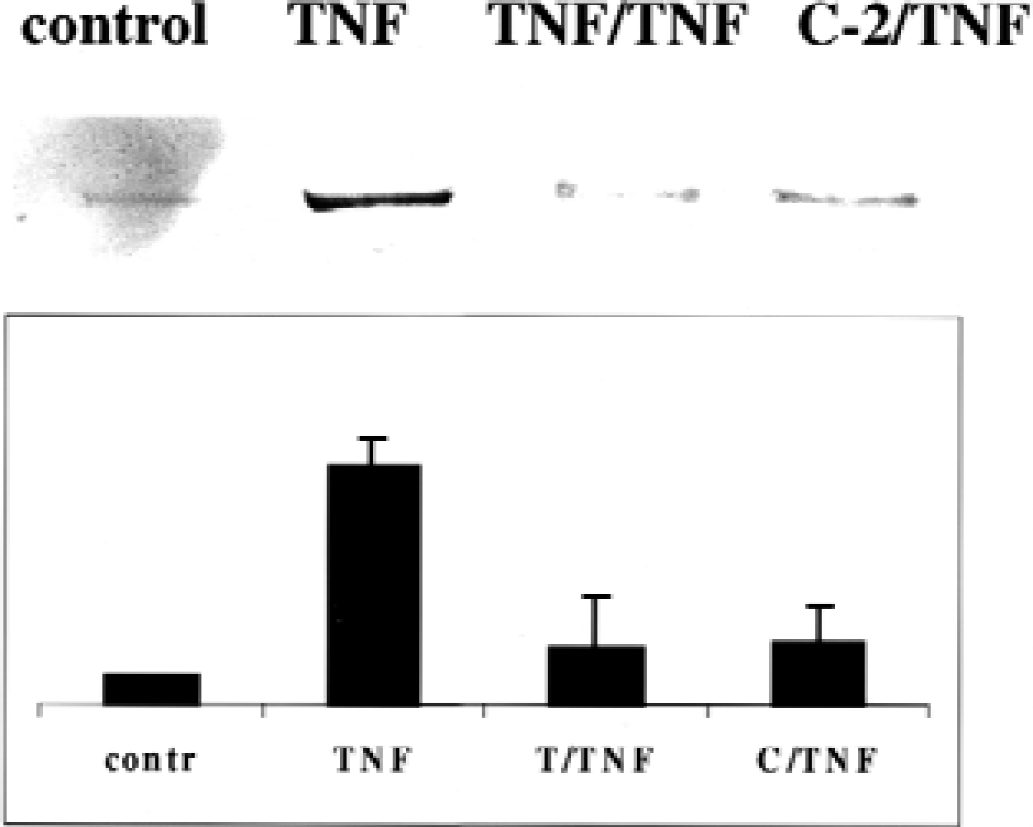
Effect of tumor necrosis factor alpha (TNF-α)/ceramide preconditioning on interaction of p65 with p300. Naïve and preconditioned astrocytes were activated with TNF-α for 30 minutes. At the end of incubation, nuclear extracts were prepared and immunoprecipitated with p65-specific antibody; p300 in precipitates was identified by Western blotting with p300-specific antibody. Control, control untreated astrocytes; TNF, astrocytes activated with TNF-α without preconditioning; TNF/TNF, astrocytes activated with TNF-α after preconditioning with TNF-α; C-2/TNF, astrocytes activated with TNF-α after preconditioning with C-2 ceramide. Representative gel and results of densitometry are shown. Each bar represents mean ± SD of three experiments.
Inhibition of p65 phosphorylation and PKA activity in tolerant cells
Ghosh and collaborators have recently presented evidence that p65 interaction with p300 depends on phosphorylation of p65 on serine 276 by PKA (Zhong et al., 1998). They have previously shown that the PKA catalytic subunit is a part of the NF-κB/IκB/PkA cytoplasmic complex and that its activation and resulting phosphorylation of p65 is cAMP independent (Zhong et al., 1997). Based on this evidence, we hypothesized that PKA-dependent phosphorylation of p65 could be crucial for p65-p300 interaction and ICAM-1 transcription in TNF-α–activated astrocytes. Indeed, TNF-α activation of astrocytes caused a time-dependent accumulation of the phosphorylated form of p65, which was immunoprecipitated with antibody directed against phosphoserine and identified on Western blots with anti-p65 antibody (Fig. 4A). Maximum phosphorylation was observed between 15 and 25 minutes after TNF-α addition. Comparison of p65 phosphorylation in naive and tolerant cells 15 minutes after TNF-α addition confirmed that p65 phosphorylation was inhibited after preconditioning with TNF-α or ceramide (Fig. 4B). Specific PKA inhibitor H-89 (Zhong et al., 1997) inhibited p65 phosphorylation (Fig. 4B) and also downregulated ICAM-1 transcription to the same low levels as were found in both TNF-α- and ceramide-preconditioned cells (Fig. 5A). Astrocyte resistance to TNF-α activation, although dependent on PKA, was independent of cAMP levels because addition of forskolin at concentrations previously shown to upregulate cAMP and activate cytosolic cAMP-dependent PKA (Zhong et al., 1997) failed to restore ICAM-1 mRNA synthesis in TNF-α- or ceramide-preconditioned astrocytes (Fig. 5A).
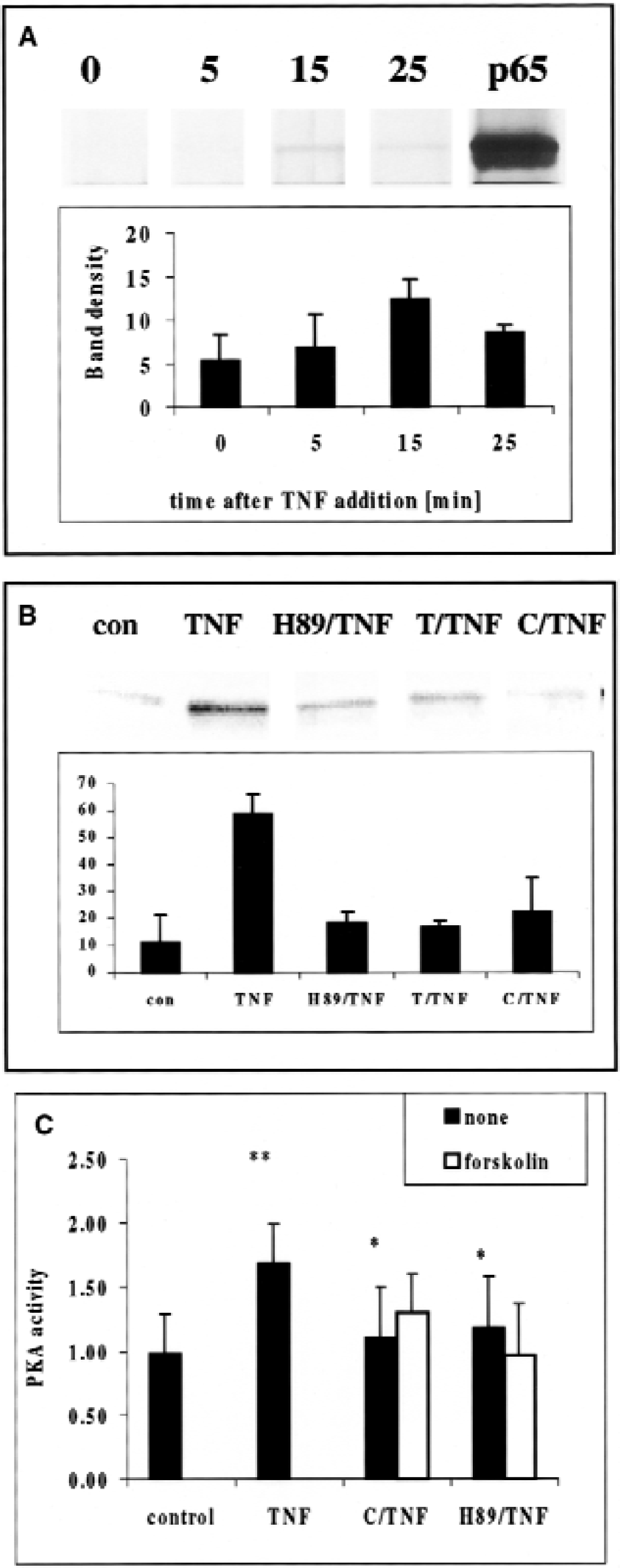
Effect of tumor necrosis factor alpha (TNF-α)/ceramide preconditioning on p65 phosphorylation and protein kinase A (PKA) activity. Whole cell lysates were immunoprecipitated with antibody directed against phosphoserine and p65 was identified on Western blots with anti-p65 antibody.
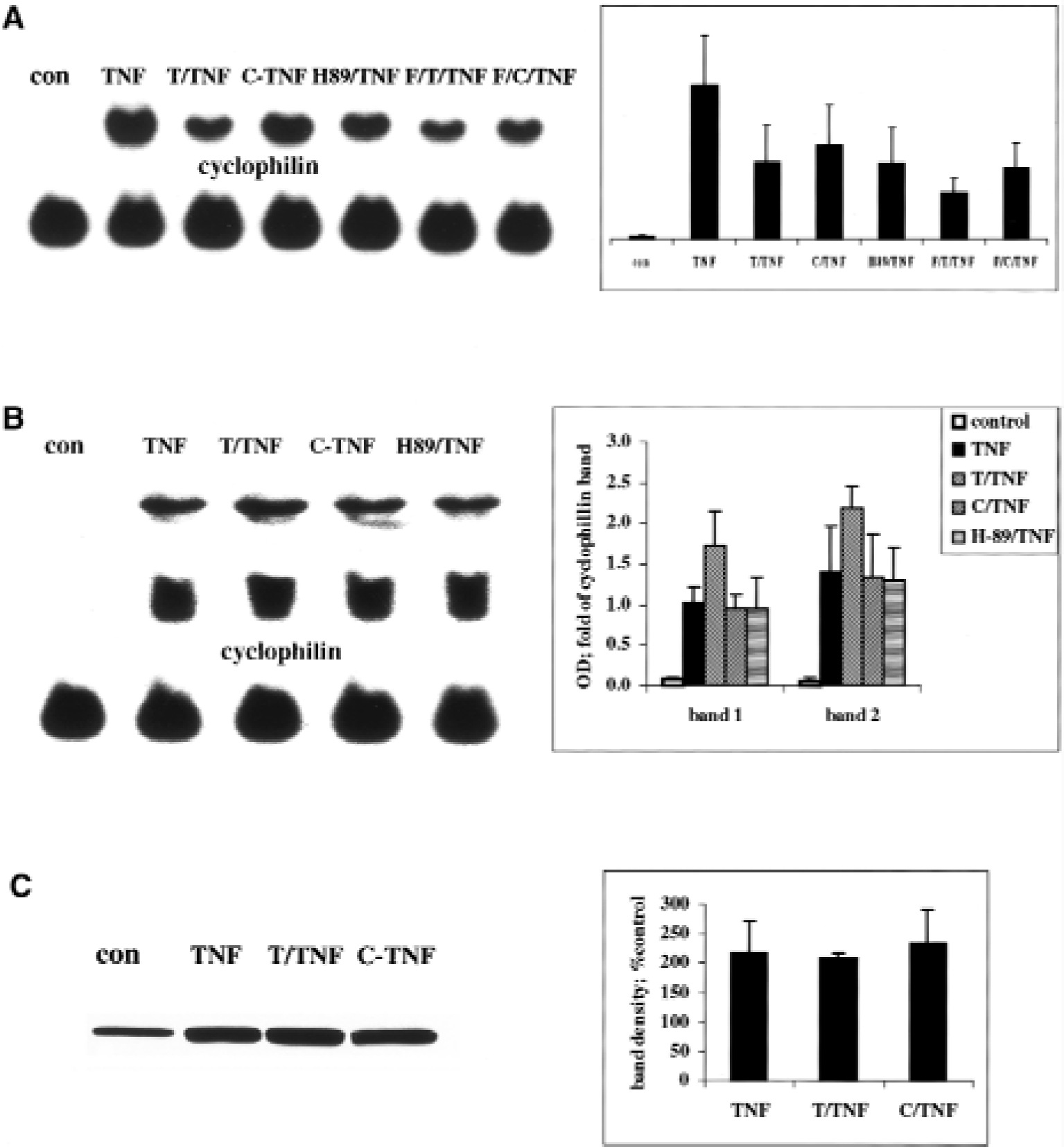
The effect of tumor necrosis factor alpha (TNF-α)/ceramide preconditioning and p65 phosphorylation on ICAM-1 and MnSOD upregulation.
To confirm the role of PKA in induction of tolerance, we have measured PKA activity, which coprecipitated with p65 in TNF-α–activated naive and ceramide-preconditioned cells. Astrocytes activated with TNF-α for 15 minutes significantly upregulated p65-associated, cAMP-independent PKA activity (from 0.99 ± 0.3 in control cells to 1.69 ± 0.3 relative units; mean ± SD, P = 0.001; paired t-test; n = 5). However, in tolerant astrocytes preconditioned with ceramide or pretreated with PKA inhibitor, H-89, no activation of p65-associated PKA activity was observed (1.1 ± 0.4 and 1.18 ± 0.4, respectively; P = 0.01 compared with naive cells activated with TNF-α). Again, addition of forskolin failed to restore PKA activity in ceramide-preconditioned and H-89–pretreated cells (Fig. 4C).
Effect of TNF-α and ceramide preconditioning on the expression of MnSOD.
In order to investigate whether the mechanism of preconditioning described here is gene-specific or affects all NF-κB–dependent stress-response genes, astrocytes were preconditioned with TNF-α or ceramide and then activated with TNF-α to evaluate expression of MnSOD, a mitochondrial enzyme demonstrated to have a beneficial effect in ischemic injury (Keller et al., 1998). Two species of MnSOD mRNA (3.8 kb and 1.3 kb) were detected on the Northern blots in accordance with previous findings (Hurt et al., 1992). In contrast to ICAM-1 activation, both, naïve and tolerant cells had similar levels of MnSOD mRNAs (Fig. 5B) and protein (Fig. 5C). Similarly PKA inhibitor H-89 had no effect on MnSOD transcription (Fig. 5B).
DISCUSSION
Cellular stress disturbs homeostatic equilibrium between the cell and its environment and triggers activation of multiple signaling pathways, which lead to a new pattern of gene expression (O'Connor et al., 2000). To resist the stress, the cell produces a wave of biochemical and genetic changes that attempt to return the cell to its original condition. This “after stress” homeostatic correction consists of at least of two mechanisms: either cytoprotective genes are activated to neutralize the effect of harmful genes (e.g., antiinflammatory cytokines versus proinflammatory cytokines, antiapoptotic genes versus proapoptotic genes, or antioxidants versus generators of free radicals), or expression of harmful genes is simply shut off by newly synthesized or activated inhibitors of transcription and/or translation. If a stressful stimulus is reapplied at the peak of the stress-controlling reaction, cells are likely to tolerate it better. This phenomenon is known as stress adaptation or preconditioning, where sublethal stress induces an adaptive response, which results in cell tolerance to a subsequent challenge that would otherwise be lethal. The best-studied stress adaptation phenomenon in mammals, ischemic preconditioning, is well documented in animal models of ischemic injury of brain (reviewed in Chen and Simon, 1997).
TNF-α is known as a key contributor to cell dysfunction and death in brain ischemia (Arvin et al., 1995; del Zoppo et al., 2000) and at the same time as an initiator of many stress-controlling protective reactions (Mattson et al., 2000; del Zoppo et al., 2000). TNF-α activates a transcription factor NF-κB that binds promoters of multiple genes, thus ensuring pleiotropic response to TNF-α. TNF-α and its intracellular messenger ceramide can be used as preconditioning agents, which render animals (Nawashiro et al., 1997; Tasaki et al., 1997) and cultured neuronal cells (Barger et al., 1995; Goodman and Mattson, 1996; Ginis et al., 1999) tolerant to oxidative stress and ischemic injury. We describe here for the first time a molecular mechanism by which TNF-α and ceramide, when acting as preconditioning stimuli, selectively curtail NF-κB–dependent transcription of a harmful gene, while preserving NF-κB–mediated transactivation of another TNF-α–dependent gene that is beneficial for the cell.
NF-κB is a heterodimer. It consists of a 65-kDa protein (p65/RelA) and a 50-kDa protein (p50) (both belong to the Rel family of proteins) that is sequestered in cytoplasm by an anchor protein, inhibitor of NF-κB (IκB). Phosphorylation of IκB on serines 32 and 36 by IκB kinase leads to its ubiquitination and degradation by proteosomal enzymes, which allows NF-κB heterodimer to translocate to the nucleus (for review see Baeuerle, 1998). In the nucleus, NF-κB dimer binds DNA consensus sequence GGGRNNYYCC (R = A or G; Y = C or T) and initiates transcription. Both subunits contain DNA-binding domains, but only p65 is capable of trans-activation (Schmitz et al., 1994).
According to our data, NF-κB translocation to the nucleus and its binding to DNA are not altered in tolerant cells. Recent reports demonstrate that NF-κB transcriptional activity could be controlled by phosphorylation of p65 (Wang and Baldwin, 1998; True et al., 2000). Tyrosine kinases (Yoza et al., 1996), casein kinase II (Bird et al., 1997), p38 MAPK (Vanden Berghe et al., 1998), phosphatidylinositol 3-kinase/Akt kinase (Sizemore et al., 1999), PKC zeta (Anrather et al., 1999), and finally PKA (Zhong et al., 1998) have been implicated in regulating of p65 trans-activation. The catalytic subunit of PKA was shown to be part of the NF-κB/IκB complex in the cytoplasm of B-cell line 70Z/3, where IκB inhibits catalytic activity of the catalytic subunit of PKA. Lipopolysaccharide-induced IκB degradation results in upregulation of cAMP-independent catalytic subunit of PKA activity and phosphorylation p65 (Zhong et al., 1997). Phosphorylation of p65 by PKA allows its interaction with the coactivator protein, CBP/p300 (Zhong et al., 1998).
p300 and its homologue, CREB-binding protein, are coactivator proteins that bind to transactivation domains of transcription factors and mediate their contact with RNA polymerase II. p300 is also a potent acetyltransferase, which can acetylate histones and transcription factors to facilitate their interaction with DNA. The p65/RelA subunit of NF-κB is among many other transcription factors that interact with p300, and p300–p65 interaction has been recently shown to regulate cell cycle progression (Perkins et al., 1997) or activation of the IL-6 gene by TNF-α (Vanden Berghe et al., 1999) in tumor cell lines. Similarly, p300 association with p65 was responsible for TNF-α–induced expression of E-selectin in human umbilical vein endothelial cells (Gerritsen et al., 1997).
In our search for a mechanism of TNF-α/ceramide-induced tolerance, we addressed the following questions: (1) whether ICAM-1 gene expression in primary astrocytes is dependent on association of p65 and p300 and whether formation p65-p300 complexes is inhibited in TNF-α–tolerant cells; (2) whether TNF-α– induced p65-p300 association is controlled by phosphorylation of p65; and, if so, whether phosphorylation of p65 is inhibited in tolerant cells; and (3) whether NF-κB–associated PKA is involved in NF-κB trans-activation of ICAM-1 and in induction of tolerance. Our data answer these three questions positively and demonstrate that adaptation to TNF-α–induced stress occurs on the level of individual gene expression at the promoter site of a gene.
The dual function of TNF-α in brain injury has been the subject of a recent review (Shohami et al., 1999), and this duality is reflected in the effects of NF-κB (Baichwal and Baeuerle, 1997; Perkins, 2000; Mattson et al., 2000b). Observations in animals also reflect this controversy. Thus, suppression of NF-κB activity in brain by a specific inhibitor resulted in DNA fragmentation (Taglialatela et al., 1998), but TNF-α receptor knockout mice, which exhibited delayed upregulation of NF-κB after traumatic brain injury, had a larger average lesion volume and blood–brain barrier breach than wild-type animals (Sullivan et al., 1999). NF-κB activity was shown to increase after brain trauma (Yang et al., 1995) and, in a model of transient focal ischemia, 72 hours after reperfusion (Schneider et al., 1999). In a rat model of permanent middle cerebral artery occlusion, activated NF-κB immunoreactivity decreased from basal levels by 2 hours after onset of ischemia and remained undetectable for up to 5 days (Botchkina et al., 1999).
If preconditioning affected gene expression at the level of IκB degradation or NF-κB translocation to the nucleus, discrimination between detrimental and neuroprotective mechanisms would be impossible, because all NF-κB–dependent genes would be inhibited. However, we demonstrate here that TNF-α–activated transcription and translation of another NF-κB–dependent gene, MnSOD (Jones et al., 1997), which has been implicated in neuroprotection (Keller et al., 1998), was not affected by TNF-α and ceramide preconditioning, or by PKA inhibition with H-89. Control of MnSOD transcription is complex and includes promoter-selective transcription factor-1, activator protein-1, activator protein-2, adenosine 3′,5′-cyclic monophosphate-regulator element binding factor (CREB), and transcription factor IID complex (Das et al., 1998). Thus, MnSOD could be activated without involvement of NF-κB (Borrelo and Demple, 1997). The other explanation, however, is that p300 interaction with p65 subunit is not essential for MnSOD upregulation. MnSOD mRNA accumulates in astrocytes to a maximal level by 6 hours after addition of TNF-α (Pahan et al., 1999), whereas ICAM-1 message peaks at 1.5 to 2 hours (Lee et al., 1998).
According to our data, p65 phosphorylation and interaction with p300 occurs during the first 30 minutes of incubation with TNF-α. Ceramide is a short-lived compound and is metabolized quickly into more complex sphingolipids such as cerebrosides, sulfatides, and sphingomyelin (Merrill et al., 1997). We have previously demonstrated that TNF-α preconditioning results in inhibition of the ICAM-1 response to subsequent TNF-α activation only when the time of the second TNF-α addition coincides with the peak of ceramide de novo synthesis (18–24 hours after addition of TNF-α). Thus, ceramide is a short-lived effector of tolerance, which targets only those genes that become transcriptionally active at the time of its release. Our recent studies demonstrate a protective effect of ceramide in cultured neurons subjected to hypoxia (Ginis et al., 1999) and in animal models of brain ischemia (Chen et al., 2001; Furuya et al., 2001; Zimmerman et al., 2001). The data presented here suggest a novel mechanism for protective effect of ceramide. Ceramide quickly changes cellular makeup by affecting transcription of those genes for which trans-activation depends on p300 interaction with NF-κB. According to current theory, transcription factors compete for scarce levels of free CBP/p300 (Sheppard et al., 1998). Thus, p300, which is not engaged in NF-κB interactions, might be used by other transcription factors, for example, CREB, which was implicated in NF-κB–independent expression of MnSOD (Kim et al., 1999). Consistent with this theory is a recent finding that ceramide can induce (rather than inhibit) MnSOD transcription in primary astrocytes (Pahan et al., 1999).
Although the recently identified ceramide-activated proline-directed serine-threonine kinase (Liu et al., 1994), or a ceramide-dependent serine-threonine phosphatase (Wolff et al., 1994) could indirectly mediate the ceramide effect on p65 phosphorylation, emerging evidence suggests a central role of atypical PKC-zeta in ceramide-induced regulation of NF-κB activity (Lozano et al., 1994; Muller et al., 1995; Galve-Roperh et al., 1997; Wang et al., 1999). In addition PKC-zeta could be activated via p21ras (Diaz-Meco et al., 1994). On binding to GTP, Ras recruits and activates downstream effectors such as RAF, PI 3-kinase, and kinase suppressor of Ras (Hanna et al., 1999). Interestingly, ceramide has been implicated in this pathway as well (Muller et al., 1998; Zhang et al., 1997). It has been shown that inhibition of either one of these pathways (PKCzeta or p21ras) inhibits p65 phosphorylation and transcriptional activity in endothelial cells (Anrather et al., 1999). To complete the picture, recent work demonstrates that activation of p21ras is required for neuronal preconditioning against ischemic injury (Gonzalez-Zulueta et al., 2000). Although the authors demonstrate that p21ras is activated by nitric oxide, one cannot exclude that ceramide produced during hypoxic preconditioning of neurons (Liu et al., 2000) is also a player in this pathway.