
Abstract
Oxidative stress is a major brain injury mechanism after ischemic stroke. 12/15-lipoxygenase (12/15-LOX) is a key mediator of oxidative stress, contributing to neuronal cell death and vascular leakage. Nonetheless, the mechanism leading to its upregulation is currently unknown. We show here that Signal Transducers and Activators of Transcription (STATs), specifically STAT6 and possibly STAT1, increase transcription of 12/15-LOX in neuronal cells. Both p-STAT6 and −1 bound to specific STAT binding sites in the mouse 12/15-LOX promoter. Small interfering RNA (siRNA) knockdown showed STAT6 to be the dominant regulator, reducing 12/15-LOX promoter activation and cell death in oxidatively stressed HT22 cells. STAT6 siRNA efficiently prevented the increase of 12/15-LOX in murine primary neurons, both after induction of oxidative stress and after oxygen-glucose deprivation. Early activation of STAT6 and STAT1 in mice was consistent with a role in regulating 12/15-LOX in focal ischemia. Brains of human stroke patients showed increased p-STAT6 and p-STAT1 in the peri-infarct region, along with 12/15-LOX and markers of apoptosis. These results link STAT6 and STAT1 to the 12/15-LOX damage pathway and suggest disregulation of STAT-dependent transcription as injury mechanism in stroke. Selectively targeting STATs may thus be a novel therapeutic approach to reducing brain injury after a stroke.
INTRODUCTION
In the healthy brain, an intricate network of pro- and antioxidative proteins and peptides maintains a redox balance inside the cell, preventing cellular injury but allowing for redox-dependent signaling events. After an ischemic stroke this redox network breaks down. The ensuing oxidative stress leads to neuronal death and various forms of cerebral pathology including neurovascular injury, Blood–Brain barrier disruption, edema, and hemorrhage.1,2 Components of the redox network include the antioxidant glutathione, which decreases after ischemia, while pro-oxidative enzymes including those of the arachidonic acid cascade are upregulated. In particular, 12/15-lipoxygenase (12/15-LOX) is elevated in both mouse and human brain after an ischemic stroke, 3 and contributes to delayed cell death in the area surrounding the core infarct.4–6 Inhibition of 12/15-LOX reduces infarct size, edema, and Blood–Brain barrier leakage, and is being investigated as a novel approach to stroke therapy.3,5–8
One of the key regulators of this redox homeostatic network, along with the hypoxia-inducible factors, is the Signal Transducers and Activators of Transcription (STAT) family of proteins. 9 Phosphorylation at specific tyrosine residues and subsequent dimerization is required for STAT activation; the dimers then translocate to the nucleus, where they bind to their target gene's promoter region. 10 STAT-dependent gene regulation has been implicated in a number of pathologies, including several forms of cancer and polycystic kidney disease.11,12 In the ischemic mouse brain after experimental stroke, loss of STAT3 has been shown to downregulate protective manganese-dependent superoxide dismutase,2,13 and STAT3 is generally seen as neuroprotective. Conversely, STAT1 is activated and contributes to ischemic injury. 14 For STAT6, the situation is less clear. While changes in STAT6 phosphorylation after ischemia in rodents have been documented (see discussion for details),15–17 possible functional consequences were not investigated in those studies.
Surprisingly, although the elevation of 12/15-LOX levels in neurons and ischemic cortex is a critical factor contributing to neuronal death and brain injury after cerebral stroke, its transcriptional regulation in the brain has not been elucidated. We investigated here a possible regulation of 12/15-LOX through STATs, with a specific focus on STATs 1 and 6. Our findings here show that predominantly STAT6, but possibly also STAT1 contributes to the upregulation of 12/15-LOX in oxidatively stressed neuronal cells, and also coincide with increased 12/15-LOX in the infarcted human brain in vivo.
MATERIALS AND METHODS
Human Brain Tissue Samples
Samples were taken from two patients with ischemic stroke, who were previously described in. 3 Briefly, patient I was a 77-year-old female who suffered from an ischemic stroke, with hemorrhagic transformation after thrombolytic treatment. The second patient (patient II) was a 59-year-old male with a history of hypertension and diabetes mellitus type II. He suffered from an acute ischemic stroke due to severe right carotid stenosis (atherothrombotic stroke). The patient did not receive tissue plasminogen activator, and upon autopsy showed no signs of neurodegenerative disease. Samples for immunohistochemistry were immediately fixed with 4% paraformaldehyde and kept at − 80° until use. Samples for western blotting were immediately frozen, then homogenized in cell Lysis buffer and stored at − 80° until use. The study was approved by the Ethics Committee of the Hospital Vall d'Hebron [PR(HG)85/04]. Informed consent was acquired from relatives before the autopsy.
Animals
All experiments with animals were performed in accordance with National Institutes of Health guidelines, the Arrive guidelines, and were approved by the Massachusetts General Hospital Institutional Animal Care and Use of Laboratory Animals Committee. C57Bl6 male mice (25 to 30 g, 12 weeks old) for mouse transient focal ischemia and C57Bl6 female mice (14 days timed pregnant) for mouse primary cortical neurons cultures were purchased from Charles River Laboratories (Wilmington, MA, USA).
Focal Cerebral Ischemia
Male mice (25 to 30 g) were subjected to 45 minutes of transient focal cerebral ischemia and reperfusion. The mice were anesthetized with 1.5% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. Rectal temperature was controlled at 37° with a feedback heating pad. A Silicon rubber-coated monofilament (Doccol, Sharon, MA, USA) was introduced into the right common carotid artery. Filament size was 6-0, diameter was 0.09 to 0.11 mm, length was 20 mm. After 45 minutes of middle cerebral artery occlusion, blood flow was restored by withdrawal of the nylon suture. To confirm adequate induction of focal ischemia and successful reperfusion, regional blood flow was measured before, during, and after middle cerebral artery occlusion by Laser-Doppler flowmetry (3 mm lateral to bregma). Their relative cerebral blood flow (% of basal) was reduced to around 20% of the baseline, and restored to near baseline after removal of the filament in all mice we used for this study. Sham-operated mice did not undergo the filament insertion into the middle cerebral artery. The total number of animals used for this study was five mice for sham and nine mice for middle cerebral artery occlusion group.
Primary Cortical Neuron Culture
The protocol followed was adapted from a standard procedure described previously18,19 with minor modifications. Cortical neurons were prepared from brains of C57Bl6 mouse embryos prepared on embryonic day 15, plated on coated dishes with poly-D-lysine, and cultured in DMEM (Invitrogen, San Diego, CA, USA) containing 10% FBS, penicillin (50 U/mL), and streptomycin (50 μg/mL). The next day, they were either used in the glutamate-induced oxidative stress model, or switched to neurobasal medium and matured in vitro (see below).
Glutamate-Induced Oxidative Stress Model In Vitro Glutathione depletion was induced in mouse primary cortical neurons or in HT22 cells 1 day after seeding by treatment with 10mmol/L glutamate, and oxidative glutamate toxicity in cells was measured by lactate dehydrogenase (LDH) release to detect cell death.18,20
Oxygen-Glucose Deprivation and Reoxygenation Neurons were cultured in a 12-well plate (Greiner Bio-One, Monroe, NC, USA) in Neurobasal medium with 2% B27 supplement (Invitrogen) until 7 days after seeding. The neurons were then transfected with 10 nmol/L small interfering RNA (siRNA) for 12 hours, followed by 2 hours of oxygen-glucose deprivation (OGD) and 12 hours of reoxygenation (OGD/R). For this purpose, the medium was replaced with DMEM without glucose and other supplements, and the cells were placed in a gas-tight humidified chamber (Heidolph, incubator 1000, Brinkmann Instruments, Westbury, NY, USA) at 37°, which contained an anaerobic gas mixture (90% N2, 5% H2, and 5% CO2). After 2 hours of OGD, the cell medium was changed back to Neurobasal medium with glucose and 2% B27 and cells were reoxygenated for 12 hours. For each of three experiments, all treatment conditions were performed in duplicate in a single 12-well plate, and cells were imaged with equal settings for fluorescence detection. For measurement of signal intensity in each image, the NIH ImageJ program was used, and fluorescence intensity was normalized to the control value. Separate cohorts of cells were used for measurement of cell death using the LDH assay.
Small Interfering RNA Transfection
The siRNA probes targeted to mouse STAT1 or 6 and mouse 12/15-LOX were purchased from Qiagen (Valencia, CA, USA). The target sequences for the mouse-specific STAT1 siRNA mixture were as follows: ATGCATCTTACTGAAGGTGAA (SI02710729), ATGAGTTGGTTTAATATATAT (SI02735054), CCAATGCTCTATCAAACTATA (SI00183547), and TCCTATTATT ATTTAATATAA (SI02668862). The target sequences for the mouse-specific STAT6 siRNA mixture were as follows: CCAGAAGATCTTCAACGACAA (SI00183596), CACAGGAGAGATCATGAACAA (SI026688869), CTCGAATGT GATACAACTGTA (SI00183575), and CTGGAGAAGCCCAGAAACAAA (SI00183589). The target sequences for the mouse-specific 12/15-LOX siRNA mixture were as follows: AAGCTTCTAGTTCCTCACCTA (SI00896644), CTGGCAAGTCATGAATCGGTA (SI04421158), AAGCCTTAATAGAGTCTAATA (SI00896630), and ACCGTTATTAACTTCCCTAAA (SI00896637). Nontargeting, scrambled siRNA (SI03650318) was used as a control in all siRNA transfection experiments. Primary cortical neurons grown on 24-well plates (3×10 5 cells/well) or 6-well plates (2×10 6 cells/plate) previously coated with poly-D-lysine were transfected for 24 hours with 10 nmol/L siRNA per well using HiPerFect Transfection Reagent (Qiagen) according to the manufacturer's guidelines.
Immunofluorescent Staining
The primary cortical neurons subjected to OGD/R were fixed with 4% formaldehyde in PBS and then were incubated with 0.1% Triton X-100 for 15 minutes at room temperature. The cells were washed two times with Tris-buffered saline and blocked with PBS containing 3% bovine serum albumin (BSA) for 1 hour at room temperature, and incubated overnight at 4° with primary antibodies used at 1:50 dilution in PBS containing 0.1% Tween and 0.3% BSA. Sheep polyclonal antiserum to 15-LOX-1 (kindly provided by Dr J Cornicelli, Pfizer) and rabbit polyclonal STAT6 (sc-621, Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used. Colocalization studies were performed with both primary antibodies incubated simultaneously in the same well. The cells were washed with PBS containing 0.1% Tween and incubated at room temperature for 1 hour with the following secondary antibodies (1:100 dilution; Alexa fluor 488 affini pure anti-sheep IgG; Rhodamine (TRITC)-conjugated donkey anti-rabbit IgG, Jackson ImmunoResearch, West Grove, PA, USA) in PBS containing 0.1% Tween and 0.3% BSA. Cells were imaged using a Nikon Eclipse Ti-S fluorescence microscope or a Zeiss LSM5 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Cell Death Assay
Cell viability was quantified by a standard measurement of LDH release using LDH kit (Roche Diagnostics, Indianapolis, IN, USA). The general procedure was followed using the manufacturer's guidelines.
Immunohistochemistry
Human brain tissue samples for immunohistochemistry were taken from the peri-infarct cortex on the ipsilateral side as well as the corresponding contralateral cortex, and immediately fixed with 4% paraformaldehyde and kept at − 80° until use. Frozen sections (20 fjm thick) were prepared with 0.1% Triton X-100 for 15 minutes at room temperature and blocked with PBS containing 3% BSA for 1 hour at room temperature, and incubated overnight at 4° with primary antibodies used at 1:50 dilution in PBS containing 0.1% Tween and 0.3% BSA. 12/15-LOX is a rabbit polyclonal antibody directed against the C terminus of mouse and human 12/15-LOX, and affinity purified against the same peptide used for immunization, characterized in Pekcec et al 19 and Yigitkanli et al; 3 MDA2 is a mouse monoclonal antibody directed against malondialdehyde-modified lysine residues (a kind gift from Dr J Witztum, University of California, San Diego). 21 All other antibodies (mouse monoclonal p-STAT1(Y701), sc-8394; rabbit polyclonal p-STAT6 (Y641), sc-11762-R; goat polyclonal apoptosis-inducing factor (AIF), sc-9416) used were from Santa Cruz Biotechnology. Colocalization studies were performed with both primary antibodies incubated simultaneously in the same section. The sections were washed with PBS containing 0.1% Tween and incubated at room temperature for 1 hour with the following secondary antibodies (1:100 dilution; Fluorescein (FITC)-conjugated donkey anti-mouse IgG; Rhodamine (TRITC)-conjugated donkey anti-rabbit IgG, and Fluorescein (FITC)-conjugated donkey antigoat IgG, Jackson ImmunoResearch) in PBS containing 0.1% Tween and 0.3% BSA. Brain sections were imaged using a Nikon Eclipse Ti-S fluorescence microscope or a Zeiss LSM5 confocal microscope (Carl Zeiss).
Western Blot Analysis
Whole-cell protein extractions were obtained from the cerebral cortex of ipsilateral hemisphere or from the primary cerebral cortical neurons. Equal amount of protein sample was run on a SDS gel, subsequently transferred onto a nitrocellulose membrane, and immunoblotted. The primary antibodies used were monoclonal or polyclonal antibodies against p-STAT1 (Y701); sc-8394, p STAT6 (Y641); sc-111762-R, STAT1; sc-464, and STAT6; sc-621 (1:1,000; Santa Cruz Biotechnology), β-actin (1:5,000; Sigma-Aldrich, St. Louis, MO, USA), and 12-LOX (1:1,000; ab23678, Abcam, Cambridge, MA, USA). The signal was then detected with horseradish peroxidase-conjugated IgG using an enhanced chemiluminescent substrate, ECL kit (Thermo Scientific, Rockford, IL, USA).
Real Time RT-PCR Analysis
Total RNA was isolated from the ipsilateral hemisphere with the RNeasy Plus Mini Kit (Qiagen). To generate cDNA, M-MLV reverse transcriptase and random primers (Invitrogen) were used. For real-time RT-PCR analysis, Fast SYBR Green master mix (Applied Biosystem, Foster City, CA, USA) was used. The following different set of primer sequences (5′-3′) were used: mouse 12/15-LOX; CTT CCT TCT GGA TGG GAT CA and GGT GGG GTA GAC CCA GTT TT; CCT GGT TCT GCA ACT GGA TT and AGT TCC TCC TCC CTG TGG TT; CGA AAT CGC TGG TCT ACA GG and CGT GGT TGA AGA CTC TCA AGG; CAG GGA TCG GAG TAC ACG TT and GAT TGT GCC ATC CTT CCA GT; CAC TGC GCA GCA CTC TTC CAT CC and CAC CAT AAC AGC CTG GCG TCT GC. As a control, mouse GAPDH was used with the following primer sequences (5′-3′): AAG GTC ATC CCA GAG CTG AA and ATG TAG GCC ATG AGG TCC AC. The mixtures were subjected to real-time RT-PCR on an Applied Biosystem 7000 Real-Time PCR System.
Chromatin Immunoprecipitation Assay
Chromatin isolation and chromatin immunoprecipitation (ChIP) assay were performed according to the manufacturer's guidelines using a EZ-ZymeTM Chromatin prep kit and a EZ-ChIPTM kit (Upstate, Temecula, CA, USA). Briefly, mouse primary cerebral cortical neurons (9.5 × 10 6 cells/dish) were seeded on 10-cm cell culture dish pre-coated with poly-D-lysine. In all, 10 mmol/L of glutamate was treated for 3 hours, and then cells were fixed with 1% formaldehyde. Each soluble chromatin was isolated and digested using EZ-Zyme lysis buffer and EZ-Zyme enzymatic cocktail after fixation. The diluted chromatin solution with ChIP dilution buffer was precleared, and then the precleared chromatin solution was divided and used in immunoprecipitation assays with phospho-STAT1 or 6 antibodies. After multiple washing, the antibody-protein-DNA complex was eluted. After reversal crosslink incubation, protein and RNA were removed by proteinase K and RNase A. Purified DNA was used as a template for PCR with primers specific for several putative STAT-binding sites on the mouse 12/15-LOX promoter. The sequences of the PCR primers used are F1 forward, 5′-GCA CCT ACT CTG GTA CTG-3′, R1 reverse, 5′-CTT GCT ACA CAC TCT TGC C-3′; F2 forward, 5′-GGC AAG AGT GTG TAG CAA G-3′, R2 reverse, 5′-TTA CTC CAG CTC GAA CTG-3′; F3 forward, 5′-GGC AGT TCG AGC TGG AGT-3′, R3 reverse, 5′-GGG CTC TCC GTG AGT AGT AAG-3′; F4 forward, 5′-CTT ACT ACT CAC GGA GAG CCC-3′, R4 reverse, 5′-GGC TTT ACT GGA GAG ACA GAC-3′; F5 forward, 5′-GTC TGT CTC TCC AGT AAA GCC-3′, R5 reverse, 5′-CCC TTG ATT TCT CTC CCT ATG C-3′.
Transient Transfection and Luciferase Activity Assay
HT22 cells were cultured in 24-well plates (2.5×10 4 cells/well) and transfected with 15-LOX-1-luciferase reporter DNA using Lipofectamine (Invitrogen). The luciferase reporter DNA construct containing 15-LOX-1 promoter region was kindly gifted from Dr Liu at Department of medicine, Karolinska University Hospital Solna and Karolinska Institutet, Stockholm, Sweden. 22 A 1,081-bp fragment of the 15-LOX-1 promoter region containing several STAT binding sites was ligated into pGL3-basic (Promega, Madison, WI, USA). Two hundred fifty nanograms of pGL3-15-LOX-1 promoter reporter DNA were used per well. After 24 hours of incubation, the cells were treated with 10 nmol/L of siRNA for STAT1 or 6 or non-targeting siRNA per well for 24 hours. Cells were treated with 10mmol/L of glutamate for 24 hours and subsequently analyzed for luciferase activity. A Luciferase Assay System (Promega) was used to detect luciferase activity from cell lysates, according to the manufacturer's guidelines.
Statistical Analysis
Data are presented as mean ± s.e.m. Statistical significance was determined using Student's t-test when comparing two groups, or one-way ANOVA followed by post hoc testing, as indicated. Differences were considered as statistically significant at P < 0.05.
RESULTS
Phosphorylated STAT1 and STAT6 Are Recruited onto the Mouse 12/15-Lipoxygenase Promoter in Response to Glutamate-Induced Oxidative Stress
Previous studies have shown a crucial role for 12/15-LOX in cell death caused by glutamate-induced oxidative stress in immature primary neurons.20,23,24 In this model, freshly isolated primary neurons, which do not express functional glutamate receptors and are thus not subject to excitotoxicity, are treated with millimolar levels of glutamate. This leads to glutathione depletion, 25 followed by lipoxygenase activation and death of the cell. 23 To clarify whether STAT1 or STAT6 has a role as transcriptional regulator of 12/15-LOX gene expression, we examined recruitment of STAT1 and STAT6 onto the mouse 12/15-LOX promoter using a ChIP assay in mouse primary cortical neurons. The 12/15-LOX promoter in mouse has not been published to date. We identified a putative promoter region by its homology with the human 12/15-LOX promoter, and found several canonical p-STAT binding motifs (TTCNNNGAA or TTNNNNNAA). The mouse 12/15-LOX promoter was then divided into five consecutive domains (Figure 1A), and probed with the corresponding primer sets to perform the ChIP assay. Binding of p-STAT1 and p-STAT6 was detected using phosphorylation-specific antibodies for STAT6 (Tyrosine-641) and STAT1 (Tyrosine-701), in homogenates from control- or glutamate-treated primary neurons. After 3 hours of glutamate challenge, p-STAT1 and p-STAT6 bound strongly to domains III, IV, and V. The signal in control-treated cells was considerably lower, supporting a role for p-STAT1 and p-STAT6 in regulating 12/15-LOX under glutamate-induced oxidative stress (Figure 1B). Binding to domains I and II was not detected, despite the presence of a putative STAT binding site in the second domain. Incubation with non-specific mouse IgG abolished the signal, while PCR analysis with 1% of input DNA confirmed equal loading in control and glutamate-treated samples.
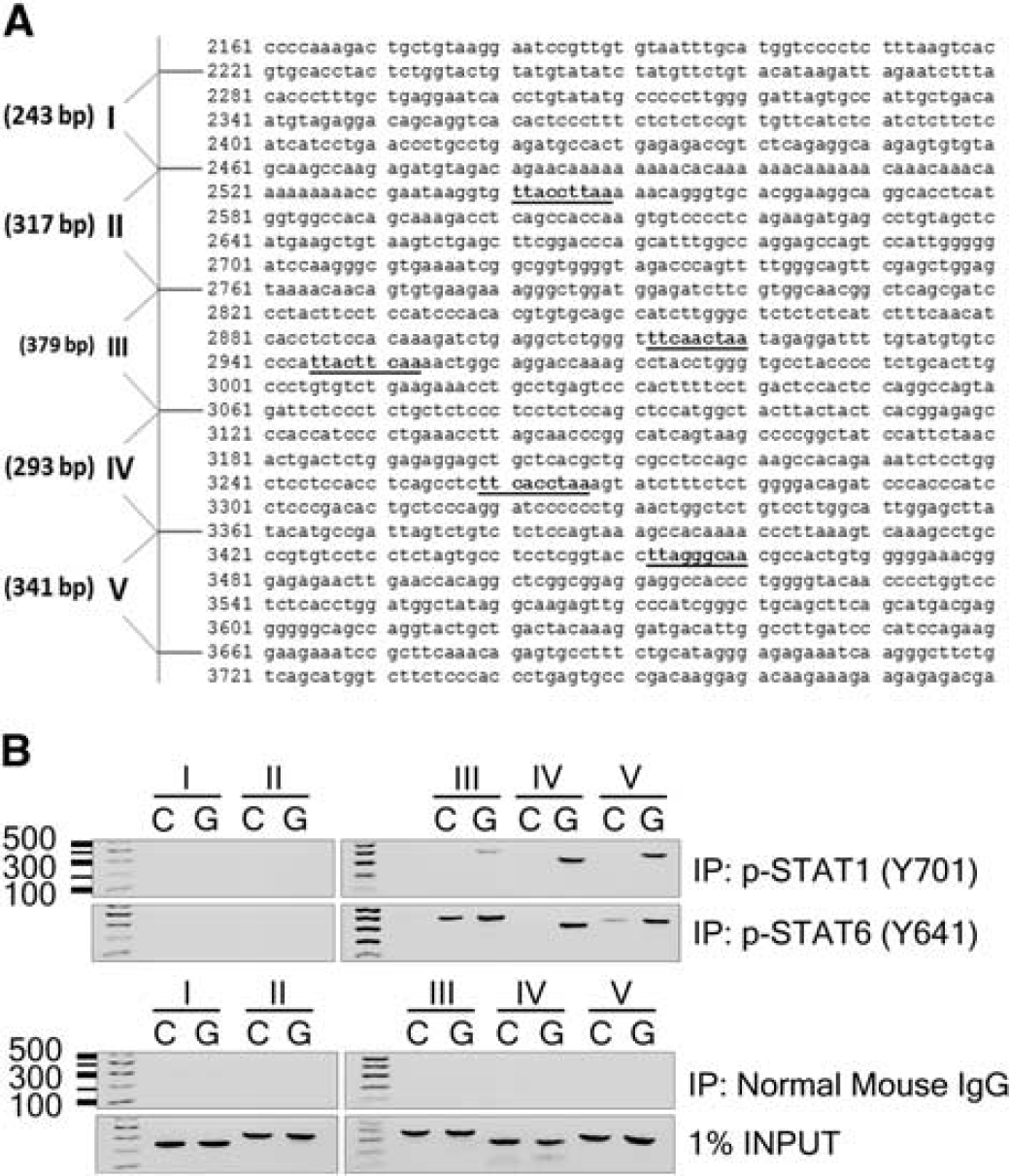
STAT1 and STAT6 as transcriptional regulators of 12/15-lipoxygenase (12/15-LOX) under oxidative stress. (
STAT1 and STAT6 Activate the 12/15-Lipoxygenase Promoter and Contribute to Cell Death in Murine HT22 Neuronal Cells
To confirm the upregulation of 12/15-LOX under glutamate oxidative stress, we performed a luciferase assay in the mouse hippocampal cell line HT22, which is also known to undergo 12/15-LOX dependent oxidative glutamate toxicity.23,26,27 Twenty-four hours after transfection with the pGL3-15-LOX-1 luciferase DNA construct containing the human 12/15-LOX promoter (a kind gift from Dr C Liu, Karolinska Institute 22 ), glutamate treatment for 24 hours led to an increase in luciferase luminescence, suggesting promoter activation (Figure 2A). We then used siRNAs directed toward STAT1 and STAT6 to investigate STAT involvement in 12/15-LOX upregulation. Transfection of HT22 cells for 24 hours with two different STAT1 siRNAs attenuated the increase of luciferase signal seen after glutamate treatment, although the reduction was not statistically significant (Figure 2B). STAT6 siRNA was more effective, leading to a significant decrease in luciferase signal. Finally, we determined the effect of the STAT-specific siRNAs on the survival of HT22 cells treated with glutamate by measuring cytosolic LDH released into the medium. Glutamate treatment led to massive release of LDH, signifying cell death, and this was reduced by siRNAs to either STAT1 or STAT6 (Figure 2C). Taken together these results show that targeting both STAT1 and STAT6 may be neuroprotective, although knockdown of STAT6 appears to overall have the greater effect.
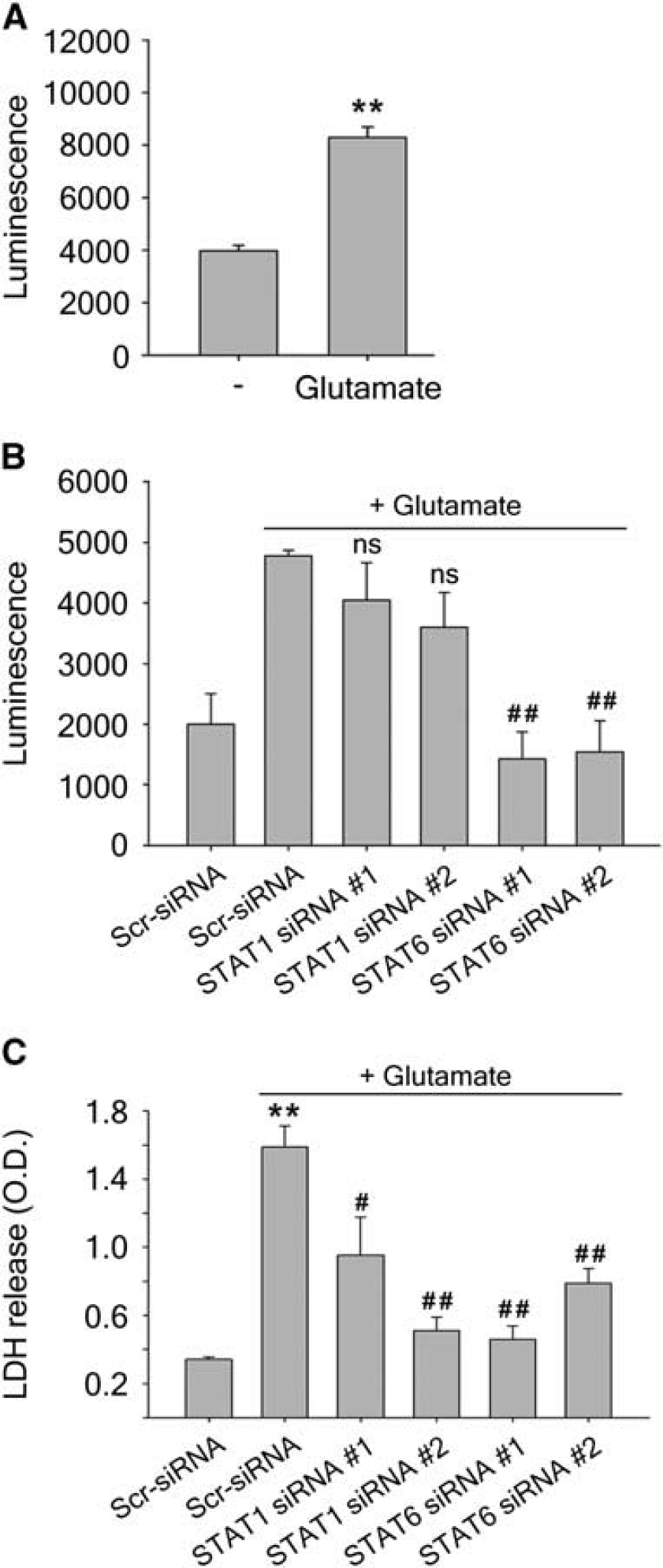
(
STAT1 and STAT6 Activation Contribute to Increased Levels of 12/15-Lipoxygenase Protein in Oxidatively Stressed Primary Neurons
To investigate the effect of STAT activation on 12/15-LOX expression on the protein level, we compared changes in expression over time after glutamate challenge by western blotting. We observed a gradual increase over time for 12/15-LOX (Figure 3A), which was preceded by increases in phosphorylation of STAT1 and STAT6 (Figure 3B), consistent with a role for these STATs in upregulating 12/15-LOX. To more directly gauge these effects, we used STAT1- and STAT6-specific siRNAs. These largely prevented the glutamate-induced increase of 12/15-LOX (Figures 3C and 3D).
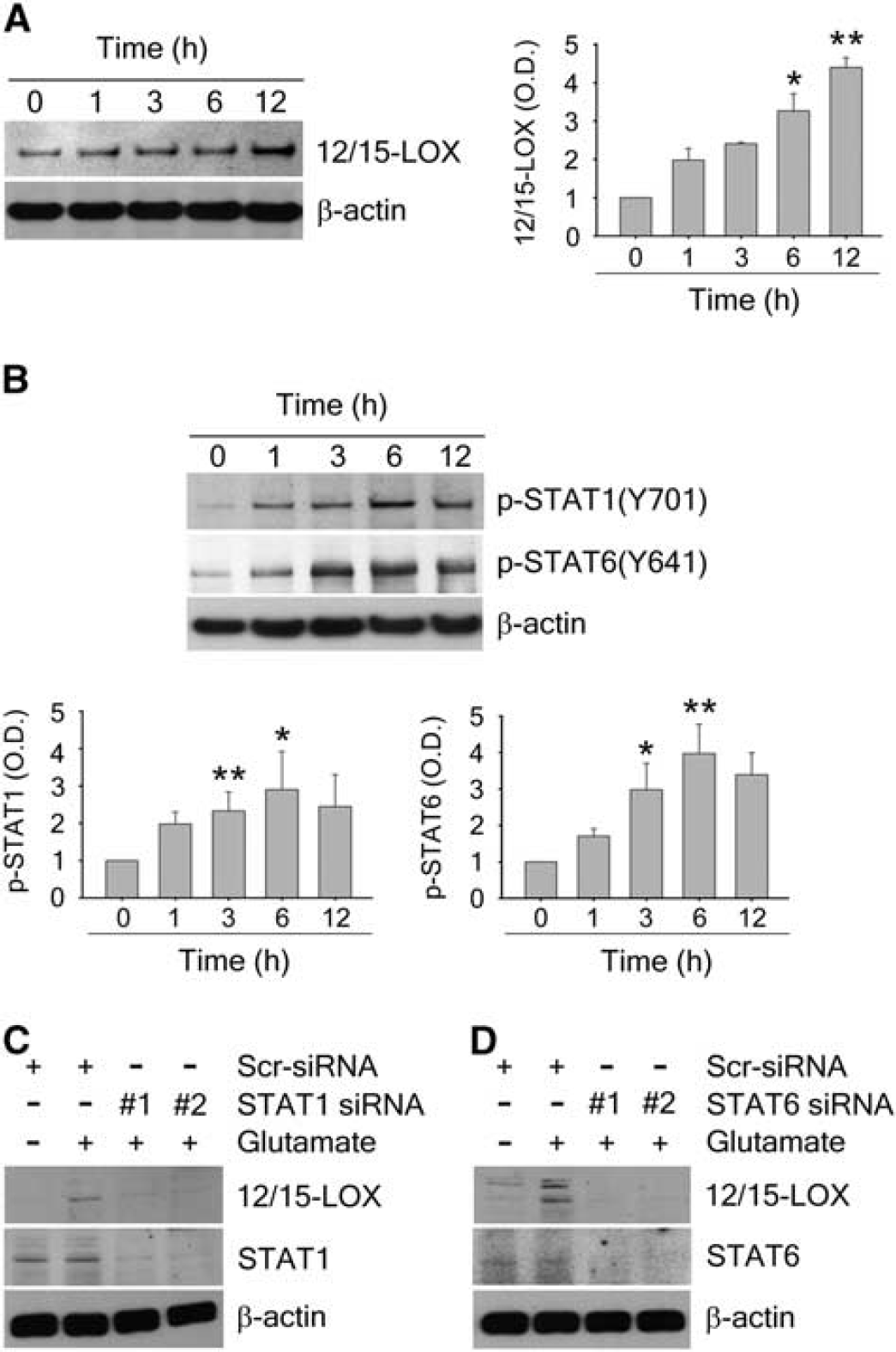
12/15-lipoxygenase (12/15-LOX) upregulation via STAT1 and STAT6 in cortical neurons treated with glutamate. (
STAT6 Regulates Increased 12/15-Lipoxygenase After Oxygen-Glucose Deprivation and Reoxygenation in Primary Neurons
A frequently used model of in vitro ischemia is OGD, followed by reoxygenation. To determine whether STAT-dependent regulation of 12/15-LOX occurs during OGD/R, we subjected mouse primary neurons to 2 hours of OGD and 12 hours of reoxygenation. While immunofluorescence showed only little 12/15-LOX staining in normoxic neurons transfected with a scrambled control siRNA, levels of 12/15-LOX were higher after OGD/R (Figure 4, left top and middle panels). In cells pretreated with STAT6-specific siRNA, the signal for STAT6 was clearly diminished, indicating efficient knockdown. These cells also showed reduced 12/15-LOX staining, confirming that increased 12/15-LOX depends on changes in STAT6. In these experiments, STAT1 knockdown was less effective, showing a clear reduction of 12/15-LOX in only one of three experiments (results not shown). Quantitative evaluation of the immunofluorescent images from three independent experiments confirmed both the increase of 12/15-LOX after OGD/R and the significant reduction in 12/15-LOX signal when STAT6 is knocked down (Figure 4B). To determine whether STAT6 knockdown affects viability after OGD/R in these cells, we treated a separate cohort of cells, and measured survival as LDH content in cell lysates, normalized to control-treated cells (Figure 4C).
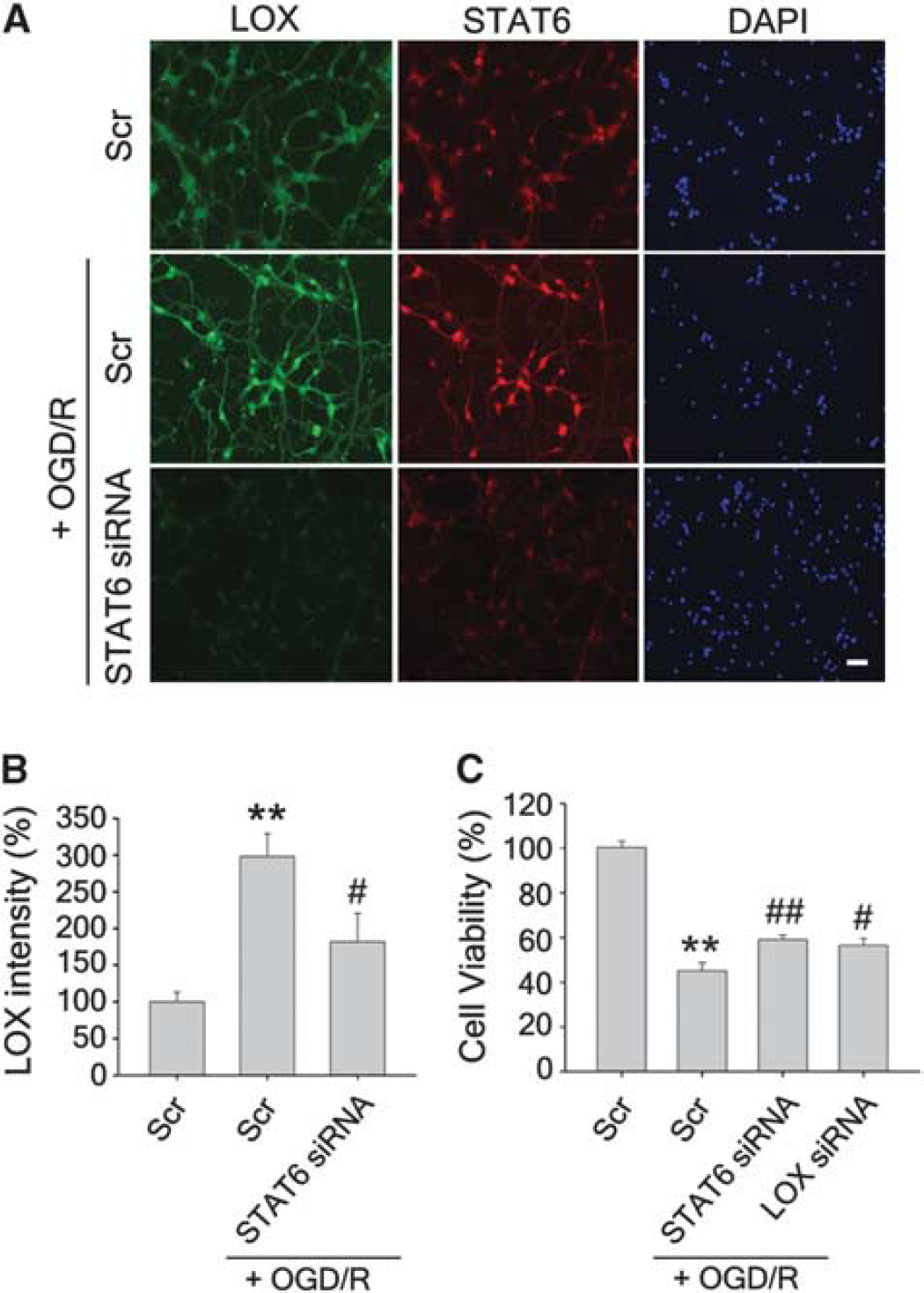
Increased 12/15-lipoxygenase (12/15-LOX) in primary cortical neurons subjected to oxygen-glucose deprivation and reoxygenation (OGD/R) is attenuated by knockdown of STAT6. (
Increase of 12/15-Lipoxygenase mRNA After Ischemia/Reperfusion in Mice Is Preceded by STAT Activation
In a mouse model of experimental stroke, we previously detected a time-dependent increase of 12/15-LOX in the peri-infarct cortex over 24 hours. 4 Expression of this enzyme is both transcriptionally and translationally regulated,22,28–31 so we decided to probe the mRNA levels of 12/15-LOX in ischemic mouse tissue. After transient focal ischemia with 45 minutes occlusion and 24 hours of reperfusion (Figure 5A), 12/15-LOX mRNA was significantly increased in the ischemic hemisphere as judged by real-time RT-PCR, indicating a transcriptional upregulation of 12/15-LOX in the ischemic mouse brain. Western blotting confirmed an early upregulation of p-STAT1 and p-STAT6, already at 1 and 3 hours of reperfusion after 45 minutes ischemia (Figures 5B and 5C). These findings are consistent with a role for STAT activation in regulating 12/15-LOX.
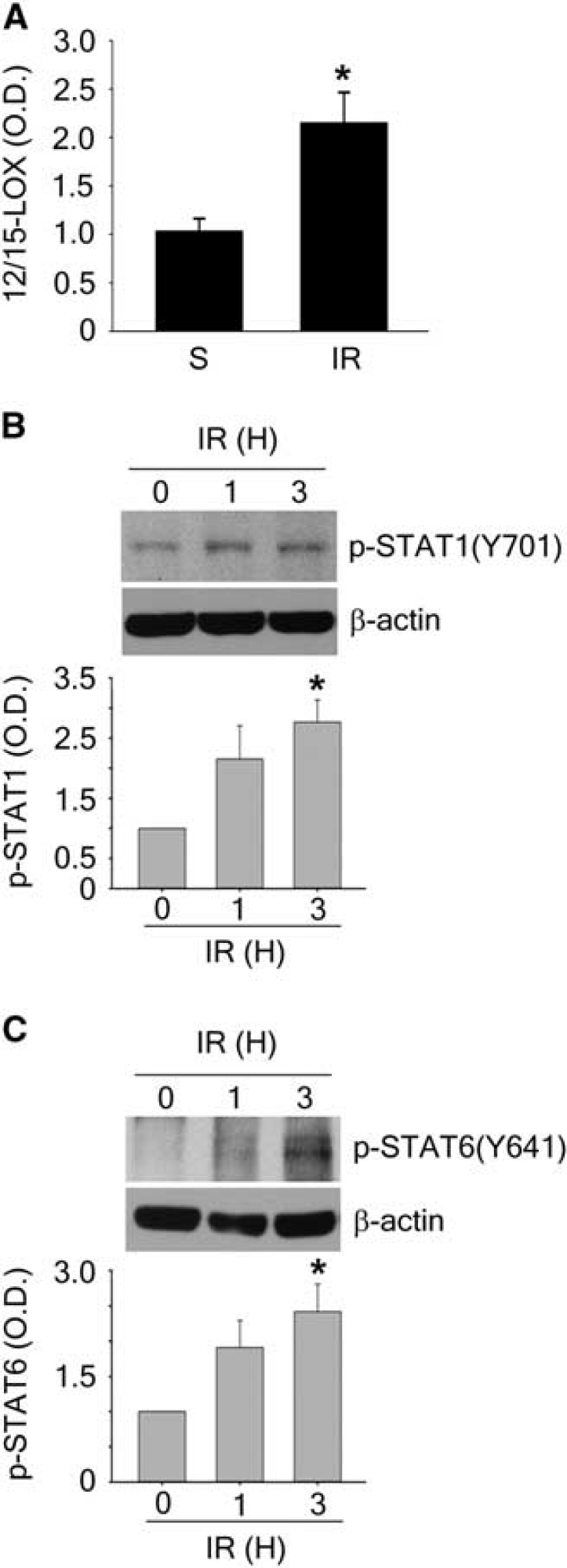
12/15-lipoxygenase (12/15-LOX) mRNA is increased after ischemia-reperfusion, which is accompanied by early phosphorylation of STAT1 and STAT6. (
Increased Levels of Activated p-STAT1 and p-STAT6 in Human Stroke
To determine the relevance of our findings for stroke in humans, we queried brain sections taken from stroke patients. As a measure of STAT activity in the brain after ischemia, we used antibodies to detect p-STAT1 and p-STAT6 in brain sections of a 77-year-old female patient with ischemic stroke. 3 Western blotting showed increased levels of p-STAT1 and p-STAT6, respectively, in the sample from the peri-infarct region compared with contralateral cortex (Figure 6A). These findings, which reflect increased activity of both of these transcriptional activators, were confirmed by immunohistochemistry (Figure 6B). From previous studies in mice and humans, we know that 12/15-LOX is increased in the peri-infarct region as well, coincident with the AIF.3–6 To determine a possible link to the disease state of the corresponding cells, we performed colocalization studies with sections taken from the peri-infarct cortex. Double labeling with the corresponding antibodies showed that p-STAT6 and p-STAT1 are increased in the same cells; p-STAT6 colocalizes with the oxidative stress marker MDA2, and with AIF; the same cells that show increased AIF also show high levels of 12/15-LOX; and the 12/15-LOX-positive cells are also positive for p-STAT1 (Figure 6C, top to bottom). Finally, both p-STAT6 and AIF showed partial nuclear colocalization, as shown by overlay with the DNA dye DAPI (Figure 6D, top panel). This is consistent with the transcriptional activity of STAT6, as well as the pro-apoptotic nuclear effects of AIF. 32 To confirm the validity of these findings, we also investigated the brain of a second patient, a 59-year-old male. Again, we found a colocalization of 12/15-LOX with p-STAT1, also with partial overlap with the nuclear DAPI dye (Figure 6D, bottom panel). Taken together, these findings document increased levels of p-STAT6 and p-STAT1 in the ischemic peri-infarct area, which coincide with increased 12/15-LOX and the marker for non caspase-related apoptosis, AIF.
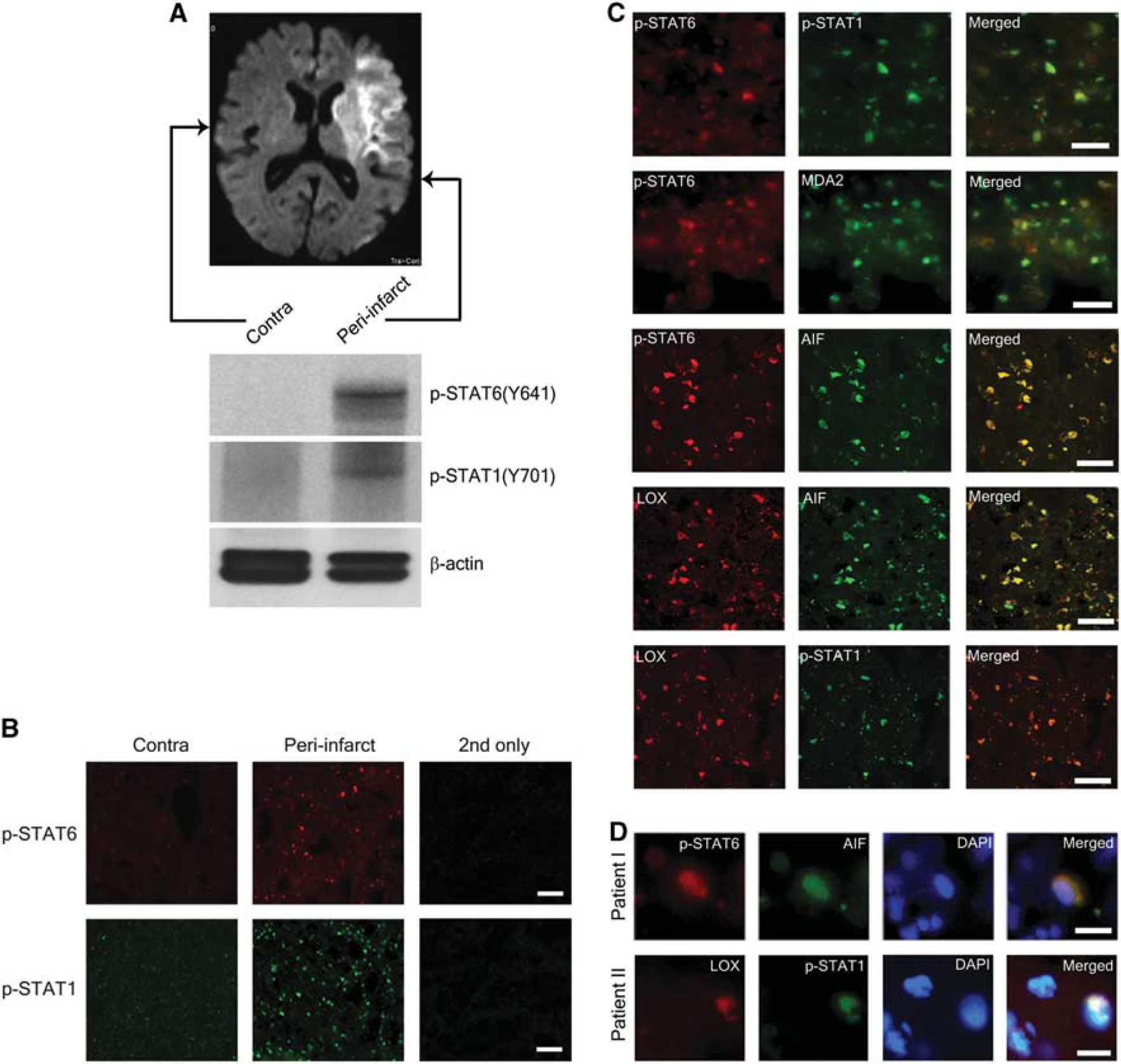
STAT6 and STAT1 phosphorylation is increased in the peri-infarct cortex of a human stroke patient, and colocalizes with 12/15-lipoxygenase (12/15-LOX). (
DISCUSSION
Oxidative stress is a major injury mechanism in ischemic strokes, and 12/15-LOX is one of its central mediators. This study for the first time shows how 12/15-LOX is upregulated via activation of STAT6 and STAT1 in cultured neurons, and in vivo after stroke.
12/15-LOX is an enzyme with great destructive potential due to its ability to directly oxidize phospholipids and damage intracellular organelles.33–35 Consequently, it is subject to several layers of regulation including transcriptional, translational, and posttranslational control mechanisms that keep the enzyme activity in check. 36 After a catastrophic event such as an ischemic stroke, this regulatory network breaks down, leading to increased 12/15-LOX protein levels. The catabolic activity of 12/15-LOX is further enhanced by increases in intracellular calcium, while the major antioxidant glutathione decreases, both of which are hallmarks of ischemic injury. The 12/15-LOX protein is found mostly in neurons and endothelial cells of the peri-infarct cortex after transient focal ischemia in mice, as well as ischemic stroke in humans. Our group and others have previously shown that blocking 12/15-LOX activity is powerfully neuroprotective, qualifying 12/15-LOX as a novel drug target for stroke. An alternative to block the enzymatic activity of 12/15-LOX might be to prevent its upregulation in the first place, which could potentially be similarly neuroprotective. Identifying the mode of regulation that contributes to increased levels of 12/15-LOX in the ischemic brain appeared to be a promising first step in this direction. Transcriptional regulation of 12/15-LOX in neuronal cells and the ischemic brain has to our knowledge not been investigated to date. Our study focused on the jak/stat pathway, because STATs are involved in a variety of redox regulatory steps. STAT1 has previously been implicated in causing brain injury in a mouse model of cerebral ischemia. 14 Early phosphorylation of STAT1 at Tyr701 was found 30 minutes after 2 hours transient focal ischemia in mice. STAT1 knockout mice featured reduced infarct sizes, along with reduced activation of caspase-3 and increased levels of protective phosphorylated AKT. 14 In proteomics studies, STAT6 phosphorylation was also reported to be increased in mice after experimental stroke,15,16 but with unknown consequences. In rats on the protein level, phosphorylated STAT6 was found to be increased in the penumbra compared with the infarct core, consistent with our current findings, but less so than on the contralateral side of the brain. 17 STAT6 regulates 12/15-LOX in airway epithelial cells and macrophages in response to IL-4 and IL-13,22,29–31,37 but neural cells had not been investigated in this context. These findings suggested that STAT1 and STAT6 might be promising candidates as transcriptional regulators of 12/15-LOX.
In the brain of a human stroke patient where we had previously found increased 12/15-LOX in the peri-infarct region, we now found that both p-STAT6 and p-STAT1 were increased as well (Figures 6A and 6B), and immunohistochemistry showed 12/15-LOX colocalized with p-STAT1 in what appeared to be mostly neurons (Figure 6C). We could not test the colocalization of 12/15-LOX with p-STAT6 in this case, because both antibodies were of rabbit origin. Due to the extensive overlap of 12/15-LOX with the same markers that p-STAT6 colocalized with, we nonetheless assume a mostly overlapping expression pattern. Moreover, p-STAT6 colocalized with the injury markers MDA2 and AIF, indicating that these are damaged cells. These increases were similarly found in mice after transient focal ischemia, where both mRNA levels of 12/15-LOX (Figure 5A), and phosphorylation of STAT6 and STAT1 were increased (Figures 5B and 5C). A direct binding of p-STAT1 and p-STAT6 to distinct regions of the 12/15-LOX promoter was then shown by ChIP analysis in primary cortical neurons subjected to oxidative stress (Figure 1). This was accompanied by an increase in 12/15-LOX promoter activity, which was somewhat reduced by cotransfection with STAT1-specific siRNA, and abolished by STAT6 siRNA (Figure 2B). Overall, the p-STAT6 appears to be a much stronger regulator of 12/15-LOX activation than STAT1. Consistent with a role for these STATs as transcriptional regulators of 12/15-LOX, increased phosphorylation of STAT6 and STAT1 temporally preceded increased 12/15-LOX protein in the oxidative glutamate toxicity model (Figures 3A and 3B), and STAT1 as well as STAT6 siRNAs prevented the increased protein levels of 12/15-LOX (Figures 3C and 3D). This was confirmed for STAT6 in the commonly used model of OGD/R, where the increased immunofluorescence signal after OGD/R was attenuated by prior transfection with STAT6 siRNA, whereas the effects of STAT1 siRNA were less consistent in this model. Significant protection against cell death was also provided in the OGD/R model by STAT6 knockdown, comparable to knockdown of 12/15-LOX itself (Figure 4C). Both of these knockdowns did not provide complete protection however, suggesting other triggers of cell death besides 12/15-LOX operate in the OGD/R model. Finally, cell death in the oxidative glutamate toxicity model of oxidative stress was reduced by siRNAs directed against STAT1 or STAT6 (Figure 2C). Taken together, these results clearly show the transcriptional regulation of 12/15-LOX via STAT6. How important the role of STAT1 is in regulating 12/15-LOX remains to be established in further studies.
While we have thus shown conclusively that 12/15-LOX is regulated by these STATs, suggesting that they may be viable targets for neuroprotection, several caveats need to be considered. (1) While we have focused here on transcriptional regulation via STAT phosphorylation, transcriptional silencers such as GATA-6 and hypermethylation of CpG islands in the 12/15-LOX promoter may also have a role in determining the amount of 12/15-LOX protein. In addition, translational control via RNA-binding proteins including hnRNP K and hnRNP E1 is also known for 12/15-LOX, 28 and should be investigated. The increased 12/15-LOX mRNA levels point to a dominant effect of transcriptional events, however. (2) Both STAT1 and STAT6 will likely regulate additional genes in the ischemic brain, including the NADPH oxidases NOX1 and NOX4, 38 and the cyclooxygenase COX-2. 39 Furthermore, AKT phosphorylation was increased, and caspase-3 cleavage decreased in STAT1 knockouts subjected to focal ischemia, 14 and the impact of 12/15-LOX on these events should be explored. However, this broad spectrum of STAT-dependent regulatory activity may also facilitate reining in several damage pathways that can otherwise contribute to ischemic oxidative stress, apoptosis, and inflammatory reactions after a stroke. (3) Because both STAT6 and STAT1 can contribute to upregulation of 12/15-LOX, both of these STATs may need to be targeted, although in our experiments STAT6 appeared to be the more crucial target. Unfortunately, very few STAT-specific inhibitors are available at present. 40 (4) As always when considering acute stroke therapy, timing is a major issue. Our findings concerning early activation of STAT6 and STAT1, which confirm earlier reports, suggest that the time window after onset of stroke may be fairly limited. Nonetheless, our study highlights the need for development of more STAT-selective inhibitors, which can be used to investigate specific effects ofeach STAT and may lead to new approaches to stroke therapy.
Footnotes
JEJ and KvL planned and directed the experiments. JEJ performed the in vitro and cell culture experiments. HK and YL performed the focal ischemia experiments. AY helped with the gene expression experiments. JM supplied the human brain sections and helped with interpretation of the results. JEJ, EHL, and KvL evaluated the results and, with input from all authors, wrote the article.
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Drs C Liu, D Xu, and J Sjöberg of the Karolinska Institute, Sweden, for the human 15-LOX promoter construct. The authors thank Dr JL Witztum for a kind gift of the MDA2 antibody, and Dr J Cornicelli for the sheep antiserum to 15-LOX. Support from the National Institutes of Health (R01 NS069939 and R01 NS049430 to KvL) is gratefully acknowledged.