
Abstract
Soluble epoxide hydrolase (sEH) contributes to cardiovascular disease, including stroke, although the exact mechanism remains unclear. While primarily a cytosolic enzyme, sEH can translocate into peroxisomes. The relevance of this for stroke injury is not understood. We tested the hypothesis that sEH-mediated injury is tied to the cytoplasmic localization. We found that a human sEH variant possessing increased affinity to peroxisomes reduced stroke injury in sEH-null mice, whereas infarcts were significantly larger when peroxisomal translocation of sEH was disrupted. We conclude that sEH contributes to stroke injury only when localized in the cytoplasm, while peroxisomal sEH may be protective.
Keywords
INTRODUCTION
Soluble epoxide hydrolase (sEH), which inactivates neuroprotective arachidonic acid derived epoxyeicosatrienoic acid (EET), has been implicated in multiple cardiovascular diseases, including stroke. 1 Gene deletion of sEH or pharmacological inhibition of its hydrolase activity provides robust protection from experimental stroke injury.2,3 The interest in the role of sEH in stroke was further enhanced by the observation that common human single-nucleotide polymorphisms (SNPs) of sEH affect its hydrolase activity and ability to inactivate protective EET. 4 A human sEH variant that exhibits reduced hydrolase activity in vitro, the R287Q SNP, is associated with a decrease in stroke incidence. 5 The same R287Q variant improves neuronal survival after in vitro ischemia, 4 further emphasizing the relevance for sEH in stroke injury.
sEH is a complex protein, however, and the precise mechanism by which it contributes to stroke injury remains poorly understood. Besides the hydrolase moiety that inactivates EET, sEH also contains a lipid phosphatase domain, 6 whose biologic function remains less clear. In addition, recent studies suggest that sEH is not an exclusively cytosolic enzyme, as previously thought, but possesses a peroxisomal translocation sequence (PTS), which allows sEH import from the cytoplasm into the peroxisomes. 7 Interestingly, human SNPs can influence subcellular localization of sEH, such that the R287Q variant is preferentially localized in the peroxisomes, whereas the wildtype enzyme is present in both the cytoplasm and peroxisomes. 8 Cytoplasmic sEH decreases rapidly after stroke, 9 suggesting that sEH might translocate to the peroxisomes in response to ischemia as part of an endogenous protective pathway reducing cytoplasmic epoxide hydrolase activity. However, the relevance of peroxisomal translocation and subcellular localization of sEH for stroke injury has not yet been studied.
We hypothesized that translocation of cytoplasmic sEH to the peroxisomes is beneficial after stroke. To test this, we used a unique model of TAT-protein transduction domain mediated protein transfer to introduce human sEH variants with intact or defective peroxisomal targeting sequence into sEH-null mice before exposing them to experimental stroke.
MATERIALS AND METHODS
Expression and Purification of TAT-Fusion Protein
Plasmids for TAT-human sEH fusion proteins were generated by subcloning WT (TAT-sEH-WT) and mutant R287Q (TAT-sEH-287) sEH cDNAs into the pTAT2.1 vector, as previously described. 4 PCR-mediated deletion was used to remove the PTS of TAT-sEH-WT to create translocation deficient TAT-sEH-ΔPTS (Figure 1A). Deletion of the PTS was verified by sequencing. Recombinant proteins were expressed and purified as previously described. 4 Concentrations of purified fusion proteins were measured with a Bradford assay (Bio-Rad, Hercules, CA, USA). Protein purity and specificity was confirmed by immunoblotting.
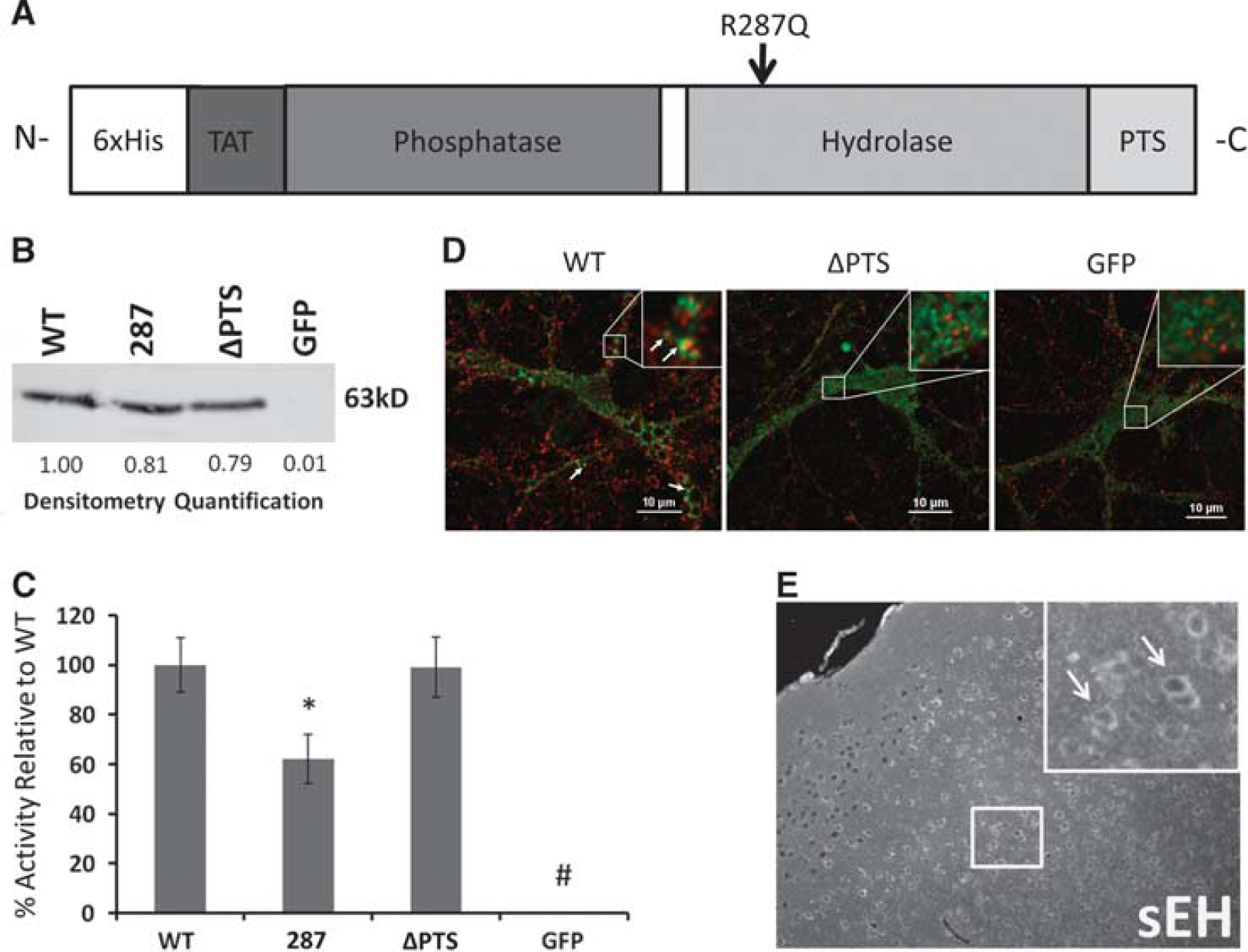
Characterization of recombinant TAT-fusion proteins. (
Immunoblot and Hydrolase Assay
Immunoblot was performed as previously described 2 with 1:250 anti-sEH primary antibody (Cayman Chemical, Ann Arbor, MI, USA) and 1:2,000 Cy5-conjugated secondary antibody (GE Healthcare, Little Chalfont, Buckinghamshire, UK) on a Typhoon imager (GE Healthcare). Densitometry analysis was performed using the ImageQuant software (GE Healthcare). Hydrolase enzyme activity of fusion proteins was quantified by EP7 assay (Cayman Chemical) as previously described. 10
Neuronal Culture and Immunocytochemistry
Mouse primary neurons were cultured as previously described. 11 On DIV 10, neurons were transduced with 0.5 μmol/L final concentration of the TAT-fusion proteins and washed and fixed 2 hours later. Peroxisomes were detected with a goat anti-PMP70 antibody (1:200; Abcam, Cambridge, MA, USA) and Alexa Fluor 488-conjugated secondary antibody (Life Technologies, Carlsbad, CA, USA), while TAT-fusion proteins were detected using a rabbit anti-6xHis antibody (1:2,500; Rockland, Limerick, PA, USA) and Alexa Fluor 594-conjugated secondary antibody (Life Technologies). Confocal images were acquired on an A1R+ microscope (Nikon, Melville, NY, USA) with laser power and detector gain held constant between images.
Animals and Treatment Groups
All experiments were performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Oregon Health and Science University. Male sEH knockout (sEHKO) mice (20 to 26 g) were obtained from our in-house colony. 3 Mice (7 to 8 per group) were randomly assigned to treatment with different TAT-sEH variants, TAT-green fluorescent protein (GFP), or vehicle. Investigators were blinded to treatment allocation. Mice received injections of TAT-fusion proteins 2 hours before 60 minutes of focal stroke, followed by infarct size analysis 24 hours later (Figure 2A). Experiments and results are reported according to the ARRIVE guidelines (www.nc3rs.org.uk).
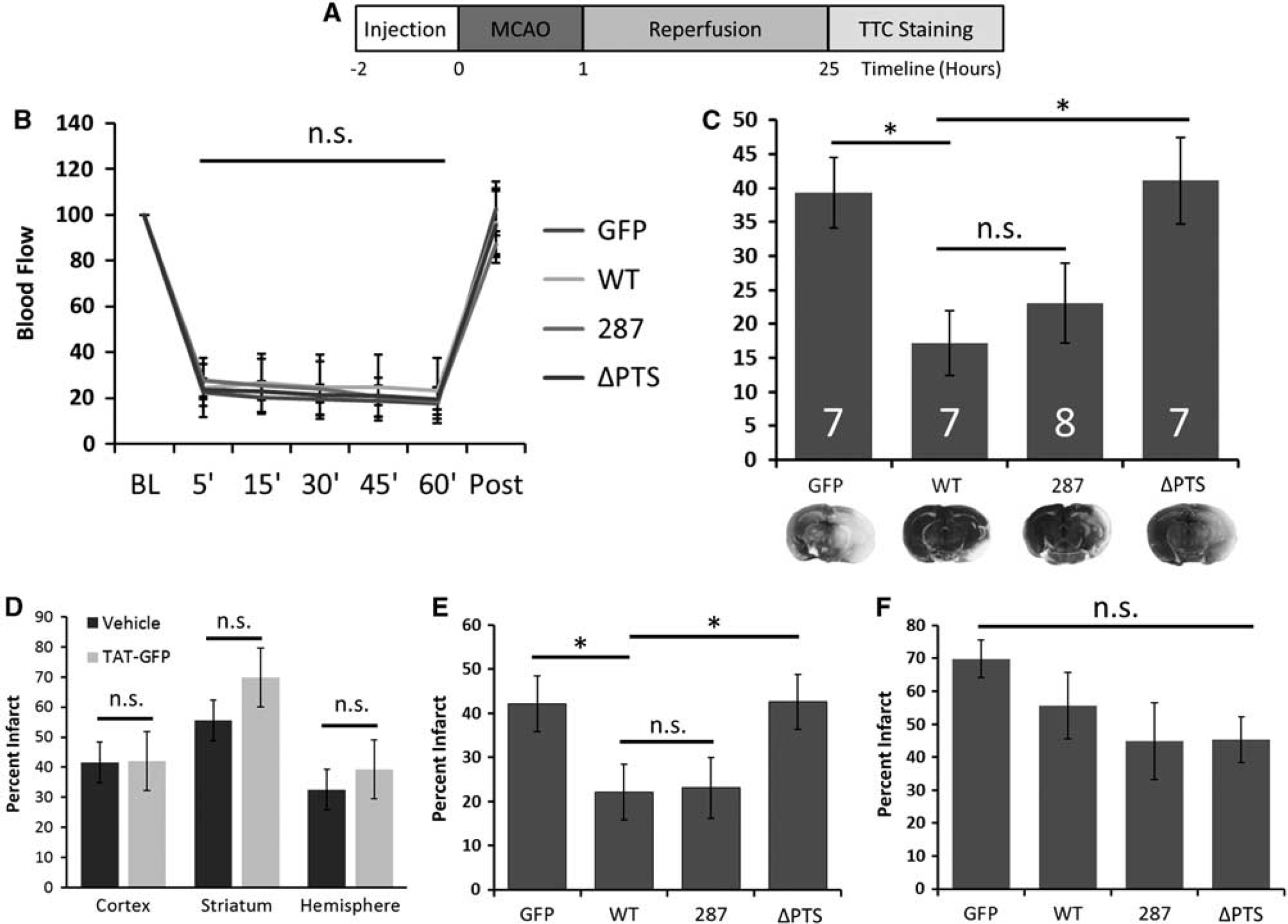
Peroxisomal localization of soluble epoxide hydrolase (sEH) protects against ischemic injury. (
TAT-Soluble Epoxide Hydrolase Injection and Middle Cerebral Artery Occlusion
Mice were anesthetized with isoflurane (1.5% to 2% by face mask). A PE-10 catheter was inserted into the right internal jugular vein and 0.5 nmol of TAT-sEH or TAT-GFP in 500 μL vehicle, or vehicle alone, was slowly injected. Rectal temperature was monitored and normothermia was maintained throughout the experiment using a heating lamp.
Ischemia was induced 2 hours after TAT-sEH injection using the intraluminal filament model of transient middle cerebral artery occlusion (MCAO). 12 A silicone-coated 6-0 nylon monofilament was inserted into the right internal carotid artery via the external carotid artery and advanced until the MCA was occluded. Ischemia was confirmed by laser-Doppler monitoring. After 60 minutes of MCAO, the occluding filament was withdrawn and reperfusion verified by laser-Doppler monitoring. Anesthesia was terminated and mice were allowed to recover. One mouse (TAT-sEH-WT) was excluded because MCAO could not be achieved.
A separate cohort of mice (n= 5 per treatment; TAT-GFP, TAT-sEH-WT, and TAT-sEH-ΔPTS) was instrumented with a PE-10 catheter in the right femoral artery before induction of MCAO to allow continuous monitoring of arterial blood pressure throughout the experiment. Blood samples were taken 30 minutes after reperfusion to measure arterial blood gases, pH, and glucose.
Infarct Size Analysis
Infarct size was measured 24 hours after MCAO using 2,3,5-triphenyltetrazolium chloride (TTC) staining and digital image analysis, as previously described. 2
Immunohistochemistry
To confirm brain penetration of systemically provided TAT-sEH fusion proteins, sEHKO mice were perfused with 4% paraformaldehyde 8 hours after injection with TAT-sEH-WT protein. Brains were paraffin embedded and 6 μm coronal sections were cut. Sections were deparaffinized and stained with sEH antibody (1:200; H-215, Santa Cruz Biotechnology, Dallas, Texas, USA), followed by biotin-labeled goat-anti-rat secondary antibody (1:150) and Cy-3 linked streptavidin (1:700, both from GE Healthcare).
Statistical Analysis
All values are mean ± s.e.m. Statistical analysis was performed using SigmaStat (Systat Software Inc., San Jose, CA, USA). Hydrolase activity of TAT-sEH fusion proteins was compared using one-way analysis of variance (ANOVA) for repeated-measures followed by SNK post hoc test. For infarct analysis, one-way ANOVA was used followed by Holm-Sidak post-hoc test.
RESULTS
Characterization of TAT-Human Soluble Epoxide Hydrolase Fusion Proteins
To test whether peroxisomal translocation is required for sEH effects on ischemic injury, we generated TAT-fusion proteins containing wildtype human sEH (TAT-sEH-WT) as well as a naturally occurring human variant sEH containing the R287Q SNP (TAT-sEH-287). The R287Q variant was previously shown to be localized preferentially in peroxisomes. 8 We also created a synthetic variant of human sEH engineered to lack the ability to translocate to the peroxisomes through deletion of its PTS (TAT-sEH-ΔPTS). We used TAT fused to GFP (TAT-GFP) as a control for unspecific effects of the TAT domain.
Immunoblotting for sEH confirmed that purity of all three recombinant TAT-sEH variants was similar (Figure 1B). Protein concentrations were adjusted according to quantification of the immunoblots to ensure that equivalent amounts of sEH protein were used in all downstream applications. Hydrolase activity assay confirmed that all three TAT-sEH fusion proteins were catalytically active (Figure 1C). As expected, and previously reported, 4 hydrolase activity of equimolar amounts of the R287Q sEH variant was only 60% of the hydrolase activity seen in WT-sEH. Deletion of the PTS, in contrast, did not affect hydrolase activity (Figure 1C).
We confirmed the subcellular localization of the different TAT-sEH variants in primary cultured neurons. We found that while TAT-sEH-WT partially colocalized with peroxisomal marker PMP70, this colocalization was absent in neurons transduced with TAT-sEH-ΔPTS, confirming that deletion of the PTS abolishes the ability of sEH to translocate to the peroxisomes (Figure 1D). 8
Finally, we confirmed that the TAT-protein transduction domain effectively transported sEH-fusion proteins across the blood–brain barrier after systemic administration. We easily detected TAT-sEH-WT in parenchymal brain cells, presumably neurons (Figure 1E), which is consistent with previous reports using TAT-mediated protein transduction to introduce neuroprotective proteins into the central nervous system. 13
Wildtype Human Soluble Epoxide Hydrolase Reduces Infarct Size in Soluble Epoxide Hydrolase Knockout Mice
Cerebral blood flow during ischemia and reperfusion was not different between treatment groups, suggesting that restoration of sEH did not alter blood flow (Figure 2B). Similarly, arterial blood pressure, blood gases, pH, and glucose levels were not different between groups (data not shown). Infarct size was significantly reduced in sEHKO mice receiving wildtype human sEH (TAT-sEH-WT), compared with mice injected with control TAT-GFP (17 ± 5% of contralateral hemisphere TAT-sEH-WT versus 39 ± 5% TAT-GFP, P < 0.05; Figure 2C). Mice treated with TAT-sEH-287 enjoyed similar reduction in infarct size as mice receiving TAT-sEH-WT (23 ± 6% of contralateral hemisphere, Figure 2C). In contrast, mice treated with translocation-deficient TAT-sEH-ΔPTS had significantly larger infarcts compared with mice treated with TAT-sEH-WT (41 ± 6% of contralateral hemisphere, P < 0.05 versus TAT-sEH-WT, Figure 2), similar to sEHKO mice treated with TAT-GFP control. To exclude a toxic effect of the TAT-construct itself, we also analyzed infarct size in animals injected with vehicle only. There was no difference in infarct size between sEHKO mice injected with vehicle versus TAT-GFP, suggesting that the TAT-construct was not toxic (hemisphere: 33 ± 4% vehicle versus 39 ± 5% TAT-GFP; cortex: 42 ± 5% vehicle versus 42 ± 6% TAT-GFP; striatum: 56 ± 5% vehicle versus 70 ± 6% TAT-GFP; P = 0.3; Figure 2D). Protection by TAT-sEH was mostly due to a reduction in infarct size in the penumbra (cortical infarction, Figure 2E, P < 0.05), rather than the infarct core (striatal infarction, Figure 2F, P = 0.2).
DISCUSSION
This study has two main findings. First, introduction of human sEH into a mouse that is genetically deficient for sEH reduces infarct size after ischemic stroke. Second, this protection requires an intact peroxisomal targeting sequence (PTS), suggesting that protection depends on the presence of sEH in the peroxisomes.
The function of sEH that is best understood is the hydration of EET, cytochrome P450 epoxygenase metabolites of arachidonic acid, to inactive DHET. Since EET are both vasodilators and anti-inflammatory mediators, an increase in net concentrations of EET is thought to account for many of the beneficial effects achieved by inhibition or deletion of sEH in cardiovascular disorders, including stroke.1–3,9 Indeed, we found previously that protection against stroke afforded by inhibition of sEH depends on EET production and can be recapitulated by infusion of EET. 2 As cytosolic levels of sEH decrease rapidly after stroke, 9 we speculated that sEH may be sequestered away from cytoplasmic EET into other cellular compartments as part of an endogenous protective mechanism. The recent description of a PTS in sEH raised the possibility that cytoplasmic sEH may move to the peroxisomes after ischemia.
We used an sEH variant without a PTS to selectively restore cytoplasmic sEH in sEHKO mice to test the hypothesis that peroxisomal localization of sEH reduces ischemic injury. Surprisingly, transduction with this sEH variant did not increase stroke injury beyond controls treated with nonfunctional TAT-GFP or with vehicle. Hydrolase activity of TAT-sEH-ΔPTS is intact, which should have increased infarct size if cytoplasmic hydrolase activity was the main contributor to stroke injury. In contrast, transduction of TAT-sEH-WT with an intact PTS into sEHKO mice significantly reduced infarct size, contrary to expectations. This suggests that translocation of sEH from the cytoplasm to the peroxisomes is indeed protective after stroke, but not by passive sequestration of sEH away from its cytoplasmic substrate EET, but rather by an active protective function of sEH within the peroxisomes.
Peroxisomes are at the center of reactions such as β-oxidation of fatty acids that produce large amounts of ROS and thus harbor several antioxidant systems, such as catalase and superoxide dismutase. Oxidative stress during ischemia/reperfusion may increase production of highly reactive epoxides from long-chain fatty acid in the peroxisomes. Rapid detoxification of these fatty acid epoxides by peroxisomally localized sEH may be necessary to limit injury after ischemia.
Our study has limitations. We introduced TAT-sEH constructs into an sEH knockout background. This allowed us to selectively compare different sEH variants in the absence of confounding wildtype enzyme, unmasking consequences of differential subcellular localization of sEH variants that were not detected in our previous work using sEH variants on a wildtype background. 4 However, the chronic deprivation of sEH in the knockout background may have produced effects that differ from a normal mouse, including downregulation of EET downstream signaling pathways. Other epoxides hydrolyzed by sEH, such as omega-3 epoxides, 14 may also be altered in this system. Despite these limitations, we believe that our observation of a protective sEH effect adds to the field and sheds additional light on the complex functions of this enzyme.
Our study describes for the first time a set of circumstances in which sEH has beneficial and protective functions. Our observation that sEH can reduce stroke injury adds to the field, as sEH so far has been viewed exclusively as a detrimental enzyme. Furthermore, this is the first study to examine the functional significance of the sEH PTS. Our observation is timely, as such incomplete understanding of sEH functional relevance may have contributed to the recent failure of the first clinical trial of sEH inhibition to treat hypertension. 15 We hope that our novel findings will help understand the complex function of sEH in stroke and spark further investigations to unravel the significance of subcellular localization of sEH for ischemic injury toward successful translation into clinical practice in the future.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.